
The Conservation Genetics of Iris lacustris (Dwarf Lake Iris), a Great Lakes Endemic


Abstract
1. Introduction
2. Results
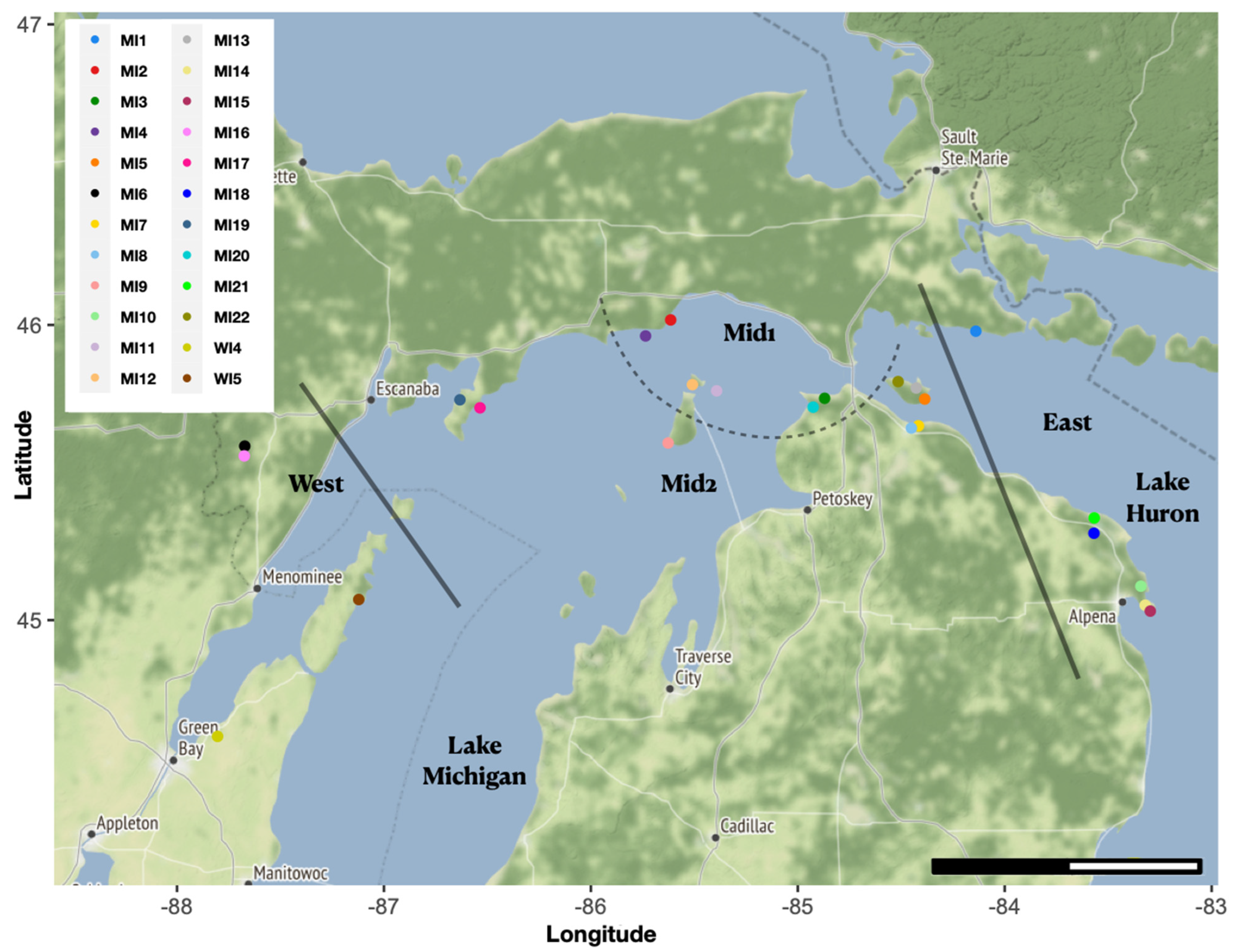
2.1. DNA Sequencing and Polyploid Filtering
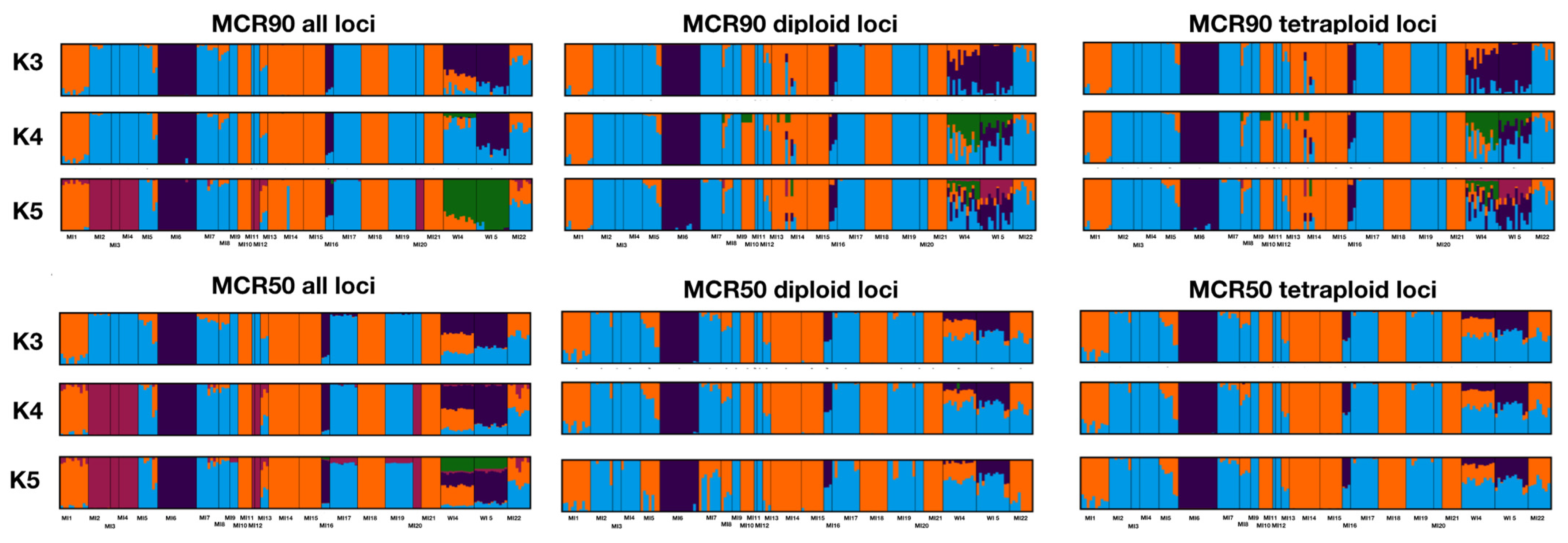
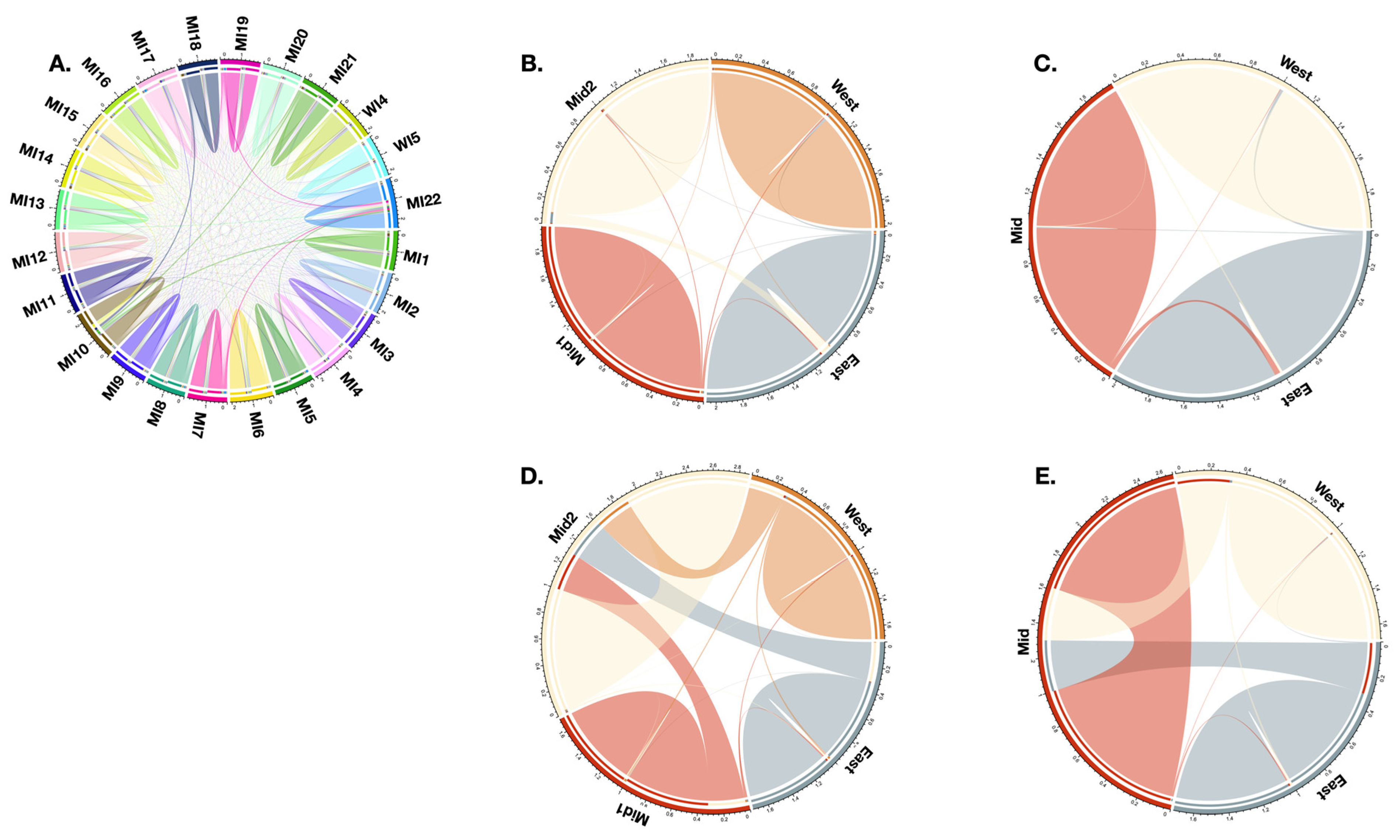
2.2. Population Genomics
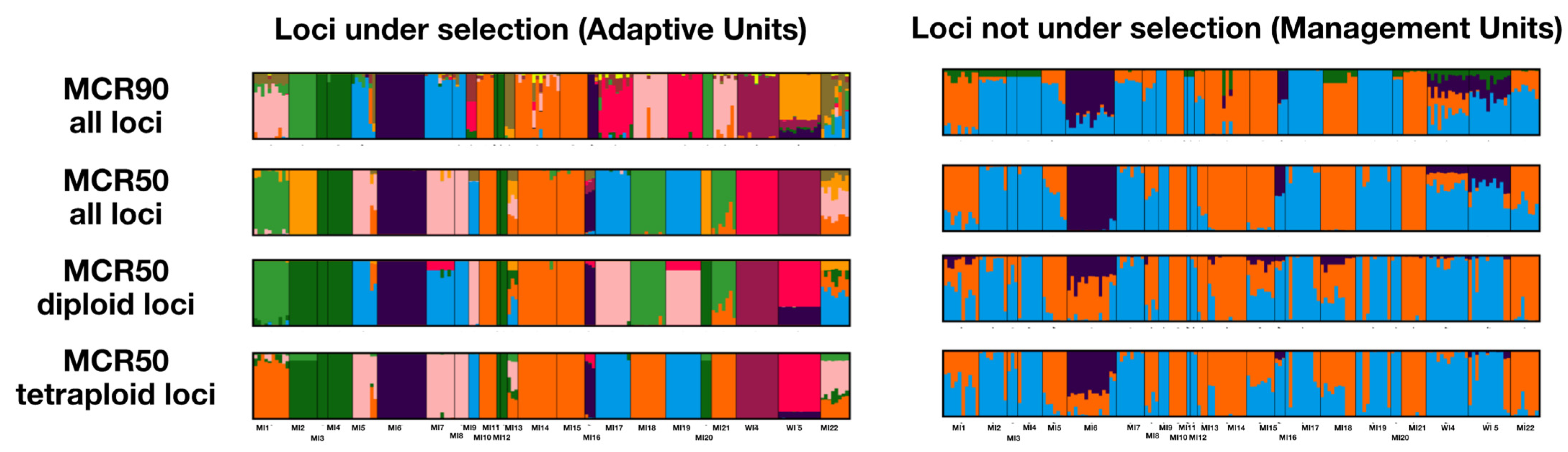
2.3. Conservation Units
3. Discussion
3.1. Population Structure and Genetic Diversity
3.2. Migration and Demography
3.3. Subsetting Diploid and Tetraploid Loci
3.4. Conservation Genetics of I. lacustris
4. Materials and Methods
4.1. Plant Material
4.2. DNA Sequencing
4.3. Polyploidy Filtering
4.4. Population Genomics
4.5. Conservation Units
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nuttall, T. The Genera of North American Plants: And a Catalogue of the Species, to the Year 1817; D. Heartt: Philadelphia, PA, USA, 1817; Volume 1. [Google Scholar]
- U.S. Fish and Wildlife Service. Status Review—Dwarf Lake Iris (Iris lacustris); East Lansing Field Office: East Lansing, MI, USA, 2022; p. 10.
- Voss, E.G. Michigan Flora, 3rd ed.; Cranbrook Institute of Science: Bloomfield Hills, MI, USA; University of Michigan Herbarium: Ann Arbor, MI, USA, 1972; Volume 1, p. 488. [Google Scholar]
- Brotske, V. Pollination, Seed Dispersal, Germination, and Seedling Survival in the Federally Threatened Dwarf Lake Iris (Iris Lacustris). Master’s Thesis, University of Wisconsin-Green Bay, Green Bay, WI, USA, 2018. [Google Scholar]
- U.S. Fish and Wildlife Service. 5-Year Review Dwarf Lake Iris (Iris lacustris); U.S. Fish and Wildlife Service: East Lansing, MI, USA, 2011; p. 21.
- Van Kley, J.E.; Wujek, D.E. Habitat and ecology of Iris lacustris (the dwarf lake iris). Mich. Bot. 1993, 32, 209–222. [Google Scholar]
- State of Michigan. State Facts and Symbols. Available online: https://www.michigan.gov/som/about-michigan/state-facts-and-symbols (accessed on 15 January 2023).
