
Population Structure and Genetic Diversity of Two-Rowed Barley Accessions from Kazakhstan Based on SNP Genotyping Data

 ,
,
Abstract
1. Introduction
2. Results
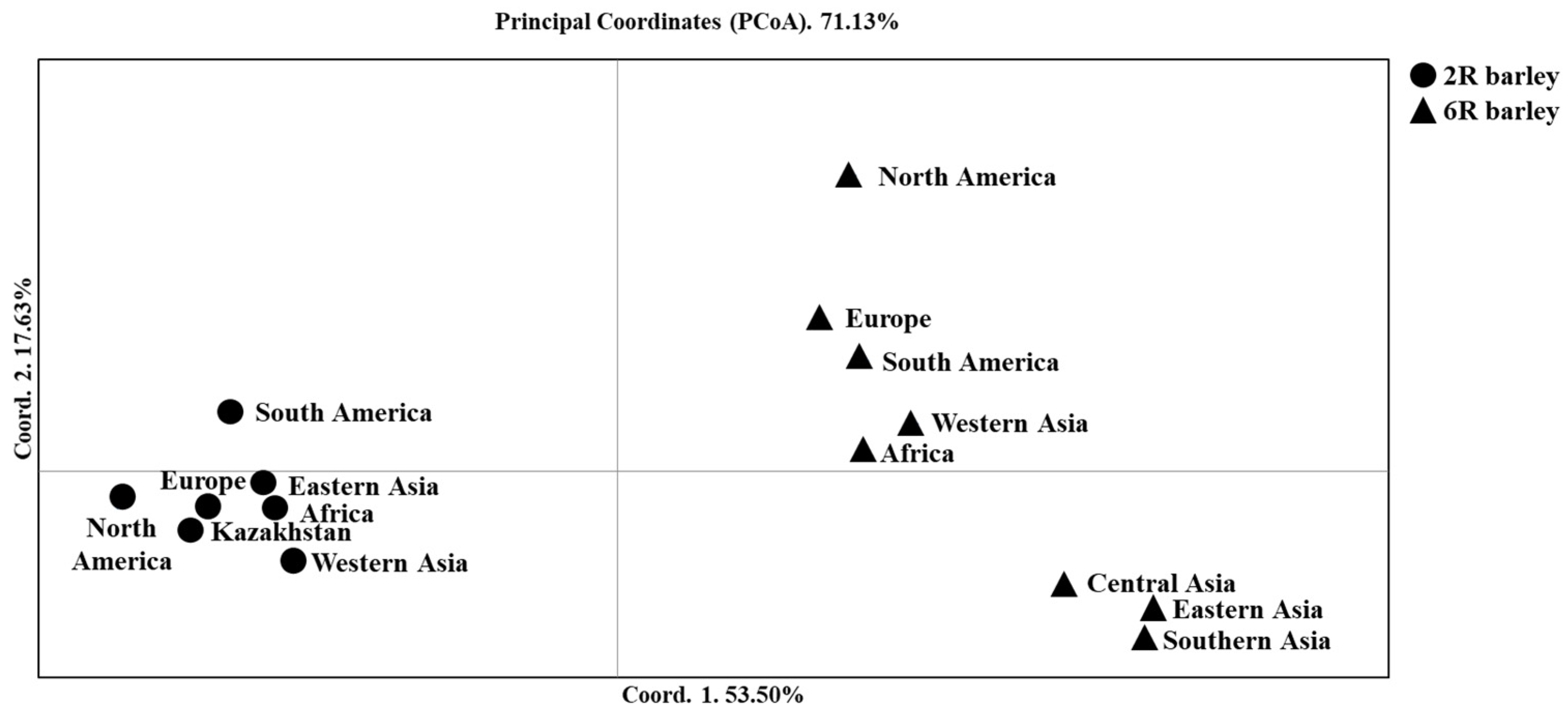
2.1. The Relationship of Two- and Six-Rowed Barley Accessions Based on SNP Marker Analysis
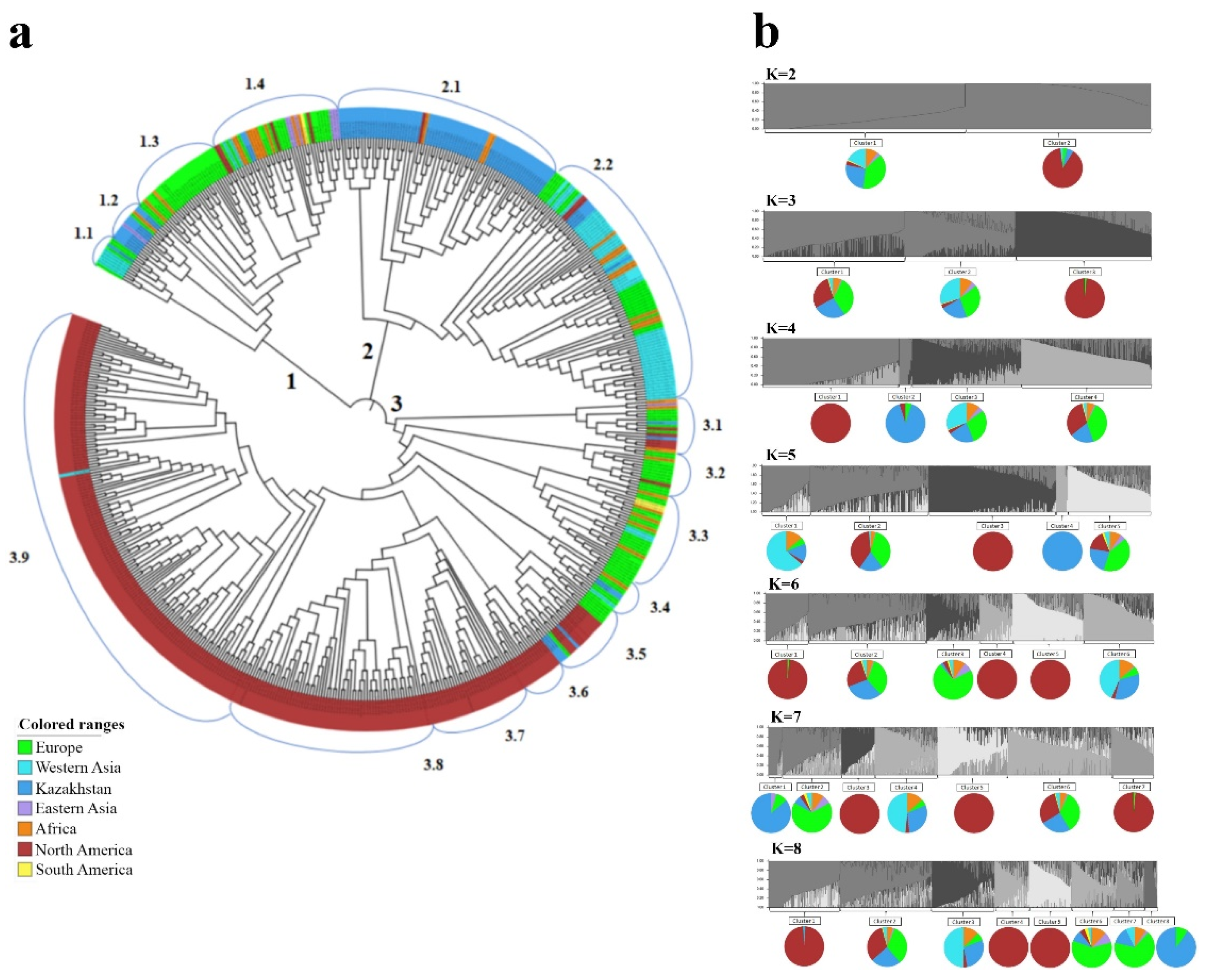
2.2. The Clusterization Analysis of Accessions from Kazakhstan in the Two-Rowed Barley Collection
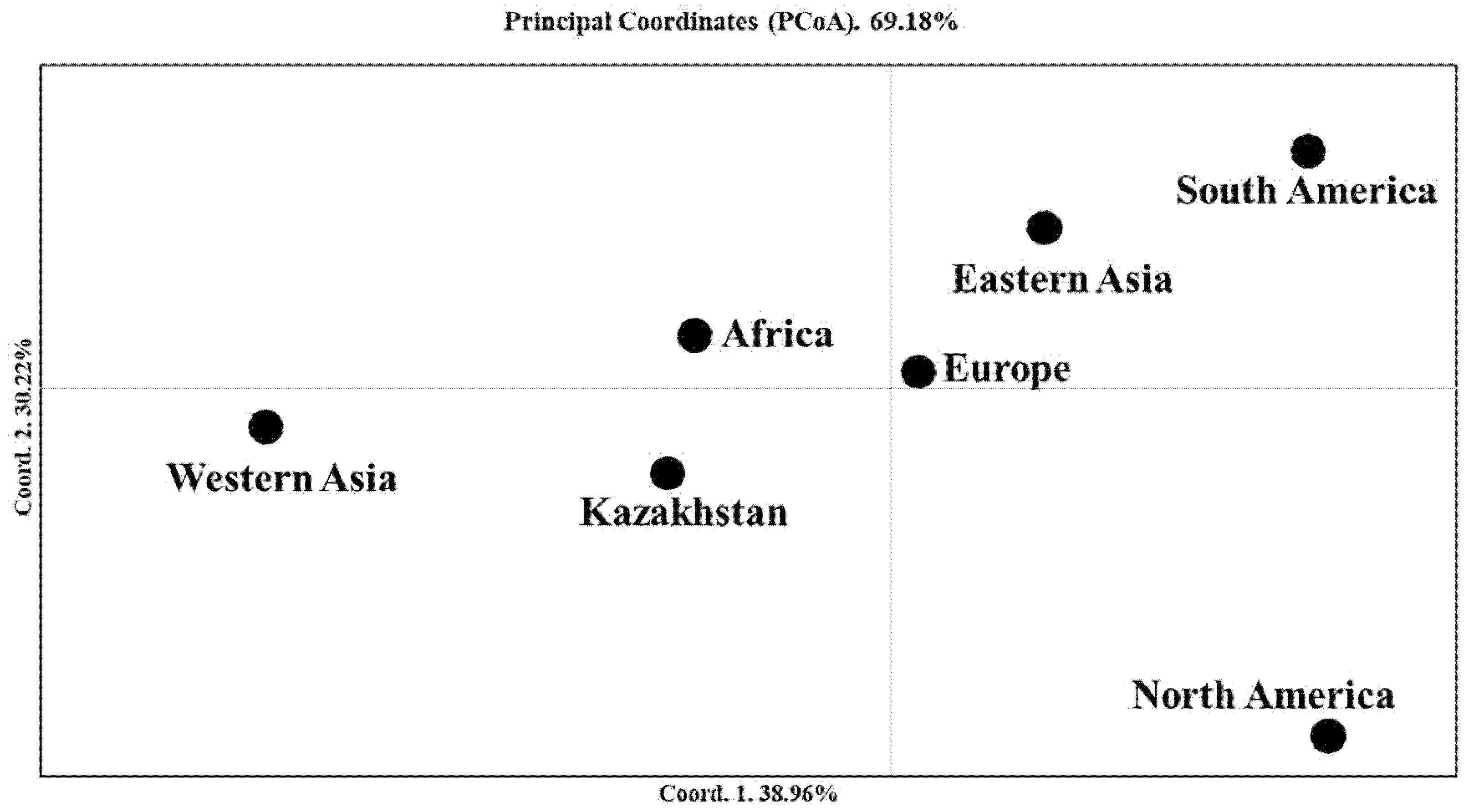
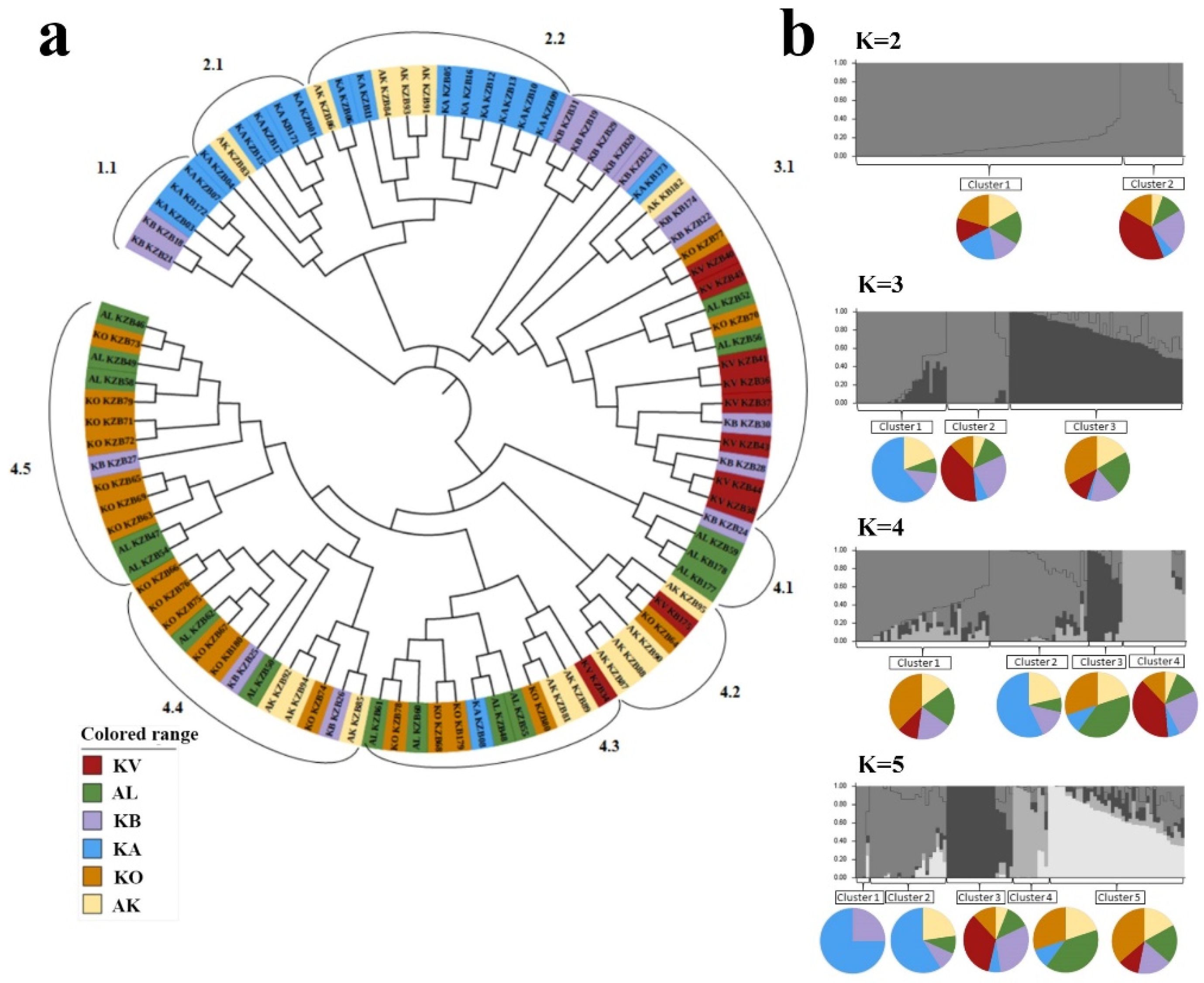
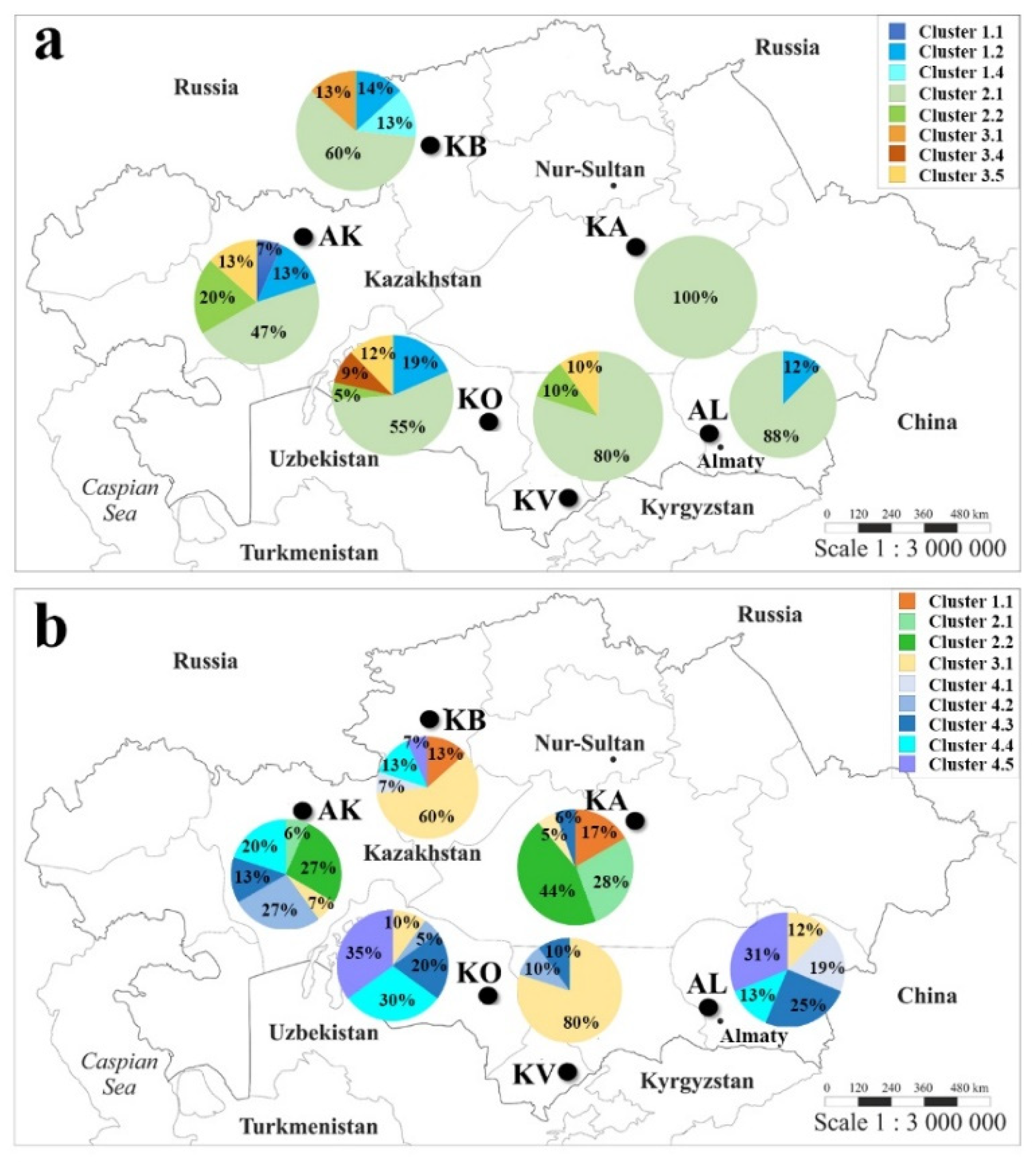
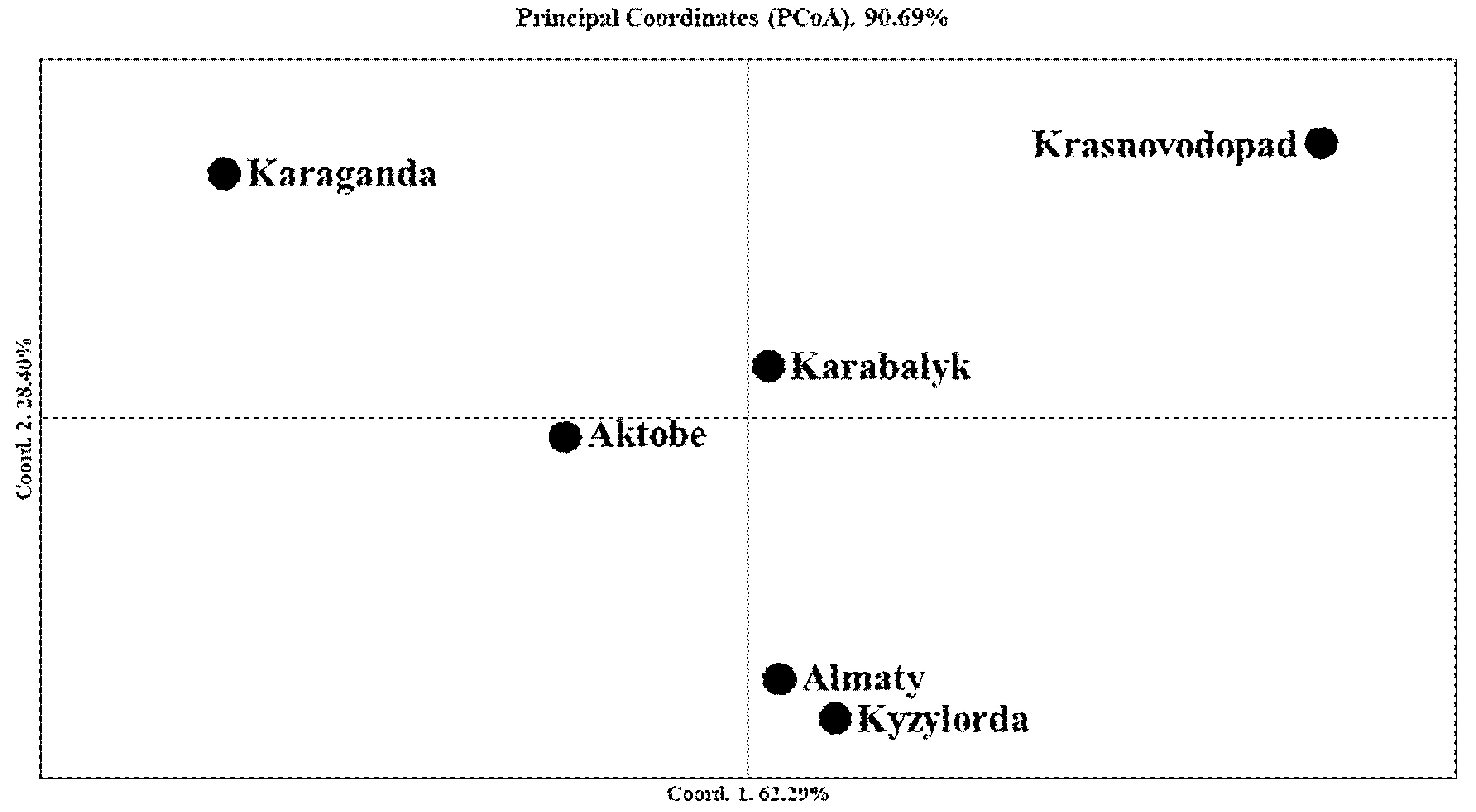
2.3. The Genetic Geography of Two-Rowed Barley Accessions from Six Breeding Programs of Kazakhstan According to the Illumina SNP Array
3. Discussion
3.1. The Clusterization Analysis of the Two-Rowed Barley Collection
3.2. The Genetic Geography of Kazakhstan Barley Accessions
3.3. The Mining of SNP Markers Associated with Plant Adaptation in Different Regions of Kazakhstan
4. Materials and Methods
4.1. Barley SNP Genotyping Sources
4.2. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Food and Agriculture Organization of the United Nations (FAO). Available online: http://www.fao.org/faostat/en/#data/QC (accessed on 21 May 2021).
- Turuspekov, Y.; Sariev, B.; Chudinov, V.; Sereda, G.; Tokhetova, L.; Ortaev, A.; Tsygankov, V.; Doszhanov, M.; Volis, S.; Abugalieva, S. Genotype×environment interaction patterns for grain yield of spring barley in different regions of Kazakhstan. Russ. J. Genet. 2013, 49, 196–205. [Google Scholar] [CrossRef]
- Genievskaya, Y.; Almerekova, S.; Sariev, B.; Chudinov, V.; Tokhetova, L.; Sereda, G.; Ortaev, A.; Tsygankov, V.; Blake, T.; Chao, S.; et al. Marker-trait associations in two-rowed spring barley accessions from Kazakhstan and the USA. PLoS ONE 2018, 13, e0205421. [Google Scholar] [CrossRef]
- Almerekova, S.; Sariev, B.; Abugalieva, A.; Chudinov, V.; Sereda, G.; Tokhetova, L.; Ortaev, A.; Tsygankov, V.; Blake, T.; Chao, S.; et al. Association mapping for agronomic traits in six-rowed spring barley from the USA harvested in Kazakhstan. PLoS ONE 2019, 14, e0221064. [Google Scholar] [CrossRef] [PubMed]
- Turuspekov, Y.; Ormanbekova, D.; Rsaliev, A.; Abugalieva, S. Genome-wide association study on stem rust resistance in Kazakh spring barley lines. BMC Plant Biol. 2016, 16, 13–21. [Google Scholar] [CrossRef]
- Harlan, J.R.; Zohary, D. Distribution of wild wheats and barley. Science 1966, 153, 1074–1080. [Google Scholar] [CrossRef]
- Nevo, E.; Beiles, A.; Zohary, D. Genetic resources of wild barley in the Near East: Structure, evolution and application in breeding. Biol. J. Linn. Soc. 1986, 27, 355–380. [Google Scholar] [CrossRef]
- Nevo, E. Genome evolution of wild cereal diversity and prospects for crop improvement. Plant Genet. Resour. 2006, 4, 36–46. [Google Scholar] [CrossRef]
- Nevo, E. Evolution of Wild Barley and Barley Improvement. In Advance in Barley Sciences; Zhang, G., Li, C., Liu, X., Eds.; Springer: Dordrecht, The Netherlands, 2013; pp. 1–23. ISBN 978-94-007-4682-4 . [Google Scholar] [CrossRef]
- Vavilov, N.I. The phytogeographical basis for plant breeding. In Vavilov NI Origin and Geography of Cultivated Plants; Dorofeyev, V.F., Ed.; Cambridge University Press: Cambridge, UK, 1935. [Google Scholar]
- Zohary, D.; Hopf, M.; Weiss, E. Domestication of Plants in the Old World: The origin and Spread of Domesticated Plants in Southwest Asia, Europe, and the Mediterranean Basin, 4th ed.; Oxford University Press Inc.: New York, NY, USA, 2012; ISBN 978-0-19-954906-1. [Google Scholar]
- Dai, F.; Nevo, E.; Wu, D.; Comadran, J.; Zhou, M.; Qiu, L.; Chen, Z.; Beiles, A.; Chen, G.; Zhang, G. Tibet is one of the centers of domestication of cultivated barley. Proc. Natl. Acad. Sci. USA 2012, 109, 16969–16973. [Google Scholar] [CrossRef] [PubMed]
- Pankin, A.; Altmüller, J.; Becker, C.; von Korff, M. Targeted resequencing reveals genomic signatures of barley domestication. New Phytol. 2018, 218, 1247–1259. [Google Scholar] [CrossRef]
- von Bothmer, R.; van Hintum, T.; Knüpffer, H.; Sato, K. Diversity in Barley (Hordeum vulgare); Elsevier: Amsterdam, Netherlands, 2003; ISBN 0 444 505857. [Google Scholar]
- Knüpffer, H. Triticeae genetic resources in ex situ genebank collections. In Genetics and Genomics of the Triticeae; Feuillet, C., Muehlbauer, G.J., Eds.; Springer: New York, NY, USA, 2009; pp. 31–79. ISBN 978-0-387-77488-6. [Google Scholar]
- Bockelman, H.E.; Valkoun, J. Barley germplasm conservation and resources. In Barley: Production, Improvement, and Uses; Ullrich, S.E., Ed.; Wiley-Blackwel: Oxford, UK, 2011; pp. 144–159. ISBN 978-0-8138-0123-0. [Google Scholar]
- Cunha Alves, A.A.; Azevedo, V. Embrapa Network for Brazilian Plant Genetic Resources Conservation. Biopreservation Biobanking 2018, 16, 350–360. [Google Scholar] [CrossRef]
- Azough, Z.; Kehel, Z.; Benomar, A.; Bellafkih, M.; Amri, A. Predictive Characterization of ICARDA Genebank Barley Accessions sing FIGS and Machine Learning. In Proceedings of the 15th Intelligent Environments Workshop, Rabat, Morocco, 24–27 June 2019; Muñoz, A., Ouhbi, S., Minker, W., Eds.; IOS Press BV: Amsterdam, Netherlands, 2019; pp. 121–129. Available online: https://hdl.handle.net/20.500.11766/10493 (accessed on 6 August 2021).
- Takeya, M.; Yamasaki, F.; Uzuhashi, S.; Aoki, T.; Sawada, H.; Nagai, T.; Tomioka, K.; Tomooka, N.; Sato, T.; Kawase, M. NIASGBdb: NIAS Genebank databases for genetic resources and plant disease information. Nucleic Acids Res. 2011, 39, D1108–D1113. [Google Scholar] [CrossRef][Green Version]
- Nagel, M.; Vogel, H.; Landjeva, S.; Buck-Sorlin, G.; Lohwasser, U.; Scholz, U.; Börner, A. Seed conservation in ex situ genebanks—Genetic studies on longevity in barley. Euphytica 2009, 170, 5–14. [Google Scholar] [CrossRef]
- González, M.Y.; Philipp, N.; Schulthess, A.W.; Weise, S.; Zhao, Y.; Börner, A.; Oppermann, M.; Graner, A.; Reif, L.C. Unlocking historical phenotypic data from an ex situ collection to enhance the informed utilization of genetic resources of barley (Hordeum sp.). Theor. Appl. Genet. 2018, 131, 2009–2019. [Google Scholar] [CrossRef]
- Milner, S.G.; Jost, M.; Taketa, S.; Mazón, E.R.; Himmelbach, A.; Oppermann, M.; Weise, S.; Knüpffer, H.; Basterrechea, M.; König, P.; et al. Genebank genomics highlights the diversity of a global barley collection. Nat. Genet. 2019, 51, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Loskutov, I.G. Genetic resources and main directions and results of barley and oat breeding in Russia. Agron. Res. 2010, 8, 673–679. [Google Scholar]
- National Center for Biotechnology Information (NCBI). Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 24 May 2021).
- Silvester, N.; Alako, B.; Amid, C.; Cerdeño-Tarrága, A.; Clarke, L.; Cleland, I.; Harrison, P.W.; Jayathilaka, S.; Kay, S.; Keane, T.; et al. The european nucleotide archive in 2017. Nucleic Acids Res. 2018, 46, D36–D40. [Google Scholar] [CrossRef]
- Tateno, Y. DNA data bank of Japan (DDBJ) for genome scale research in life science. Nucleic Acids Res. 2002, 30, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, G.; Karsch-Mizrachi, I.; Takagi, T.; International Nucleotide Sequence Database Collaboration. The international nucleotide sequence database collaboration. Nucleic Acids Res. 2016, 44, D48–D50. [Google Scholar] [CrossRef]
- The International Barley Genome Sequencing Consortium. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Close, T.J.; Bhat, P.R.; Lonardi, S.; Wu, Y.; Rostoks, N.; Ramsay, L.; Druka, A.; Stein, N.; Svensson, J.T.; Wanamaker, S.; et al. Development and implementation of high-throughput SNP genotyping in barley. BMC Genom. 2009, 10, 1–13. [Google Scholar] [CrossRef]
- Comadran, J.; Kilian, B.; Russell, J.; Ramsay, L.; Stein, N.; Ganal, M.; Shaw, P.; Bayer, M.; Thomas, W.; Marshall, D.; et al. Natural variation in a homolog of Antirrhinum CENTRORADIALIS contributed to spring growth habit and environmental adaptation in cultivated barley. Nat Genet. 2012, 44, 1388–1392. [Google Scholar] [CrossRef]
- Bayer, M.M.; Rapazote-Flores, P.; Ganal, M.; Hedley, P.E.; Macaulay, M.; Plieske, J.; Ramsay, L.; Russell, J.; Shaw, P.D.; Thomas, W.; et al. Development and evaluation of a barley 50k iSelect SNP array. Front. Plant Sci. 2017, 8, 1792. [Google Scholar] [CrossRef]
- Bykova, I.V.; Lashina, N.M.; Efimov, V.M.; Afanasenko, O.S.; Khlestkina, E.K. Identification of 50 K Illumina-chip SNPs associated with resistance to spot blotch in barley. BMC Plant Biol. 2017, 17, 250. [Google Scholar] [CrossRef] [PubMed]
- Wenzl, P.; Carling, J.; Kudrna, D.; Jaccoud, D.; Huttner, E.; Kleinhofs, A.; Kilian, A. Diversity Arrays Technology (DArT) for whole-genome profiling of barley. Proc. Natl. Acad. Sci. USA 2004, 101, 9915–9920. [Google Scholar] [CrossRef] [PubMed]
- Poland, J.A.; Rife, T.W. Genotyping-by-sequencing for plant breeding and genetics. The Plant Genome 2012, 5, 92–102. [Google Scholar] [CrossRef]
- Kobayashi, F.; Tanaka, T.; Kanamori, H.; Wu, J.; Katayose, Y.; Handa, H. Characterization of a mini core collection of Japanese wheat varieties using single-nucleotide polymorphisms generated by genotyping-by-sequencing. Breed. Sci. 2016, 66, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Ishikawa, G.; Ogiso-Tanaka, E.; Yanagisawa, T.; Sato, K. Development of genome-wide SNP markers for barley via reference-based RNA-Seq analysis. Front. Plant Sci. 2019, 10, 577. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Amatriaín, M.; Cuesta-Marcos, A.; Endelman, J.B.; Comadran, J.; Bonman, J.M.; Bockelman, H.E.; Chao, S.; Russell, J.; Waugh, R.; