


Transcriptome Profiling of a Soybean Mutant with Salt Tolerance Induced by Gamma-ray Irradiation

 ,
, 
Abstract
1. Introduction
2. Results
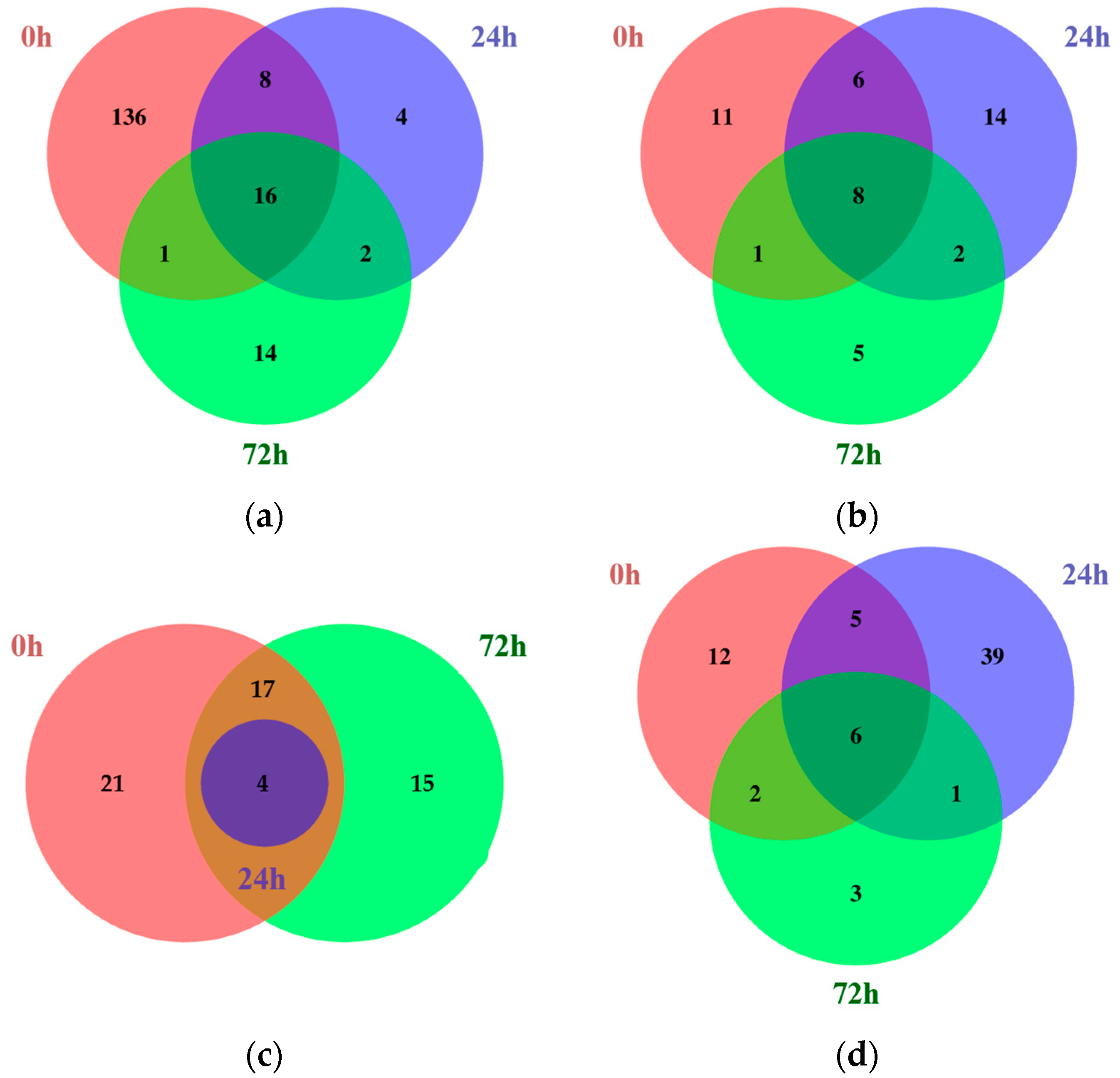
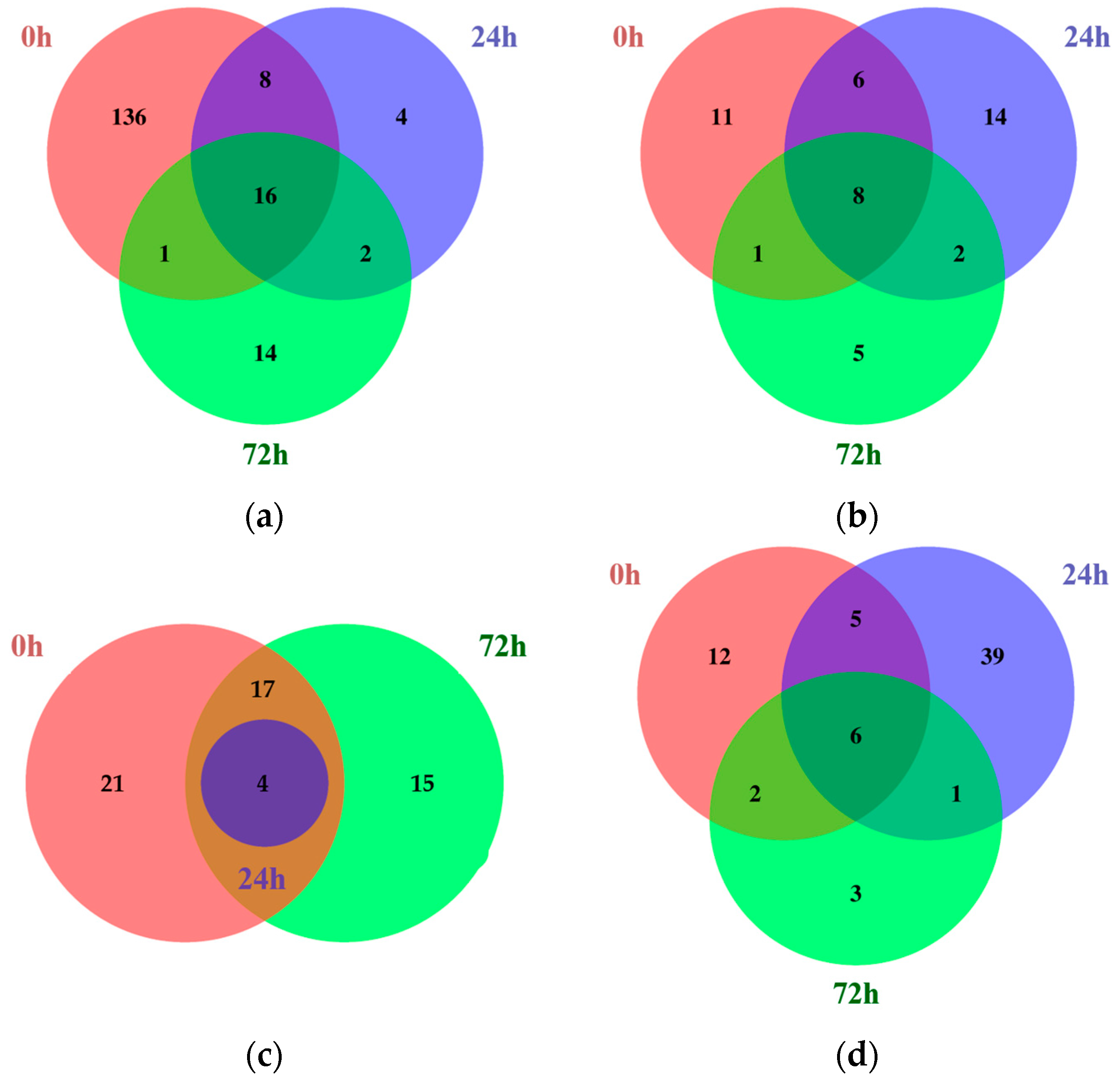
2.1. Identification of Differentially Expressed Genes Using RNA Sequencing
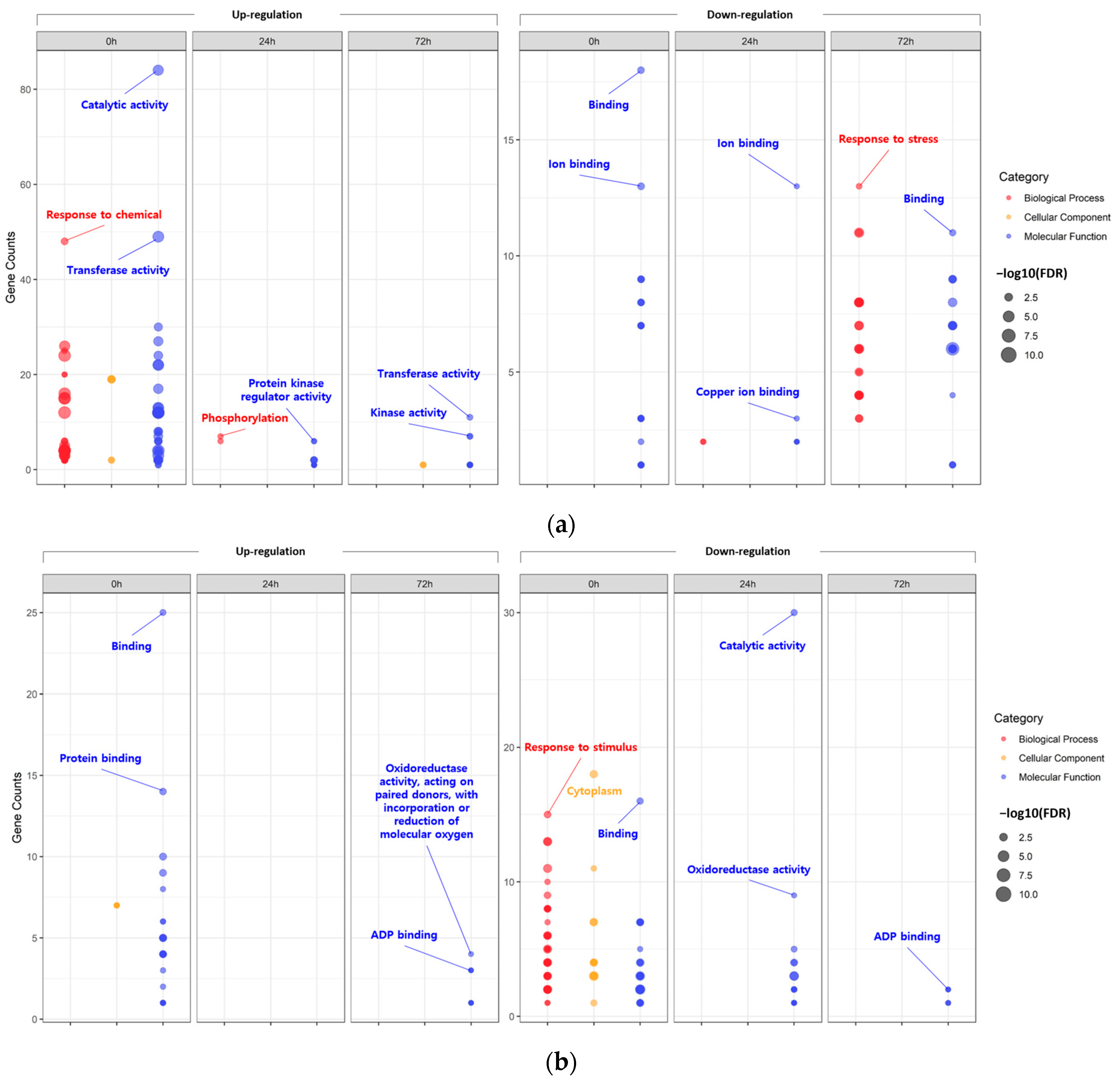
2.2. Functional Annotation of Identified DEGs Induced by Salt Stress
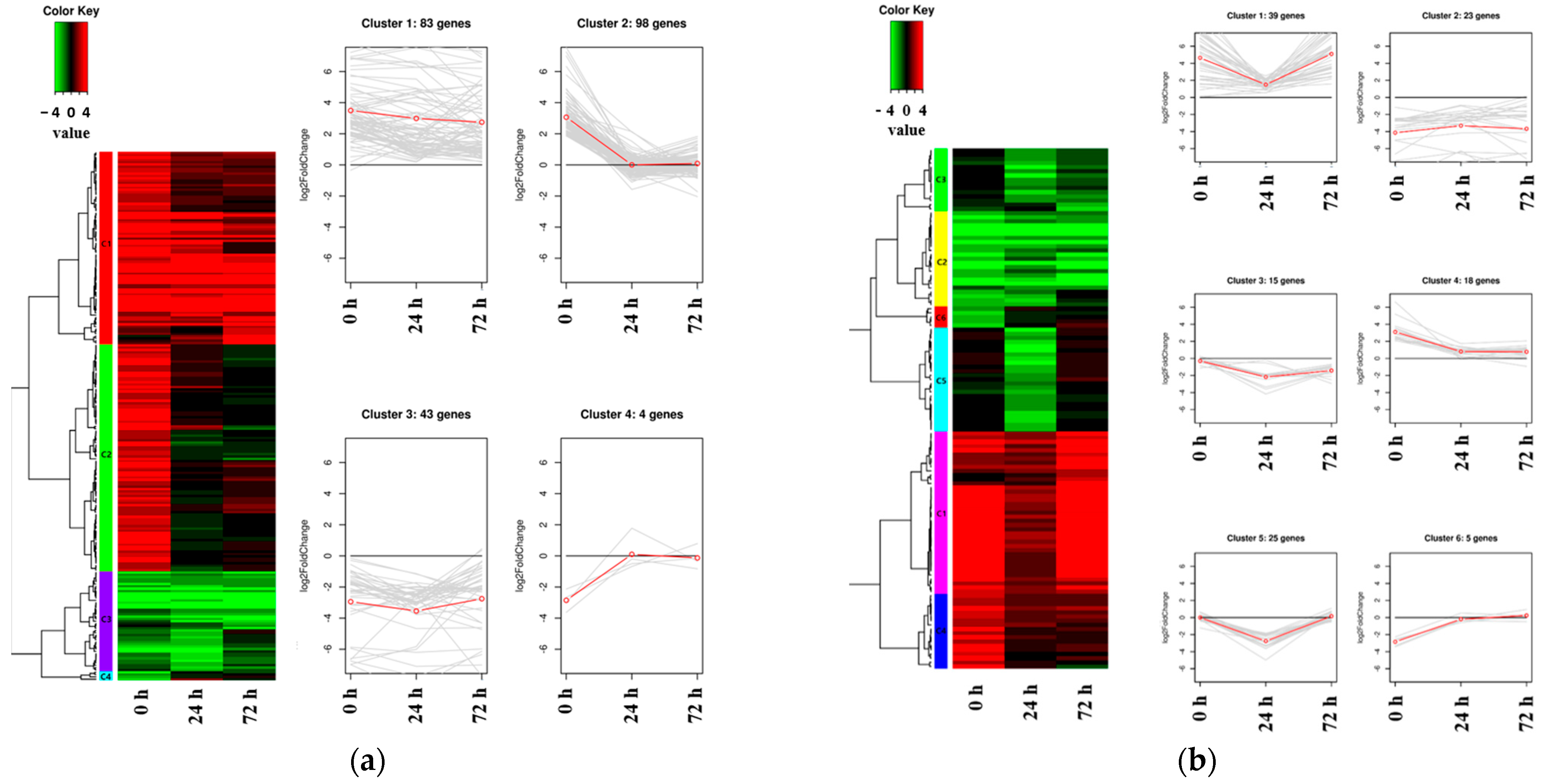
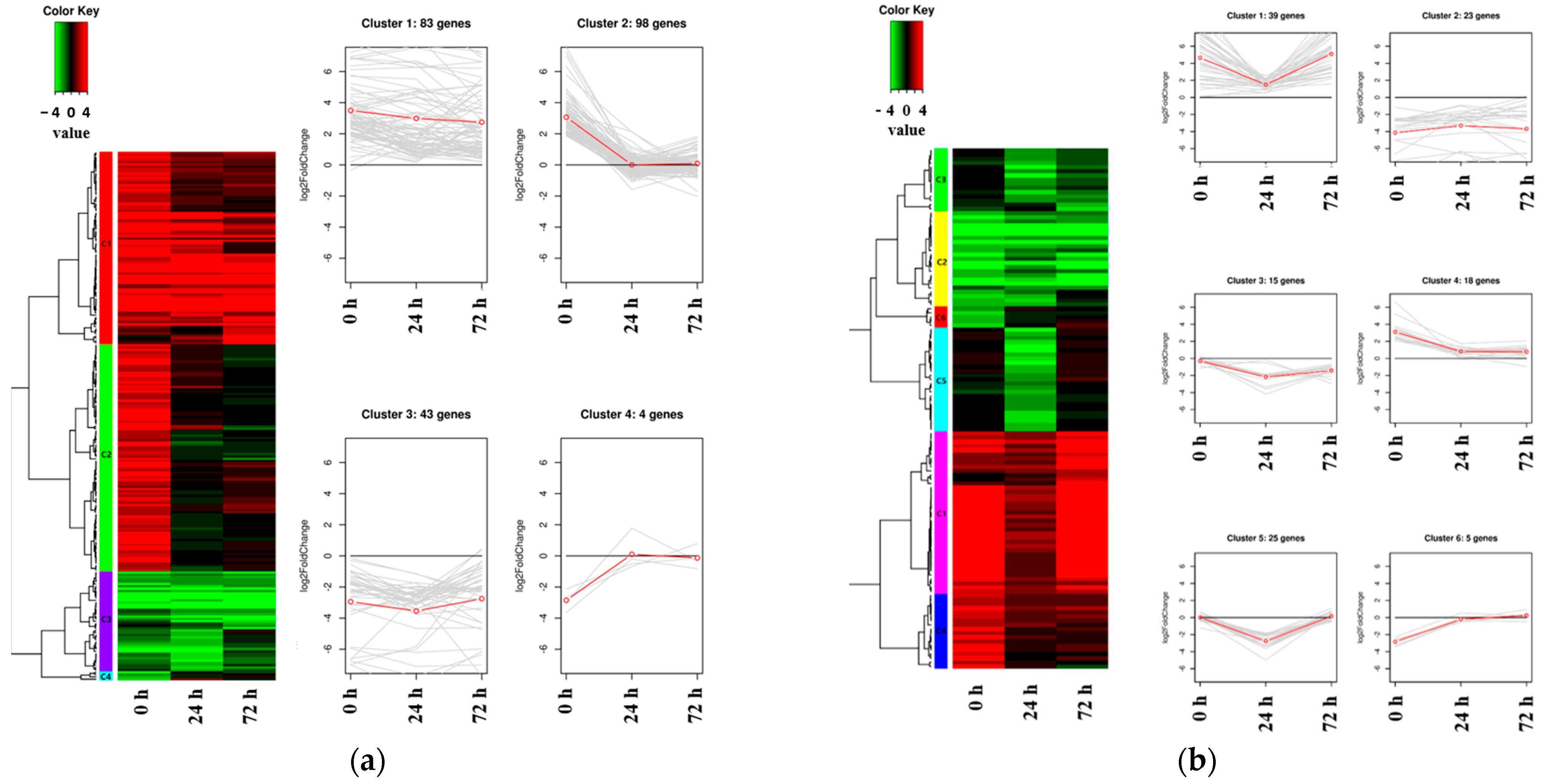
2.3. The Expression Patterns of DEGs Identified in the Salt-Tolerant Mutant
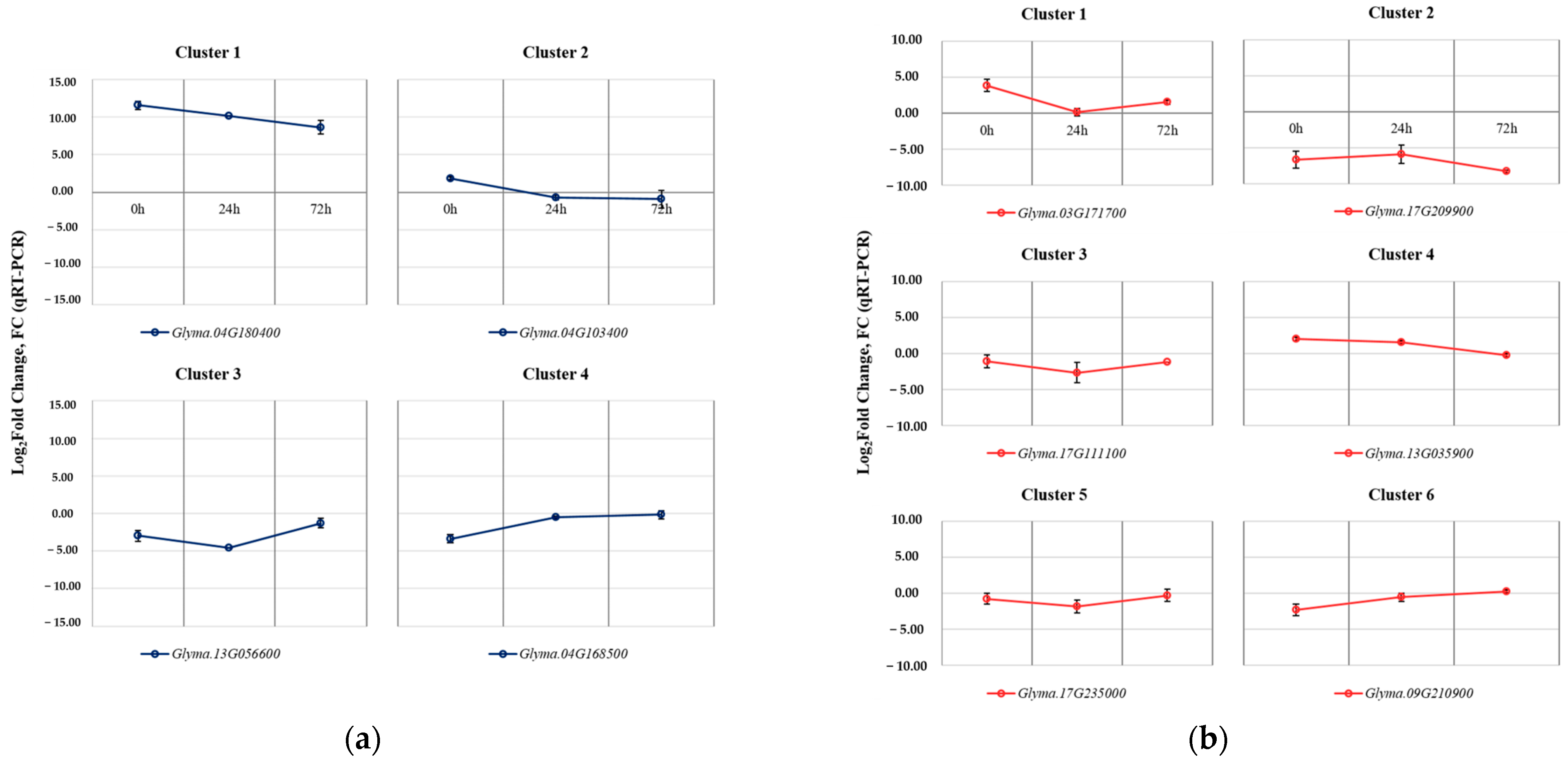
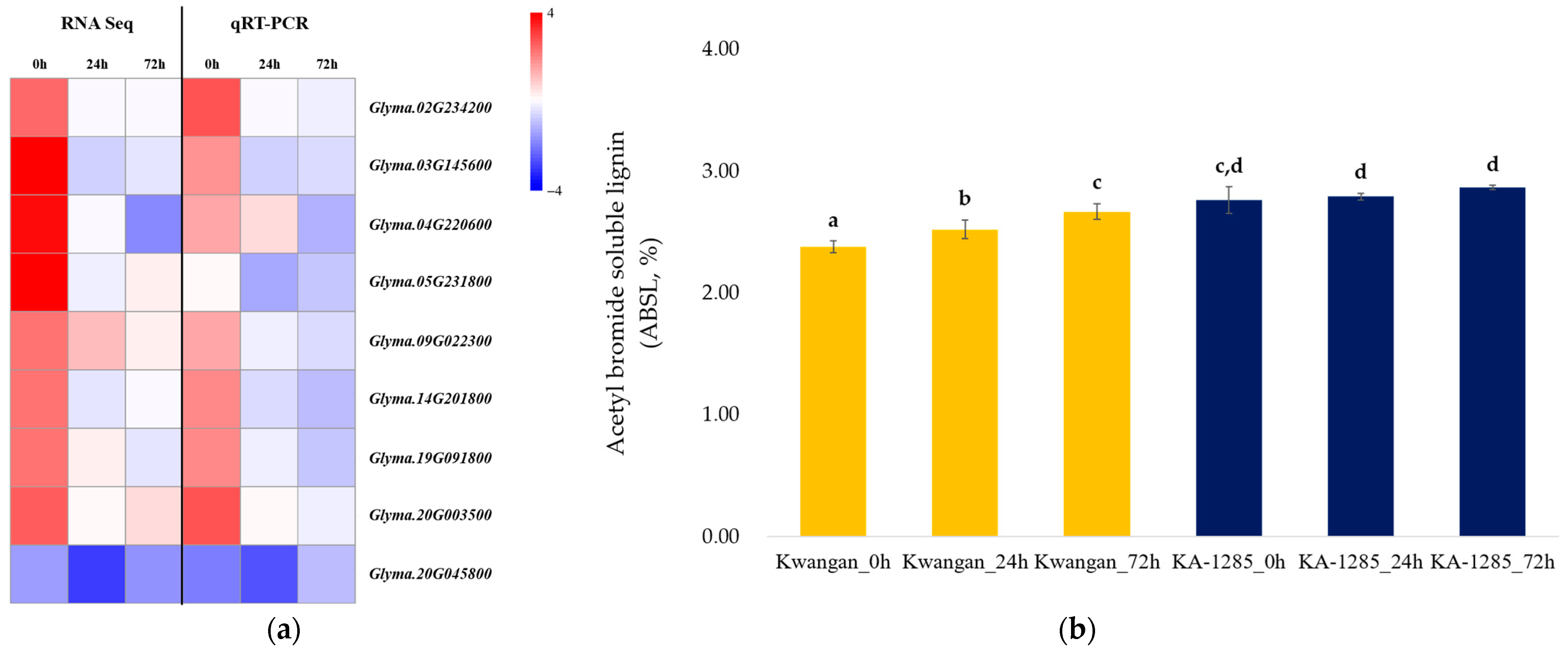
2.4. Validation of Selected DEGs
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Salt Treatment
4.2. RNA Isolation, Short-Read Sequencing, and Mapping of Clean Reads
4.3. Identification and Functional Annotation Analysis of Differentially Expressed Genes (DEGs)
4.4. Reverse Transcription followed by Quantitative Polymerase Chain Reaction (RT-qPCR)
4.5. Quantification of Acetyl Bromide Soluble Lignin (ABSL)
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Medic, J.; Atkinson, C.; Hurburgh, C.R. Current knowledge in soybean composition. J. Am. Oil Chem. Soc. 2014, 91, 363–384. [Google Scholar] [CrossRef]
- Chen, K.-I.; Erh, M.-H.; Su, N.-W.; Liu, W.-H.; Chou, C.-C.; Cheng, K.-C. Soyfoods and soybean products: From traditional use to modern applications. Appl. Microbiol. Biotechnol. 2012, 96, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, M.F.; Spina, F.; Cavallaro, S.; Bart, J. Sustainable soy biodiesel. Renew. Sustain. Energy Rev. 2013, 27, 806–852. [Google Scholar] [CrossRef]
- Food and Agriculture Organization of the United Nations. FAOSTAT Database 2021. Available online: http://www.fao.org/faostat/en/#data (accessed on 17 July 2023).
- Cao, D.; Li, Y.; Liu, B.; Kong, F.; Tran, L.S.P. Adaptive mechanisms of soybean grown on salt-affected soils. Land Degrad. Dev. 2018, 29, 1054–1064. [Google Scholar] [CrossRef]