- Simonich, M.T.; Morgan, M.D. Allozymic uniformity in Iris lacustris (dwarf lake iris) in Wisconsin. Can. J. Bot. 1994, 72, 1720–1722. [Google Scholar] [CrossRef]
- Orick, M.W. Enzyme Polymorphism and Genetic Diversity in the Great Lakes Endemic Iris lacustris Nutt. (Dwarf Lake Iris). Master’s Thesis, Eastern Michigan University, Ypsilanti, MI, USA, 1992. [Google Scholar]
- Hannan, G.L.; Orick, M.W. Isozyme diversity in Iris cristata and the threatened glacial endemic I. lacustris (Iridaceae). Am. J. Bot. 2000, 87, 293–301. [Google Scholar] [CrossRef]
- Guo, J.; Wilson, C.A. Molecular phylogeny of crested Iris based on five plastid markers (Iridaceae). Syst. Bot. 2013, 38, 987–995. [Google Scholar] [CrossRef]
- Soltis, P.S.; Soltis, D.E. The role of genetic and genomic attributes in the success of polyploids. Proc. Natl. Acad. Sci. USA 2000, 97, 7051–7057. [Google Scholar] [CrossRef] [PubMed]
- Luttikhuizen, P.C.; Stift, M.; Kuperus, P.; Van Tienderen, P.H. Genetic diversity in diploid vs. tetraploid Rorippa amphibia (Brassicaceae). Mol. Ecol. 2007, 16, 3544–3553. [Google Scholar] [CrossRef] [PubMed]
- Van de Peer, Y.; Ashman, T.-L.; Soltis, P.S.; Soltis, D.E. Polyploidy: An evolutionary and ecological force in stressful times. Plant Cell 2021, 33, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Liu, S.; Schnable, J.C.; Yeh, C.-T.E.; Wang, K.-S.; Schnable, P.S. tGBS® genotyping-by-sequencing enables reliable genotyping of heterozygous loci. Nucleic Acids Res. 2017, 45, e178. [Google Scholar] [CrossRef]
- Funk, W.C.; McKay, J.K.; Hohenlohe, P.A.; Allendorf, F.W. Harnessing genomics for delineating conservation units. Trends Ecol. Evol. 2012, 27, 489–496. [Google Scholar] [CrossRef]
- Millar, M.A.; Byrne, M. Variable clonality and genetic structure among disjunct populations of Banksia mimica. Conserv. Genet. 2020, 21, 803–818. [Google Scholar] [CrossRef]
- Edgeloe, J.M.; Severn-Ellis, A.A.; Bayer, P.E.; Mehravi, S.; Breed, M.F.; Krauss, S.L.; Batley, J.; Kendrick, G.A.; Sinclair, E.A. Extensive polyploid clonality was a successful strategy for seagrass to expand into a newly submerged environment. Proc. R. Soc. B 2022, 289, 20220538. [Google Scholar] [CrossRef] [PubMed]
- Sessa, E.B. Polyploidy as a mechanism for surviving global change. New Phytol. 2019, 221, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Fant, J.B.; Havens, K.; Keller, J.M.; Radosavljevic, A.; Yates, E.D. The influence of contemporary and historic landscape features on the genetic structure of the sand dune endemic, Cirsium pitcheri (Asteraceae). Heredity 2014, 112, 519–530. [Google Scholar] [CrossRef]
- Kincare, K.; Larson, G.J. Evolution of the Great Lakes. In Michigan Geography and Geology; Schaetzl, R.J., Darden, J.T., Brandt, D., Eds.; Pearson Custom Publishing: Boston, MA, USA, 2009; pp. 174–190. [Google Scholar]
- Larson, G.; Schaetzl, R. Origin and evolution of the Great Lakes. J. Great Lakes Res. 2001, 27, 518–546. [Google Scholar] [CrossRef]
- Chung, M.Y.; López-Pujol, J.; Lee, Y.M.; Oh, S.H.; Chung, M.G. Clonal and genetic structure of Iris odaesanensis and Iris rossii (Iridaceae): Insights of the Baekdudaegan Mountains as a glacial refugium for boreal and temperate plants. Plant Syst. Evol. 2015, 301, 1397–1409. [Google Scholar] [CrossRef]
- Clark, L.V.; Lipka, A.E.; Sacks, E.J. polyRAD: Genotype calling with uncertainty from sequencing data in polyploids and diploids. G3 Genes Genomes Genet. 2019, 9, 663–673. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Verity, R.; Nichols, R.A. Estimating the number of subpopulations (K) in structured populations. Genetics 2016, 203, 1827–1839. [Google Scholar] [CrossRef] [PubMed]
- Stift, M.; Kolář, F.; Meirmans, P.G. STRUCTURE is more robust than other clustering methods in simulated mixed-ploidy populations. Heredity 2019, 123, 429–441. [Google Scholar] [CrossRef]
- Chafin, T.K.; Regmi, B.; Douglas, M.R.; Edds, D.R.; Wangchuk, K.; Dorji, S.; Norbu, P.; Norbu, S.; Changlu, C.; Khanal, G.P. Parallel introgression, not recurrent emergence, explains apparent elevational ecotypes of polyploid Himalayan snowtrout. R. Soc. Open Sci. 2021, 8, 210727. [Google Scholar] [CrossRef]
- Salvado, P.; Aymerich Boixader, P.; Parera, J.; Vila Bonfill, A.; Martin, M.; Quélennec, C.; Lewin, J.M.; Delorme-Hinoux, V.; Bertrand, J.A.M. Little hope for the polyploid endemic Pyrenean Larkspur (Delphinium montanum): Evidences from population genomics and Ecological Niche Modeling. Ecol. Evol. 2022, 12, e8711. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.V.; Wagner, W.H., Jr. Michigan Trees. A Guide to the Trees of Michigan and the Great Lakes Region; University of Michigan Press: Ann Arbor, MI, USA, 1981. [Google Scholar]
- Walker, W.S.; Barnes, B.V.; Kashian, D.M. Landscape ecosystems of the Mack Lake burn, northern Lower Michigan, and the occurrence of the Kirtland’s warbler. For. Sci. 2003, 49, 119–139. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Schulz, M.H.; Weese, D.; Holtgrewe, M.; Dimitrova, V.; Niu, S.; Reinert, K.; Richard, H. Fiona: A parallel and automatic strategy for read error correction. Bioinformatics 2014, 30, i356–i363. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.V.; Mays, W.; Lipka, A.E.; Sacks, E.J. A population-level statistic for assessing Mendelian behavior of genotyping-by-sequencing data from highly duplicated genomes. BMC Bioinform. 2022, 23, 101. [Google Scholar] [CrossRef]
- De Meeûs, T.; Goudet, J. A step-by-step tutorial to use HierFstat to analyse populations hierarchically structured at multiple levels. Infect. Genet. Evol. 2007, 7, 731–735. [Google Scholar] [CrossRef]
- Goudet, J. HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef]
- Pina-Martins, F.; Silva, D.N.; Fino, J.; Paulo, O.S. Structure_threader: An improved method for automation and parallelization of programs structure, fastStructure and MavericK on multicore CPU systems. Mol. Ecol. Res. 2017, 17, e268–e274. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. CLUMPAK: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef]
- Li, Y.L.; Liu, J.X. STRUCTURESELECTOR: A web-based software to select and visualize the optimal number of clusters using multiple methods. Mol. Ecol. Resour. 2018, 18, 176–177. [Google Scholar] [CrossRef]
- Puechmaille, S.J. The program STRUCTURE does not reliably recover the correct population structure when sampling is uneven: Subsampling and new estimators alleviate the problem. Mol. Ecol. Resour. 2016, 16, 608–627. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- R Developement Core Team. A Language and Environment for Statistical Computing. 2009. Available online: http://www.R-project.org (accessed on 5 January 2023).
- Caye, K.; Jay, F.; Michel, O.; François, O. Fast inference of individual admixture coefficients using geographic data. Ann. Appl. Stat. 2018, 12, 586–608. [Google Scholar] [CrossRef]
- Bradburd, G.S.; Coop, G.M.; Ralph, P.L. Inferring continuous and discrete population genetic structure across space. Genetics 2018, 210, 33–52. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Ahmed, I. adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef]
- Foll, M.; Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [PubMed]
- Mussmann, S.M.; Douglas, M.R.; Chafin, T.K.; Douglas, M.E. BA3-SNPs: Contemporary migration reconfigured in BayesAss for next-generation sequence data. Methods Ecol. Evol. 2019, 10, 1808–1813. [Google Scholar] [CrossRef]
- Wilson, G.A.; Rannala, B. Bayesian inference of recent migration rates using multilocus genotypes. Genetics 2003, 163, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Collin, F.D.; Durif, G.; Raynal, L.; Lombaert, E.; Gautier, M.; Vitalis, R.; Marin, J.M.; Estoup, A. Extending approximate Bayesian computation with supervised machine learning to infer demographic history from genetic polymorphisms using DIYABC Random Forest. Mol. Ecol. Resour. 2021, 21, 2598–2613. [Google Scholar] [CrossRef] [PubMed]
- Collin, F.-D.; Estoup, A.; Marin, J.-M.; Raynal, L. Bringing ABC inference to the machine learning realm: AbcRanger, an optimized random forests library for ABC. In Proceedings of the JOBIM 2020, Montpellier, France, 30 June 2020. [Google Scholar]
- Byun, K.; Chiu, C.-M.; Hamlet, A.F. Effects of 21st century climate change on seasonal flow regimes and hydrologic extremes over the Midwest and Great Lakes region of the US. Sci. Total Environ. 2019, 650, 1261–1277. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Populations Sampled | Number of Individuals Sampled | Four Population Clusters in Analyses | Three Population Clusters in Analyses | Management Units (All Loci) | Management Units (Diploid and Tetraploid Loci) | Adaptive Units |
|---|---|---|---|---|---|---|
| MI1 | 10 | East | East | 1 | 1 | 1 |
| MI2 | 3 | Mid1 | Mid | 2 | 2 | 2 |
| MI3 | 8 | Mid1 | Mid | 2 | 2 | 3 |
| MI4 | 7 | Mid1 | Mid | 2 | 2 | 3 |
| MI5 | 7 | Mid2 | Mid | 3 | 3 | 4 |
| MI6 | 14 | West | West | 4 | 4 | 5 |
| MI7 | 8 | Mid2 | Mid | 2 | 2 | 4 |
| MI8 | 4 | Mid2 | Mid | 3 | 3 | 4 |
| MI9 | 3 | Mid2 | Mid | 2 | 2 | 6 |
| MI10 | 5 | East | East | 1 | 3 | 7 |
| MI11 | 1 | Mid1 | Mid | 2 | 2 | 3 |
| MI12 | 2 | Mid1 | Mid | 2 | 2 | 3 |
| MI13 | 3 | Mid2 | Mid | 3 | 3 | 4 |
| MI14 | 13 | East | East | 1 | 3 | 7 |
| MI15 | 8 | East | East | 1 | 1 | 7 |
| MI16 | 3 | West | West | 4 | 2 | 5 |
| MI17 | 10 | Mid2 | Mid | 2 | 2 | 6 |
| MI18 | 10 | East | East | 1 | 1 | 1 |
| MI19 | 10 | Mid2 | Mid | 2 | 2 | 6 |
| MI20 | 3 | Mid1 | Mid | 2 | 2 | 2 |
| MI21 | 7 | East | East | 1 | 3 | 1 |
| MI22 | 8 | Mid2 | Mid | 3 | 3 | 4 |
| WI4 | 12 | West | West | 4 | 2 | 8 |
| WI5 | 12 | West | West | 4 | 2 | 9 |
