
Licorice Germplasm Resources Identification Using DNA Barcodes Inner-Variants

 ,
,
Abstract
:1. Introduction
2. Results
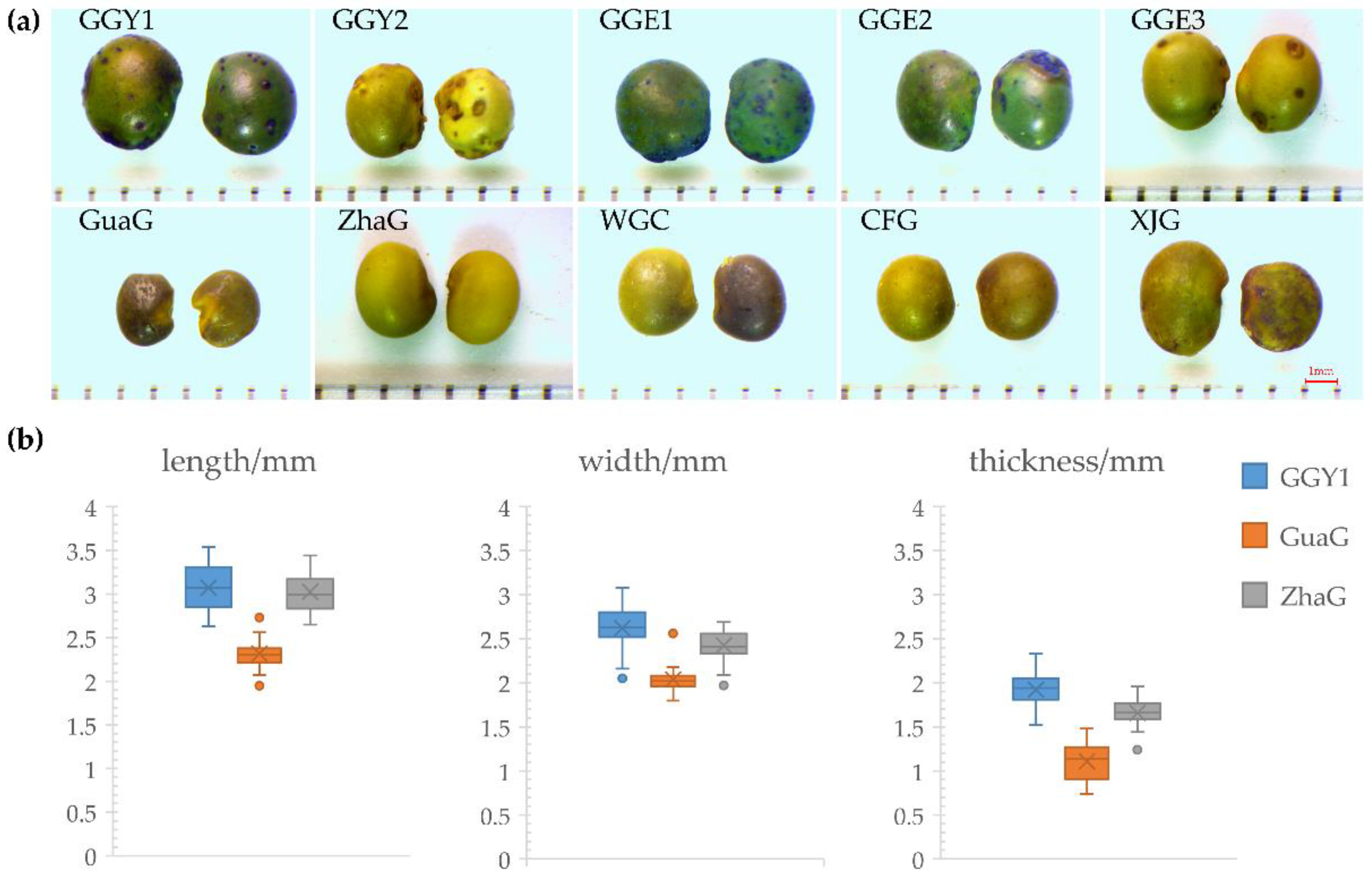
2.1. Morphological Characteristics of Licorice Seeds
2.2. Sanger Sequencing of DNA Barcoding
2.3. Full-Length Amplicon Sequencing of ITS2, psbA-trnH, and ITS
3. Discussion
4. Materials and Methods
4.1. Sample Collection of Licorice Seeds
4.2. Total DNA Extraction, PCR Amplification and Sanger Sequencing
4.3. NGS and SMRT Sequencing
4.4. Sequencing Results and Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Y.; Liu, C.; Zeng, B.; Fan, B.; Li, P.; Xu, T.; Liu, T. Research progress on germplasm resources of Glycyrrhizae Radix et Rhizoma. Chin. Tradit. Herb. Drugs 2013, 44, 3593–3598. [Google Scholar] [CrossRef]
- Commission Pharmacopoeia Commission (Ed.) Pharmacopoeia of the People’s Republic of China; China Medical Science Press: Beijing, China, 2020; Volume I, p. 88.