- Xu, X.-y.; Fan, R.; Zheng, R.; Li, C.-m.; Yu, D.-y. Proteomic analysis of seed germination under salt stress in soybeans. J. Zhejiang Univ. Sci. B 2011, 12, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Neha, A.; Ashok, K.; Sanjay, A.; Alka, S. Evaluation of soybean (Glycine max L.) cultivars under salinity stress during early vegetative growth. Int. J. Curr. Microbiol. Appl. Sci. 2015, 4, 123–134. [Google Scholar]
- Zhang, Y.; Ding, J.; Wang, H.; Su, L.; Zhao, C. Biochar addition alleviate the negative effects of drought and salinity stress on soybean productivity and water use efficiency. BMC Plant Biol. 2020, 20, 1–11. [Google Scholar] [CrossRef]
- Silva, B.R.S.d.; Lobato, E.M.S.G.; Santos, L.A.d.; Pereira, R.M.; Batista, B.L.; Alyemeni, M.N.; Ahmad, P.; Lobato, A.K.d.S. How Different Na+ Concentrations Affect Anatomical, Nutritional Physiological, Biochemical, and Morphological Aspects in Soybean Plants: A Multidisciplinary and Comparative Approach. Agronomy 2023, 13, 232. [Google Scholar] [CrossRef]
- Zhu, J.-K. Regulation of ion homeostasis under salt stress. Curr. Opin. Plant Biol. 2003, 6, 441–445. [Google Scholar] [CrossRef]
- Arif, Y.; Singh, P.; Siddiqui, H.; Bajguz, A.; Hayat, S. Salinity induced physiological and biochemical changes in plants: An omic approach towards salt stress tolerance. Plant Physiol. Biochem. 2020, 156, 64–77. [Google Scholar] [CrossRef]
- Kronzucker, H.J.; Britto, D.T. Sodium transport in plants: A critical review. New Phytol. 2011, 189, 54–81. [Google Scholar] [CrossRef] [PubMed]
- Kronzucker, H.J.; Coskun, D.; Schulze, L.M.; Wong, J.R.; Britto, D.T. Sodium as nutrient and toxicant. Plant Soil 2013, 369, 1–23. [Google Scholar] [CrossRef]
- Mehta, P.; Jajoo, A.; Mathur, S.; Bharti, S. Chlorophyll a fluorescence study revealing effects of high salt stress on Photosystem II in wheat leaves. Plant Physiol. Biochem. 2010, 48, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Kalaji, H.M.; Bosa, K.; Kościelniak, J.; Żuk-Gołaszewska, K. Effects of salt stress on photosystem II efficiency and CO2 assimilation of two Syrian barley landraces. Environ. Exp. Bot. 2011, 73, 64–72. [Google Scholar] [CrossRef]
- Joshi, S.; Nath, J.; Singh, A.K.; Pareek, A.; Joshi, R. Ion transporters and their regulatory signal transduction mechanisms for salinity tolerance in plants. Physiol. Plant. 2022, 174, e13702. [Google Scholar] [CrossRef]
- Bohnert, H.J.; Gong, Q.; Li, P.; Ma, S. Unraveling abiotic stress tolerance mechanisms–getting genomics going. Curr. Opin. Plant Biol. 2006, 9, 180–188. [Google Scholar] [CrossRef]
- Malik, V.S. RNA Sequencing as a Tool for Understanding Biological Complexity of Abiotic Stress in Plants; Springer: Berlin/Heidelberg, Germany, 2016; Volume 25, pp. 1–2. [Google Scholar]
- Hu, J.; Zhuang, Y.; Li, X.; Li, X.; Sun, C.; Ding, Z.; Xu, R.; Zhang, D. Time-series transcriptome comparison reveals the gene regulation network under salt stress in soybean (Glycine max) roots. BMC Plant Biol. 2022, 22, 157. [Google Scholar] [CrossRef]
- Rao, S.S.; El-Habbak, M.H.; Havens, W.M.; Singh, A.; Zheng, D.; Vaughn, L.; Haudenshield, J.S.; Hartman, G.L.; Korban, S.S.; Ghabrial, S.A. Overexpression of GmCaM4 in soybean enhances resistance to pathogens and tolerance to salt stress. Mol. Plant Pathol. 2014, 15, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Gan, Z.; Wang, Y.; Lu, S.; Hou, Z.; Li, H.; Xiang, H.; Liu, B.; Kong, F.; Dong, L. The soybean gene J contributes to salt stress tolerance by up-regulating salt-responsive genes. Front. Plant Sci. 2020, 11, 272. [Google Scholar] [CrossRef]
- Zhang, M.; Cao, J.; Zhang, T.; Xu, T.; Yang, L.; Li, X.; Ji, F.; Gao, Y.; Ali, S.; Zhang, Q. A putative plasma membrane Na+/H+ antiporter GmSOS1 is critical for salt stress tolerance in Glycine max. Front. Plant Sci. 2022, 13, 870695. [Google Scholar] [CrossRef]
- Guan, R.; Qu, Y.; Guo, Y.; Yu, L.; Liu, Y.; Jiang, J.; Chen, J.; Ren, Y.; Liu, G.; Tian, L. Salinity tolerance in soybean is modulated by natural variation in G m SALT 3. Plant J. 2014, 80, 937–950. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, L.; Qu, Y.; Chen, J.; Liu, X.; Hong, H.; Liu, Z.; Chang, R.; Gilliham, M.; Qiu, L. GmSALT3, which confers improved soybean salt tolerance in the field, increases leaf Cl-exclusion prior to Na+ exclusion but does not improve early vigor under salinity. Front. Plant Sci. 2016, 7, 1485. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Guan, R.; Yu, L.; Berkowitz, O.; David, R.; Whelan, J.; Ford, M.; Wege, S.; Qiu, L.; Gilliham, M. Enhanced reactive oxygen detoxification occurs in salt-stressed soybean roots expressing GmSALT3. Physiol. Plant. 2022, 174, e13709. [Google Scholar] [CrossRef]
- Ulukapi, K.; Nasircilar, A.G. Developments of gamma ray application on mutation breeding studies in recent years. In Proceedings of the International Conference on Advances in Agricultural, Biological & Environmental Sciences, London, UK, 22–23 July 2015; pp. 31–34. [Google Scholar]
- Moon, C.Y.; Kang, B.H.; Kim, W.J.; Chowdhury, S.; Kang, S.; Lee, J.-D.; Kwon, S.-J.; Kim, H.-Y.; Lee, H.-S.; Ha, B.-K. Morpho-physiological and genetic characteristics of a salt-tolerant mutant line in soybean (Glycine max L.). Theor. Appl. Genet. 2023, 136, 166. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Boerma, H.; Villagarcia, M.; Zhou, X.; Carter, T.; Li, Z.; Gibbs, M. A major QTL conditioning salt tolerance in S-100 soybean and descendent cultivars. Theor. Appl. Genet. 2004, 109, 1610–1619. [Google Scholar] [CrossRef]
- Ha, B.-K.; Vuong, T.D.; Velusamy, V.; Nguyen, H.T.; Grover Shannon, J.; Lee, J.-D. Genetic mapping of quantitative trait loci conditioning salt tolerance in wild soybean (Glycine soja) PI 483463. Euphytica 2013, 193, 79–88. [Google Scholar] [CrossRef]
- Onyekachi, O.G.; Boniface, O.O.; Gemlack, N.F.; Nicholas, N. The effect of climate change on abiotic plant stress: A review. Abiotic Biot. Stress Plants 2019, 17, 71–83. [Google Scholar]
- Angon, P.B.; Tahjib-Ul-Arif, M.; Samin, S.I.; Habiba, U.; Hossain, M.A.; Brestic, M. How do plants respond to combined drought and salinity stress—A systematic review. Plants 2022, 11, 2884. [Google Scholar] [CrossRef]
- Corwin, D.L. Climate change impacts on soil salinity in agricultural areas. Eur. J. Soil Sci. 2021, 72, 842–862. [Google Scholar] [CrossRef]
- Chaudhry, S.; Sidhu, G.P.S. Climate change regulated abiotic stress mechanisms in plants: A comprehensive review. Plant Cell Rep. 2022, 41, 1–31. [Google Scholar] [CrossRef]
- Saito, K.; Matsuda, F. Metabolomics for functional genomics, systems biology, and biotechnology. Annu. Rev. Plant Biol. 2010, 61, 463–489. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Ding, C.; Li, W.; Wang, D.; Cui, D. Applications of metabolomics in the research of soybean plant under abiotic stress. Food Chem. 2020, 310, 125914. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Shahzad, B.; Rehman, A.; Bhardwaj, R.; Landi, M.; Zheng, B. Response of phenylpropanoid pathway and the role of polyphenols in plants under abiotic stress. Molecules 2019, 24, 2452. [Google Scholar] [CrossRef] [PubMed]
- Lim, I.; Kang, M.; Kim, B.C.; Ha, J. Metabolomic and transcriptomic changes in mungbean (Vigna radiata (L.) R. Wilczek) Sprouts Under Salin Stress. Front. Plant Sci. 2022, 13, 1030677. [Google Scholar]
- Moura, J.C.M.S.; Bonine, C.A.V.; de Oliveira Fernandes Viana, J.; Dornelas, M.C.; Mazzafera, P. Abiotic and biotic stresses and changes in the lignin content and composition in plants. J. Integr. Plant Biol. 2010, 52, 360–376. [Google Scholar] [CrossRef]
- Zagoskina, N.; Dubravina, G.; Alyavina, A.; Goncharuk, E. Effect of ultraviolet (UV-B) radiation on the formation and localization of phenolic compounds in tea plant callus cultures. Russ. J. Plant Physiol. 2003, 50, 270–275. [Google Scholar] [CrossRef]
- Xu, H.; Li, Y.; Yan, Y.; Wang, K.; Gao, Y.; Hu, Y. Genome-scale identification of soybean BURP domain-containing genes and their expression under stress treatments. BMC Plant Biol. 2010, 10, 1–16. [Google Scholar] [CrossRef]
- Zeng, A.; Chen, P.; Korth, K.L.; Ping, J.; Thomas, J.; Wu, C.; Srivastava, S.; Pereira, A.; Hancock, F.; Brye, K. RNA sequencing analysis of salt tolerance in soybean (Glycine max). Genomics 2019, 111, 629–635. [Google Scholar] [CrossRef]