| All Loci | Loci under Selection | Loci Not under Selection | ||||||
|---|---|---|---|---|---|---|---|---|
| Dataset | SNPs | Loci under Selection | StructureSelector | DAPC | StructureSelector | DAPC | StructureSelector | DAPC |
| MCR90 | 5354 | 401 | 6 | 9 | 12–14 | 13 | 3–4 | 7 |
| MCR90 diploid loci | 2106 | 29 | 4–5 | 7 | - | - | - | - |
| MCR90 tetraploid loci | 1382 | 21 | 4–5 | 6 | - | - | - | - |
| MCR50 | 344,509 | 65,075 | 5–7 | 4 | 11–13 | 10 | 3 | 1 |
| MCR50 diploid loci | 50,134 | 4311 | 3–4 | 2–3 | 9–10 | 7 | 2–3 | 1 |
| MCR50 tetraploid loci | 82,237 | 6939 | 3–4 | 2–3 | 8 | 9 | 3 | 1 |
| MCR90 All Loci | MCR90 Diploid Loci | MCR90 Tetraploid Loci | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Population | HO | HS | HT | FIS | HO | HS | HT | FIS | HO | HS | HT | FIS |
| MI1 | 0.0586 | 0.0516 | 0.0516 | −0.1365 | 0.064 | 0.0519 | 0.0519 | −0.2325 | 0.0548 | 0.0462 | 0.0462 | −0.1875 |
| MI2 | 0.0503 | 0.0411 | 0.0411 | −0.2224 | 0.0538 | 0.0417 | 0.0417 | −0.2883 | 0.0532 | 0.0431 | 0.0431 | −0.2329 |
| MI3 | 0.0472 | 0.0307 | 0.0307 | −0.5394 | 0.0521 | 0.0322 | 0.0322 | −0.6191 | 0.0474 | 0.0301 | 0.0301 | −0.5768 |
| MI4 | 0.0581 | 0.0451 | 0.0451 | −0.2873 | 0.0651 | 0.0479 | 0.0479 | −0.3582 | 0.0628 | 0.0497 | 0.0497 | −0.2624 |
| MI5 | 0.0558 | 0.0532 | 0.0532 | −0.047 | 0.052 | 0.0428 | 0.0428 | −0.2161 | 0.051 | 0.041 | 0.041 | −0.2444 |
| MI6 | 0.0957 | 0.0704 | 0.0704 | −0.3593 | 0.1043 | 0.0742 | 0.0742 | −0.4064 | 0.0933 | 0.0675 | 0.0675 | −0.3826 |
| MI7 | 0.054 | 0.049 | 0.049 | −0.1021 | 0.0519 | 0.042 | 0.042 | −0.2372 | 0.0559 | 0.0449 | 0.0449 | −0.2455 |
| MI8 | 0.0655 | 0.0563 | 0.0563 | −0.1631 | 0.0677 | 0.0573 | 0.0573 | −0.1816 | 0.067 | 0.0555 | 0.0555 | −0.2074 |
| MI9 | 0.0594 | 0.0401 | 0.0401 | −0.4814 | 0.0586 | 0.0373 | 0.0373 | −0.5714 | 0.0673 | 0.0442 | 0.0442 | −0.5217 |
| MI10 | 0.0612 | 0.0477 | 0.0477 | −0.2842 | 0.0554 | 0.043 | 0.043 | −0.289 | 0.0515 | 0.0383 | 0.0383 | −0.3455 |
| MI11 | 0.0475 | - | - | - | 0.0527 | - | - | - | 0.0499 | - | - | - |
| MI12 | 0.0522 | 0.0385 | 0.0385 | −0.3578 | 0.0592 | 0.0411 | 0.0411 | −0.4413 | 0.0551 | 0.0433 | 0.0433 | −0.2749 |
| MI13 | 0.0557 | 0.0447 | 0.0447 | −0.2463 | 0.0508 | 0.0409 | 0.0409 | −0.2434 | 0.0543 | 0.0401 | 0.0401 | −0.3551 |
| MI14 | 0.0535 | 0.0488 | 0.0488 | −0.0981 | 0.0542 | 0.0448 | 0.0448 | −0.2105 | 0.0497 | 0.0415 | 0.0415 | −0.1989 |
| MI15 | 0.0573 | 0.0551 | 0.0551 | −0.0395 | 0.059 | 0.0516 | 0.0516 | −0.1434 | 0.0589 | 0.0502 | 0.0502 | −0.1724 |
| MI16 | 0.0961 | 0.0694 | 0.0694 | −0.384 | 0.0956 | 0.0661 | 0.0661 | −0.4464 | 0.0795 | 0.0541 | 0.0541 | −0.4686 |
| MI17 | 0.0671 | 0.062 | 0.062 | −0.0827 | 0.0609 | 0.0488 | 0.0488 | −0.2478 | 0.0647 | 0.0524 | 0.0524 | −0.2357 |
| MI18 | 0.0639 | 0.0567 | 0.0567 | −0.1277 | 0.0672 | 0.0542 | 0.0542 | −0.2401 | 0.0643 | 0.0534 | 0.0534 | −0.2054 |