- Yang, R.; Li, W.D.; Yuan, B.C.; Ma, Y.-S.; Zhou, S.; Liu, C.S.; Liu, Y. Simultaneous determination of 18 α-glycyrrhizic acid and 18 β-glycyrrhizic acid in three licorice samples from different origin by HPLC. Chin. J. Pharm. Anal. 2016, 36, 1065–1071. [Google Scholar] [CrossRef]
- Zhang, Q.; Ye, M. Chemical analysis of the Chinese herbal medicine Gan-Cao (licorice). J. Chromatogr. A 2009, 1216, 1954–1969. [Google Scholar] [CrossRef]
- Zheng, Y.; Wei, J.; Leng, K.; Tao, W.; Fang, S.; Peng, G.; Duan, J. Research Advances in Resource Chemistry and Utilization of Genus Glycyrrhiza. Mod. Chin. Med. 2015, 17, 1096–1108. [Google Scholar] [CrossRef]
- Fulai, Y.; Yuqiang, F.; Wenquan, W.; Qiuling, W.; Fengbo, L. Genetic variability and interrelationships of mainly quantitative traits in Glycyrrhiza uralensis cultivated population. China J. Chin. Mater. Med. 2011, 36, 2457–2461. [Google Scholar] [CrossRef]
- Xie, L.B.; Lu, J.H.; Li, X.L.; Zhang, Y.; Wei, T.; Li, X.Y. The Cross Compatibility and Hybrid Seed Vigor among Three Glycyrrhiza Species. Plant Divers. Resour. 2014, 36, 342–348. [Google Scholar] [CrossRef]
- Chen, C.N.; Lu, J.H.; Li, X.Y.; Zhou, L.L.; Xie, L.B.; Li, X.L.; Song, F. Inheritance analysis and discovery of chloroplast paternal inheritance in interspecific crossing of Glycyrrhiza. Guihaia 2017, 37, 162–168. [Google Scholar] [CrossRef]
- Wei, S.-L.; Wang, W.-Q.; Wang, J.-Y.; Liu, C.-L.; Liu, F.-B.; Sun, M.-B.; Zhang, R.-F. Geographic variation and ecological mechanism of growth characteristics and glycyrrhizin content of annual Gancao (Radix Glycyrrhizae). J. Beijing Univ. Tradit. Chin. Med. 2012, 35, 406–411. [Google Scholar]
- Shengli, W.; Wenquan, W.; Changli, L.; Jiyong, W.; Ruifeng, Z.; Minbin, S. Analysis of broad-sense heritability and genetic correlation of production and content of glycyrrhizin of annual Glycyrrhiza uralensis. China J. Chin. Mater. Med. 2012, 37, 553–557. [Google Scholar] [CrossRef]
- Zhang, N. Identification and Application of Chinese Herbal Seed and Seedling Based on DNA Barcoding Technology. Master’s Thesis, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China, 2016. [Google Scholar]
- De Vere, N.; Rich, T.C.; Trinder, S.A.; Long, C. DNA barcoding for plants. Methods Mol. Biol. 2015, 1245, 101–118. [Google Scholar] [CrossRef]
- Kress, W.J.; Erickson, D.L. DNA barcodes: Methods and protocols. Methods Mol. Biol. 2012, 858, 3–8. [Google Scholar] [CrossRef]
- Nneji, L.; Adeola, A.; Ayoola, A.; Oladipo, S.; Wang, Y.; Malann, Y.; Anyaele, O.; Nneji, I.; Rahman, M.; Olory, C. DNA barcoding and species delimitation of butterflies (Lepidoptera) from Nigeria. Mol. Biol. Rep. 2020, 47, 9441–9457. [Google Scholar] [CrossRef]
- Hu, H.; Shen, X.; Liao, B.; Luo, L.; Xu, J.; Chen, S. Herbgenomics: A stepping stone for research into herbal medicine. Sci. China Life Sci. 2019, 62, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yao, H.; Han, J.; Xin, T.; Pang, X.; Shi, L.; Luo, K.; Song, J.; Hou, D.; Shi, S.; et al. Principles for molecular identification of traditional Chinese materia medica using DNA barcoding. China J. Chin. Mater. Med. 2013, 38, 141–148. [Google Scholar] [CrossRef]
- Hao, J.; Liu, G.; Liu, Y.; An, L.; Jin, W.; Yuan, S. Research Progress of DNA Molecular Marker Technology in Germplasm Identification of Glycyrrhiza Plants. Chin. J. Mod. Appl. Pharm. 2019, 36, 2485–2490. [Google Scholar] [CrossRef]
- Shang, X.; Deng, T.; Zeng, Y.; Zheng, S.; Wang, J.; Yang, L.; Lu, Z.; Wang, K. Study on Identification of Two Kinds Licorice Seeds Adulterant. Mod. Chin. Med. 2019, 21, 204–207. [Google Scholar] [CrossRef]
- Song, M.; Zhang, Y.; Lin, Y.; Tu, Y.; Ma, X.; Sun, W.; Xiang, L.; Jiao, W.; Liu, X. Identification of Plantaginis Semen based on ITS2 and psbA-trnH sequences. China J. Chin. Mater. Med. 2014, 39, 2227–2232. [Google Scholar] [CrossRef]
- Zhang, G.; Jin, Y.; Jia, J.; Song, J.; Shi, L.; Chen, S. Identification of Notopterygium seeds using DNA barcoding method. China J. Chin. Mater. Med. 2016, 41, 390–395. [Google Scholar] [CrossRef]
- Shi, L.; Jin, Y.; Zhao, C.; Xie, L.; Liu, J. Species Identification of Anemarrhenae Rhizoma Seeds by DNA Barcoding Technique. Chin. J. Exp. Tradit. Med. Formulae 2018, 24, 21–27. [Google Scholar] [CrossRef]
- Mu, W.; Yu, J.; Zhao, Q.; Shi, M.; Jin, Y.; Xie, H.; Liu, J.; Shi, L. Identification of the Seeds of Akebiae Caulis, a Chinese Medicinal Material by DNA Barcoding Technique. World Chin. Med. 2020, 15, 1271–1274. [Google Scholar] [CrossRef]
- Jiajia, Z.; Haiming, L.; Ting, L. Study on characteristics and identifyindexes of three kinds of medicine licorice seeds. J. Gansu Agric. Univ. 2012, 4, 68–72. [Google Scholar] [CrossRef]
- Wen-bin, L.; Lin, L.; Jie, C.; Xiao-yu, C.; Jun-ling, H.; Wen-quan, W. Research progress on genetic diversity of three medicinal licorice species. J. Chin. Med. Mater. 2019, 42, 463–469. [Google Scholar] [CrossRef]
- Cheng, X.; Su, X.; Chen, X.; Zhao, H.; Bo, C.; Xu, J.; Bai, H.; Ning, K. Biological ingredient analysis of traditional Chinese medicine preparation based on high-throughput sequencing: The story for Liuwei Dihuang Wan. Sci. Rep. 2014, 4, 5147. [Google Scholar] [CrossRef]
- Singer, E.; Bushnell, B.; Coleman-Derr, D.; Bowman, B.; Woyke, T. High-resolution phylogenetic microbial community profiling. ISME J. 2016, 10, 2020–2032. [Google Scholar] [CrossRef]
- Lo, Y.T.; Li, M.; Shaw, P.C. Identification of constituent herbs in ginseng decoctions by DNA markers. Chin. Med. 2015, 10, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Liu, C.; Zheng, X.; Wu, L.; Liu, Z.; Liao, B.; Shi, Y.; Li, X.; Xu, J.; Chen, S. Full-Length Multi-Barcoding: DNA Barcoding from Single Ingredient to Complex Mixtures. Genes 2019, 10, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabanal, F.; Mandáková, T.; Soto-Jiménez, L.; Greenhalgh, R.; Parrott, D.; Lutzmayer, S.; Steffen, J.; Nizhynska, V.; Mott, R.; Lysak, M.; et al. Epistatic and allelic interactions control expression of ribosomal RNA gene clusters in Arabidopsis thaliana. Genome Biol. 2017, 18, 75. [Google Scholar] [CrossRef] [PubMed]
- Salim, D.; Gerton, J. Ribosomal DNA instability and genome adaptability. Chromosome Res. 2019, 27, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Havlová, K.; Dvořáčková, M.; Peiro, R.; Abia, D.; Mozgová, I.; Vansáčová, L.; Gutierrez, C.; Fajkus, J. Variation of 45S rDNA intergenic spacers in Arabidopsis thaliana. Plant Mol. Biol. 2016, 92, 457–471. [Google Scholar] [CrossRef]
- Sims, J.; Sestini, G.; Elgert, C.; von Haeseler, A.; Schlögelhofer, P. Sequencing of the Arabidopsis NOR2 reveals its distinct organization and tissue-specific rRNA ribosomal variants. Nat. Commun. 2021, 12, 387. [Google Scholar] [CrossRef] [PubMed]
- Pontvianne, F.; Abou-Ellail, M.; Douet, J.; Comella, P.; Matia, I.; Chandrasekhara, C.; Debures, A.; Blevins, T.; Cooke, R.; Medina, F.; et al. Nucleolin is required for DNA methylation state and the expression of rRNA gene variants in Arabidopsis thaliana. PLoS Genet. 2010, 6, e1001225. [Google Scholar] [CrossRef] [Green Version]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; deWaard, J.R. Biological identifications through DNA barcodes. Proc. Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Xie, C.; Liu, C.; Song, J.; Yao, H.; Luo, K.; Zhu, Y.; Gao, T.; Pang, X.; Qian, J.; et al. Species Identification of Medicinal Pteridophytes by a DNA BarcodeMarker, the Chloroplast psbA-trnHIntergenic Region. Biol. Pharm. Bull. 2010, 33, 1919–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajibabaei, M.; Janzen, D.H.; Burns, J.M.; Hallwachs, W.; Hebert, P.D.N. DNA barcodes distinguish speciesof tropical Lepidoptera. Proc. Natl. Acad. Sci. USA 2006, 103, 968–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Wang, W.; Yang, N.; Guo, B.; Zhang, S.; Yang, R.; Yuan, Y.; Yu, J.; Hu, S.; Sun, Q.; et al. DNA barcoding provides distinction between Radix Astragali and its adulterants. Sci. China Life Sci. 2010, 53, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Nneji, L.; Adeola, A.; Mustapha, M.; Oladipo, S.; Djagoun, C.; Nneji, I.; Adedeji, B.; Olatunde, O.; Ayoola, A.; Okeyoyin, A.; et al. DNA Barcoding Silver Butter Catfish (Schilbe intermedius) Reveals Patterns of Mitochondrial Genetic Diversity Across African River Systems. Sci. Rep. 2020, 10, 7097. [Google Scholar] [CrossRef]
- Consortium for the Barcode of Life, Plant Working Group. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 1994, 106, 51–52. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Pang, X.; Liao, B.; Yao, H.; Song, J.; Chen, S. An authenticity survey of herbal medicines from markets in China using DNA barcoding. Sci. Rep. 2016, 6, 18723. [Google Scholar] [CrossRef]
- Lan, W.; Wei, S.; Bo, W.; Zhao, H.; Li, Y.; Cai, S.; Li, X.; Zhu, Y.; Hui, Y.; Song, J. An integrated system for identifying the hidden assassins in traditional medicines containing aristolochic acids. Sci. Rep. 2015, 5, 11318. [Google Scholar] [CrossRef]
- Chen, S.; Pang, X.; Yao, H.; Han, J.; Luo, K. Identification System and Perspective for DNA Barcoding Traditional Chinese Materia Medica. World Sci. Technol. Mod. Tradit. Chin. Med. Mater. Med. 2011, 13, 747–754. [Google Scholar] [CrossRef]
- Liao, B.; Hu, H.; Xiao, S.; Zhou, G.; Sun, W.; Chu, Y.; Meng, X.; Wei, J.; Zhang, H.; Xu, J.; et al. GPGD, an integrated and mineable genomics database for traditional medicines from major pharmacopoeias. Sci. China Life Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.J.; Hu, H.Y.; Gao, H.; Liu, X.; Chen, S.L. DNA barcoding and rapid identification of the precious herb Herba Anoectochili. Chin. J. Nat. Med. 2019, 17, 738–745. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, H.; Ning, K.; Bai, H. Advances in Quality Evaluation of Traditional Chinese Medicine by DNA Barcoding. Food Drug 2013, 15, 295–299. [Google Scholar] [CrossRef]
- Qing, Z.; Hong-bo, X.; Hong-ling, Z.; Chun-ying, Z.; Li-de, X.; Lin-chun, S.; Jin-xin, L. Advances in identification of traditional Chinese medicinal materials seeds using DNA barcoding technology. Chin. Tradit. Herb. Drugs 2019, 50, 3471–3476. [Google Scholar] [CrossRef]
- Wen-bin, L.; Xing-pu, S.; Cai-xia, C.; Lin, L.; Bin-bin, Y.; Ting-wei, D.; Jun-ling, H.; Wen-quan, W. Establishment of Seed and Seedling Grading Standard of Glycyrrhiza uralensis Fisch. Mod. Chin. Med. 2020, 22, 243–249. [Google Scholar] [CrossRef]
- Song, W.; Qiao, X.; Chen, K.; Wang, Y.; Ji, S.; Feng, J.; Li, K.; Lin, Y.; Ye, M. Biosynthesis-Based Quantitative Analysis of 151 Secondary Metabolites of Licorice To Differentiate Medicinal Glycyrrhiza Species and Their Hybrids. Anal. Chem. 2017, 89, 3146–3153. [Google Scholar] [CrossRef]
- Yang, R.; Li, W.; Ma, Y.; Zhou, S.; Xue, Y.; Lin, R.; Liu, Y. The molecular identification of licorice species and the quality evaluation of licorice slices. Acta Pharm. Sin. 2017, 52, 318–326. [Google Scholar] [CrossRef]
- Kondo, K.; Shiba, M.; Nakamura, R.; Morota, T.; Shoyama, Y. Constituent properties of licorices derived from Glycyrrhiza uralensis, G. glabra, or G. inflata identified by genetic information. Biol. Pharm. Bull. 2007, 30, 1271–1277. [Google Scholar] [CrossRef] [Green Version]
- Simmler, C.; Anderson, J.; Gauthier, L.; Lankin, D.; McAlpine, J.; Chen, S.; Pauli, G. Metabolite Profiling and Classification of DNA-Authenticated Licorice Botanicals. J. Nat. Prod. 2015, 78, 2007–2022. [Google Scholar] [CrossRef] [Green Version]
- Rodionov, A.; Gnutikov, A.; Nosov, N.; Machs, E.; Mikhaylova, Y.; Shneyer, V.; Punina, E. Intragenomic Polymorphism of the ITS 1 Region of 35S rRNA Gene in the Group of Grasses with Two-Chromosome Species: Different Genome Composition in Closely Related Zingeria Species. Plants 2020, 9, 1647. [Google Scholar] [CrossRef] [PubMed]
- Shen, A.J.J.; King, J.; Scott, H.; Colman, P.; Yates, C.J. Insights into pituitary tumorigenesis: From Sanger sequencing to next-generation sequencing and beyond. Expert Rev. Endocrinol. Metab. 2019, 14, 399–418. [Google Scholar] [CrossRef] [PubMed]
- Howard, C.; Lockie-Williams, C.; Slater, A. Applied Barcoding: The Practicalities of DNA Testing for Herbals. Plants 2020, 9, 1150. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zheng, S.; Shang, X.; Deng, T.; Wang, J.; Zhao, R. Molecular Identification in Genus of Glycyrrhiza Based on DNA Barcoding. Mod. Chin. Med. 2020, 22, 207–218. [Google Scholar] [CrossRef]
- Han, E.; Yoo, J.; Chae, H.; Lee, S.; Kim, D.H.; Kim, K.J.; Kim, Y.; Kim, M. Detection of BRCA1/2 large genomic rearrangement including BRCA1 promoter-region deletions using next-generation sequencing. Clin. Chim. Acta 2020, 505, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Lucas, E.R.; Miles, A.; Harding, N.J.; Clarkson, C.S.; Lawniczak, M.K.N.; Kwiatkowski, D.P.; Weetman, D.; Donnelly, M.J. Whole-genome sequencing reveals high complexity of copy number variation at insecticide resistance loci in malaria mosquitoes. Genome Res. 2019, 29, 1250–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. Guide Methods Appl. 1990, 1990, 315–322. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1. ITS2 | variable site/bp | ||||
| haplotype | 16 | 17 | 18 | Species | Sample |