- Lin, J.S.; Huang, X.X.; Li, Q.; Cao, Y.; Bao, Y.; Meng, X.F.; Li, Y.J.; Fu, C.; Hou, B.K. UDP-glycosyltransferase 72B1 catalyzes the glucose conjugation of monolignols and is essential for the normal cell wall lignification in Arabidopsis thaliana. Plant J. 2016, 88, 26–42. [Google Scholar] [CrossRef]
- Wang, Z.; Li, H.; Wei, Z.; Sun, H.; He, Y.; Gao, J.; Yang, Z.; You, J. Overexpression of UDP-glycosyltransferase genes enhanced aluminum tolerance through disrupting cell wall polysaccharide components in soybean. Plant Soil 2021, 469, 135–147. [Google Scholar] [CrossRef]
- Wu, C.; Dai, J.; Chen, Z.; Tie, W.; Yan, Y.; Yang, H.; Zeng, J.; Hu, W. Comprehensive analysis and expression profiles of cassava UDP-glycosyltransferases (UGT) family reveal their involvement in development and stress responses in cassava. Genomics 2021, 113, 3415–3429. [Google Scholar] [CrossRef]
- Jia, Q.; Zheng, C.; Sun, S.; Amjad, H.; Liang, K.; Lin, W. The role of plant cation/proton antiporter gene family in salt tolerance. Biol. Plant. 2018, 62, 617–629. [Google Scholar] [CrossRef]
- Song, Y.; Ji, D.; Li, S.; Wang, P.; Li, Q.; Xiang, F. The dynamic changes of DNA methylation and histone modifications of salt responsive transcription factor genes in soybean. PLoS ONE 2012, 7, e41274. [Google Scholar] [CrossRef] [PubMed]
- Kiełbowicz-Matuk, A. Involvement of plant C2H2-type zinc finger transcription factors in stress responses. Plant Sci. 2012, 185, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-J.; Yin, L.-J.; Liu, Y.; Ma, J.; Zheng, J.-C.; Lan, J.-H.; Fu, J.-D.; Chen, M.; Xu, Z.-S.; Ma, Y.-Z. The ABA-induced soybean ERF transcription factor gene GmERF75 plays a role in enhancing osmotic stress tolerance in Arabidopsis and soybean. BMC Plant Biol. 2019, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, X.; Zhang, B. Transcriptome analysis and functional identification of GmMYB46 in soybean seedlings under salt stress. PeerJ 2021, 9, e12492. [Google Scholar] [CrossRef]
- Song, H.; Wang, P.; Hou, L.; Zhao, S.; Zhao, C.; Xia, H.; Li, P.; Zhang, Y.; Bian, X.; Wang, X. Global analysis of WRKY genes and their response to dehydration and salt stress in soybean. Front. Plant Sci. 2016, 7, 9. [Google Scholar] [CrossRef]