| MI19 | 0.0651 | 0.0575 | 0.0575 | −0.1324 | 0.0645 | 0.0512 | 0.0512 | −0.2604 | 0.0591 | 0.0487 | 0.0487 | −0.215 |
| MI20 | 0.0467 | 0.0343 | 0.0343 | −0.3597 | 0.0481 | 0.0351 | 0.0351 | −0.3711 | 0.0516 | 0.0365 | 0.0365 | −0.4123 |
| MI21 | 0.0624 | 0.054 | 0.054 | −0.1557 | 0.0628 | 0.0497 | 0.0497 | −0.262 | 0.0637 | 0.0512 | 0.0512 | −0.2435 |
| MI22 | 0.0543 | 0.0492 | 0.0492 | −0.1035 | 0.0464 | 0.0382 | 0.0382 | −0.2135 | 0.049 | 0.0397 | 0.0397 | −0.2343 |
| WI4 | 0.1081 | 0.0946 | 0.0946 | −0.1424 | 0.1032 | 0.0848 | 0.0848 | −0.2179 | 0.0895 | 0.0745 | 0.0745 | −0.201 |
| WI5 | 0.1033 | 0.085 | 0.085 | −0.2157 | 0.1015 | 0.0775 | 0.0775 | −0.3102 | 0.094 | 0.0734 | 0.0734 | −0.2814 |
| MI1 | MI2 | MI3 | MI4 | MI5 | MI6 | MI7 | MI8 | MI9 | MI10 | MI11 | MI12 | MI13 | MI14 | MI15 | MI16 | MI17 | MI18 | MI19 | MI20 | MI21 | MI22 | WI4 | WI5 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MI1 | - | 0.17 | 0.20 | 0.21 | 0.16 | 0.25 | 0.18 | 0.16 | 0.22 | 0.14 | 0.12 | 0.17 | 0.13 | 0.11 | 0.09 | 0.26 | 0.18 | 0.05 | 0.20 | 0.16 | 0.05 | 0.15 | 0.15 | 0.19 |
| MI2 | 0.29 | - | 0.12 | 0.12 | 0.16 | 0.24 | 0.16 | 0.18 | 0.24 | 0.20 | 0.07 | 0.11 | 0.13 | 0.16 | 0.15 | 0.27 | 0.16 | 0.19 | 0.17 | 0.00 | 0.19 | 0.11 | 0.16 | 0.20 |
| MI3 | 0.36 | 0.26 | - | −0.01 | 0.19 | 0.21 | 0.19 | 0.20 | 0.26 | 0.27 | 0.00 | 0.05 | 0.22 | 0.21 | 0.20 | 0.28 | 0.13 | 0.22 | 0.14 | 0.14 | 0.22 | 0.18 | 0.13 | 0.16 |
| MI4 | 0.37 | 0.26 | 0.01 | - | 0.19 | 0.24 | 0.18 | 0.19 | 0.20 | 0.25 | −0.08 | 0.03 | 0.19 | 0.22 | 0.20 | 0.26 | 0.14 | 0.23 | 0.15 | 0.10 | 0.23 | 0.18 | 0.16 | 0.19 |
| MI5 | 0.26 | 0.29 | 0.30 | 0.31 | - | 0.20 | 0.08 | 0.04 | 0.20 | 0.12 | 0.12 | 0.18 | 0.08 | 0.12 | 0.12 | 0.23 | 0.12 | 0.17 | 0.14 | 0.16 | 0.16 | 0.06 | 0.14 | 0.15 |
| MI6 | 0.41 | 0.39 | 0.37 | 0.39 | 0.31 | - | 0.23 | 0.19 | 0.22 | 0.24 | 0.14 | 0.19 | 0.20 | 0.24 | 0.23 | 0.13 | 0.20 | 0.25 | 0.20 | 0.21 | 0.24 | 0.22 | 0.19 | 0.20 |
| MI7 | 0.32 | 0.35 | 0.37 | 0.37 | 0.12 | 0.35 | - | 0.08 | 0.19 | 0.19 | 0.11 | 0.18 | 0.16 | 0.15 | 0.15 | 0.25 | 0.12 | 0.20 | 0.14 | 0.14 | 0.18 | 0.11 | 0.15 | 0.18 |
| MI8 | 0.31 | 0.38 | 0.41 | 0.40 | 0.07 | 0.34 | 0.14 | - | 0.18 | 0.15 | 0.04 | 0.14 | 0.10 | 0.14 | 0.12 | 0.20 | 0.11 | 0.17 | 0.13 | 0.16 | 0.16 | 0.09 | 0.12 | 0.15 |
| MI9 | 0.42 | 0.43 | 0.44 | 0.38 | 0.32 | 0.40 | 0.34 | 0.39 | - | 0.29 | 0.18 | 0.25 | 0.28 | 0.24 | 0.21 | 0.25 | 0.10 | 0.24 | 0.12 | 0.25 | 0.25 | 0.24 | 0.13 | 0.14 |
| MI10 | 0.20 | 0.34 | 0.44 | 0.42 | 0.23 | 0.38 | 0.32 | 0.29 | 0.45 | - | 0.19 | 0.25 | 0.13 | 0.03 | 0.06 | 0.28 | 0.20 | 0.13 | 0.21 | 0.21 | 0.09 | 0.12 | 0.14 | 0.19 |
| MI11 | 0.30 | 0.24 | 0.05 | −0.07 | 0.21 | 0.31 | 0.29 | 0.30 | 0.38 | 0.38 | - | −0.03 | 0.12 | 0.14 | 0.10 | 0.13 | 0.05 | 0.14 | 0.06 | 0.07 | 0.14 | 0.12 | 0.02 | 0.07 |
| MI12 | 0.33 | 0.25 | 0.07 | 0.03 | 0.26 | 0.35 | 0.34 | 0.36 | 0.41 | 0.41 | 0.01 | - | 0.17 | 0.19 | 0.15 | 0.21 | 0.13 | 0.18 | 0.13 | 0.14 | 0.20 | 0.18 | 0.09 | 0.13 |
| MI13 | 0.21 | 0.26 | 0.37 | 0.33 | 0.16 | 0.33 | 0.27 | 0.23 | 0.44 | 0.22 | 0.28 | 0.31 | - | 0.09 | 0.09 | 0.25 | 0.15 | 0.14 | 0.16 | 0.15 | 0.14 | 0.02 | 0.12 | 0.15 |
| MI14 | 0.15 | 0.31 | 0.38 | 0.39 | 0.24 | 0.40 | 0.30 | 0.28 | 0.42 | 0.04 | 0.33 | 0.36 | 0.18 | - | 0.03 | 0.26 | 0.18 | 0.10 | 0.19 | 0.15 | 0.07 | 0.09 | 0.14 | 0.19 |
| MI15 | 0.17 | 0.32 | 0.38 | 0.39 | 0.24 | 0.39 | 0.29 | 0.26 | 0.41 | 0.09 | 0.30 | 0.34 | 0.19 | 0.04 | - | 0.23 | 0.16 | 0.09 | 0.18 | 0.13 | 0.08 | 0.11 | 0.12 | 0.17 |
| MI16 | 0.43 | 0.44 | 0.44 | 0.43 | 0.32 | 0.19 | 0.37 | 0.34 | 0.42 | 0.43 | 0.30 | 0.37 | 0.39 | 0.42 | 0.39 | - | 0.20 | 0.25 | 0.20 | 0.26 | 0.26 | 0.27 | 0.14 | 0.14 |
| MI17 | 0.28 | 0.25 | 0.22 | 0.25 | 0.17 | 0.32 | 0.21 | 0.22 | 0.18 | 0.27 | 0.13 | 0.20 | 0.21 | 0.27 | 0.26 | 0.28 | - | 0.20 | 0.07 | 0.14 | 0.18 | 0.13 | 0.13 | 0.16 |
| MI18 | 0.12 | 0.33 | 0.38 | 0.40 | 0.29 | 0.41 | 0.33 | 0.32 | 0.42 | 0.17 | 0.32 | 0.35 | 0.23 | 0.14 | 0.15 | 0.41 | 0.29 | - | 0.21 | 0.18 | 0.04 | 0.16 | 0.15 | 0.21 |
| MI19 | 0.33 | 0.30 | 0.28 | 0.31 | 0.22 | 0.35 | 0.25 | 0.25 | 0.27 | 0.32 | 0.21 | 0.26 | 0.26 | 0.31 | 0.30 | 0.34 | 0.11 | 0.33 | - | 0.15 | 0.20 | 0.15 | 0.15 | 0.17 |
| MI20 | 0.29 | −0.01 | 0.29 | 0.25 | 0.27 | 0.36 | 0.33 | 0.38 | 0.45 | 0.35 | 0.24 | 0.28 | 0.29 | 0.31 | 0.31 | 0.42 | 0.22 | 0.32 | 0.28 | - | 0.18 | 0.10 | 0.12 | 0.16 |
| MI21 | 0.09 | 0.34 | 0.39 | 0.40 | 0.26 | 0.39 | 0.31 | 0.30 | 0.42 | 0.13 | 0.32 | 0.36 | 0.21 | 0.09 | 0.13 | 0.41 | 0.27 | 0.05 | 0.32 | 0.33 | - | 0.14 | 0.14 | 0.19 |
| MI22 | 0.19 | 0.22 | 0.28 | 0.30 | 0.09 | 0.33 | 0.17 | 0.15 | 0.34 | 0.17 | 0.21 | 0.26 | 0.04 | 0.15 | 0.17 | 0.36 | 0.16 | 0.22 | 0.21 | 0.21 | 0.18 | - | 0.15 | 0.18 |
| WI4 | 0.27 | 0.26 | 0.23 | 0.27 | 0.22 | 0.30 | 0.28 | 0.24 | 0.27 | 0.22 | 0.12 | 0.19 | 0.20 | 0.24 | 0.23 | 0.25 | 0.20 | 0.25 | 0.25 | 0.21 | 0.24 | 0.22 | - | 0.15 |
| WI5 | 0.33 | 0.32 | 0.24 | 0.28 | 0.26 | 0.27 | 0.30 | 0.27 | 0.27 | 0.30 | 0.16 | 0.21 | 0.26 | 0.32 | 0.30 | 0.21 | 0.23 | 0.33 | 0.27 | 0.28 | 0.31 | 0.26 | 0.24 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cohen, J.I.; Turgman-Cohen, S. The Conservation Genetics of Iris lacustris (Dwarf Lake Iris), a Great Lakes Endemic. Plants 2023, 12, 2557. https://doi.org/10.3390/plants12132557
Cohen JI, Turgman-Cohen S. The Conservation Genetics of Iris lacustris (Dwarf Lake Iris), a Great Lakes Endemic. Plants. 2023; 12(13):2557. https://doi.org/10.3390/plants12132557
Chicago/Turabian StyleCohen, James Isaac, and Salomon Turgman-Cohen. 2023. "The Conservation Genetics of Iris lacustris (Dwarf Lake Iris), a Great Lakes Endemic" Plants 12, no. 13: 2557. https://doi.org/10.3390/plants12132557
APA StyleCohen, J. I., & Turgman-Cohen, S. (2023). The Conservation Genetics of Iris lacustris (Dwarf Lake Iris), a Great Lakes Endemic. Plants, 12(13), 2557. https://doi.org/10.3390/plants12132557