| I2-i | T | G | C | G. uralensis | GGY1, GGY2, GGE1, GGE2, GGE3, WGC, CFG, XJG |
| I2-ii | C | A | A | G. glabra/inflata | GuaG, ZhaG |
| 2. ITS | variable site/bp | ||||
| haplotype | 187 | 411~413 | Species | Sample | |
| I-i | C | TGC | G. uralensis | GGY1, GGY2, GGE1, GGE2, GGE3, WGC, CFG, XJG | |
| I-ii | T | CAA | G. glabra/inflata | GuaG, ZhaG | |
| 3. psbA-trnH | variable site/bp | ||||
| haplotype | 189 | 235 | 288 | Species | Sample |
| PT-i | A | C | G | G. uralensis | GGY1, GGE1, GGE2, CFG |
| PT-ii | A | T | G | G. uralensis | GGY2, GGE3,WGC, XJG |
| PT-iii | C | T | G | G. glabra | GuaG |
| PT-iv | A | T | A | G. inflata | ZhaG |
| Total Reads | Raw Data | Merged | Filtered | Mean Length/bp |
|---|---|---|---|---|
| ITS2 | 1,387,176 | 1,246,275 | 1,199,501 | 407 |
| psbA-trnH | 1,128,832 | 1,120,013 | 1,082,034 | 389 |
| Sub Reads | Circular Consensus Sequence Filtration | Mean Length/bp | ||
| ITS | 14,540,927 | 218,721 | 736 | |
| Sample | Organ | Specie | Wild/Cultivated | Description |
|---|---|---|---|---|
| GGY1 | Seeds | G. uralensis | Cultivated | batch 1 of Market A |
| GGY2 | Seeds | G. uralensis | Cultivated | batch 2 of Market A |
| GGE1 | Seeds | G. uralensis | Cultivated | batch 1 of Market B |
| GGE2 | Seeds | G. uralensis | Cultivated | batch 2 of Market B |
| GGE3 | Seeds | G. uralensis | Cultivated | batch 2 of Market B |
| GuaG | Seeds | G. glabra | Wild | Market C |
| ZhaG | Seeds | G. inflata | Wild | Market C |
| WGC | Seeds | G. uralensis | Wild | Market C |
| CFG | Seeds | G. uralensis | Wild | Chifeng, Inner Mongolia, China |
| XJG | Seeds | G. uralensis | Wild | Xinjiang, China |
| Region | Primer Sequence | Program |
|---|---|---|
| ITS2 | ITS2-P3: YGACTCTCGGCAACGGATA ITS2-E4: RGTTTCTTTTCCTCCGCTTA [44] | 94 °C for 5 min; 94 °C for 1 min, 55 °C for 1 min, 72 °C for 1.5 min, 30 cycles; and 72 °C for 7 min |
| psbA-trnH | psbAF: GTTATGCATGAACGTAATGCTC trnHR: CGCGCATGGTGGATTCACAATCC [16] | 94 °C for 5 min; 94 °C for 1 min, 55 °C for 1 min, 72 °C for 1.5 min, 30 cycles; and 72 °C for 7 min |
| ITS | ITS4:TCCTCCGCTTATTGATATGC ITS5:GGAAGTAAAAGTCGTAACAAGG [58] | 95 °C for 3 min; 95 °C for 30 s, 55 °C for 30 s, 72 °C for 1.5 min, 35 cycles; and 72 °C for 10 min |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Guo, S.; Zheng, X.; Shen, X.; Zhang, T.; Liao, B.; He, W.; Hu, H.; Cheng, R.; Xu, J. Licorice Germplasm Resources Identification Using DNA Barcodes Inner-Variants. Plants 2021, 10, 2036. https://doi.org/10.3390/plants10102036
Liu Q, Guo S, Zheng X, Shen X, Zhang T, Liao B, He W, Hu H, Cheng R, Xu J. Licorice Germplasm Resources Identification Using DNA Barcodes Inner-Variants. Plants. 2021; 10(10):2036. https://doi.org/10.3390/plants10102036
Chicago/Turabian StyleLiu, Qianwen, Shuai Guo, Xiasheng Zheng, Xiaofeng Shen, Tianyi Zhang, Baosheng Liao, Wenrui He, Haoyu Hu, Ruiyang Cheng, and Jiang Xu. 2021. "Licorice Germplasm Resources Identification Using DNA Barcodes Inner-Variants" Plants 10, no. 10: 2036. https://doi.org/10.3390/plants10102036
APA StyleLiu, Q., Guo, S., Zheng, X., Shen, X., Zhang, T., Liao, B., He, W., Hu, H., Cheng, R., & Xu, J. (2021). Licorice Germplasm Resources Identification Using DNA Barcodes Inner-Variants. Plants, 10(10), 2036. https://doi.org/10.3390/plants10102036