- Liu, X.; Pi, B.; Du, Z.; Yang, T.; Gu, M.; Sun, S.; Yu, B. The transcription factor GmbHLH3 confers Cl−/salt tolerance to soybean by upregulating GmCLC1 expression for maintenance of anion homeostasis. Environ. Exp. Bot. 2022, 194, 104755. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Nat. Preced. 2010, 11, R106. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Jin, J.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 2020, 48, D1104–D1113. [Google Scholar] [CrossRef] [PubMed]
- Madden, T. The BLAST sequence analysis tool. In The NCBI Handbook; NCBI: Bethesda, MD, USA, 2003. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Lucas, A. Amap: Another Multidimensional Analysis Package. 2014. Available online: http://CRAN.R-project.org/package=amap (accessed on 1 February 2023).
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S. gplots: Various R programming tools for plotting data. R Package Version 2009, 2, 1. [Google Scholar]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 1–11. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Hu, R.; Fan, C.; Li, H.; Zhang, Q.; Fu, Y.-F. Evaluation of putative reference genes for gene expression normalization in soybean by quantitative real-time RT-PCR. BMC Mol. Biol. 2009, 10, 1–12. [Google Scholar] [CrossRef]
- Barnes, W.J.; Anderson, C.T. Acetyl bromide soluble lignin (ABSL) assay for total lignin quantification from plant biomass. Bio-Protocol 2017, 7, e2149. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissues | Time Points | Number of DEGs | Total DEGs | |
|---|---|---|---|---|
| Upregulated | Downregulated | |||
| Leaves | 0 h | 161 | 26 | 187 |
| 24 h | 30 | 30 | 60 | |
| 72 h | 33 | 16 | 49 | |
| Roots | 0 h | 42 | 25 | 67 |
| 24 h | 4 | 51 | 55 | |
| 72 h | 36 | 12 | 48 | |
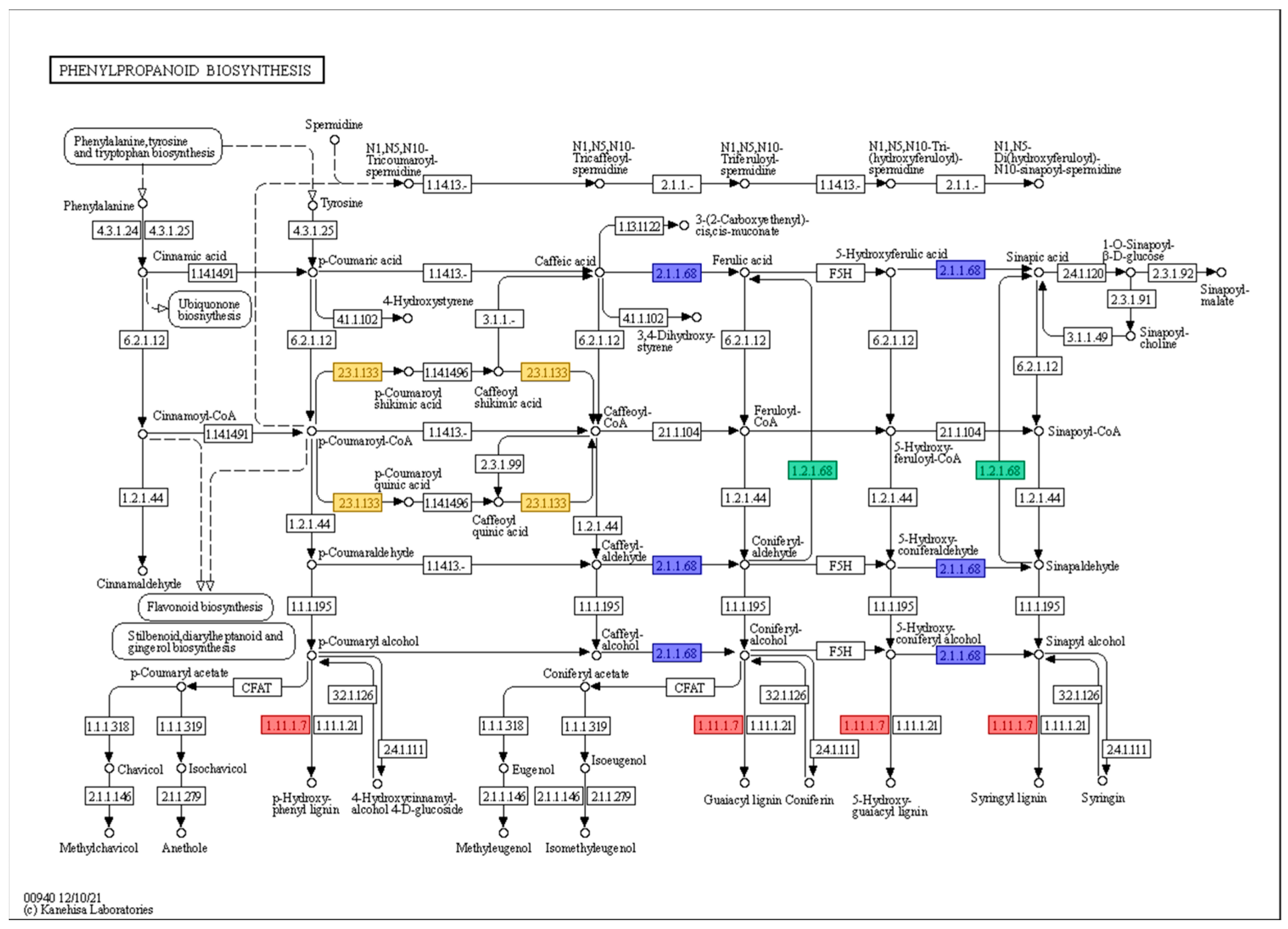
| Gene ID | Log2 (Fold Change) | Annotation | EC * Number | |||||
|---|---|---|---|---|---|---|---|---|
| Leaf | Root | |||||||
| 0 h | 24 h | 72 h | 0 h | 24 h | 72 h | |||
| Glyma.02G234200.1 | 2.51 | −0.07 | 0.02 | 1.20 | 0.51 | −0.05 | Peroxidase superfamily protein | 1.11.1.7 |
| Glyma.03G145600.1 | 4.90 | −0.69 | −0.32 | 0.19 | −0.60 | −0.38 | Peroxidase 2 | 1.11.1.7 |
| Glyma.04G220600.1 | 3.78 | 0.07 | −1.72 | 0.50 | −0.31 | −0.38 | Peroxidase superfamily protein | 1.11.1.7 |
| Glyma.05G231800.1 | 4.38 | −0.11 | 0.35 | −0.25 | −0.06 | 0.83 | Aldehyde dehydrogenase 2C4 | 1.2.1.68 |
| Glyma.09G022300.1 | 2.30 | 1.17 | 0.39 | 0.12 | 0.38 | −0.21 | Peroxidase 2 | 1.11.1.7 |
| Glyma.14G201800.1 | 2.22 | −0.30 | −0.03 | 0.30 | 0.22 | 0.27 | Peroxidase superfamily protein | 1.11.1.7 |
| Glyma.19G091800.1 | 2.36 | 0.25 | −0.28 | 0.50 | 0.12 | −0.28 | Peroxidase superfamily protein | 1.11.1.7 |
| Glyma.20G003500.1 | 2.59 | 0.23 | 0.61 | 0.26 | 0.04 | −0.01 | O-methyltransferase 1 | 2.1.1.68 |
| Glyma.20G045800.1 | −1.54 | −2.93 | −1.62 | 0.63 | −1.19 | 0.66 | Spermidine hydroxycinnamoyl transferase | 2.3.1.133 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, B.H.; Chowdhury, S.; Kang, S.-H.; Shin, S.-Y.; Lee, W.-H.; Lee, H.-S.; Ha, B.-K. Transcriptome Profiling of a Soybean Mutant with Salt Tolerance Induced by Gamma-ray Irradiation. Plants 2024, 13, 254. https://doi.org/10.3390/plants13020254
Kang BH, Chowdhury S, Kang S-H, Shin S-Y, Lee W-H, Lee H-S, Ha B-K. Transcriptome Profiling of a Soybean Mutant with Salt Tolerance Induced by Gamma-ray Irradiation. Plants. 2024; 13(2):254. https://doi.org/10.3390/plants13020254
Chicago/Turabian StyleKang, Byeong Hee, Sreeparna Chowdhury, Se-Hee Kang, Seo-Young Shin, Won-Ho Lee, Hyeon-Seok Lee, and Bo-Keun Ha. 2024. "Transcriptome Profiling of a Soybean Mutant with Salt Tolerance Induced by Gamma-ray Irradiation" Plants 13, no. 2: 254. https://doi.org/10.3390/plants13020254
APA StyleKang, B. H., Chowdhury, S., Kang, S.-H., Shin, S.-Y., Lee, W.-H., Lee, H.-S., & Ha, B.-K. (2024). Transcriptome Profiling of a Soybean Mutant with Salt Tolerance Induced by Gamma-ray Irradiation. Plants, 13(2), 254. https://doi.org/10.3390/plants13020254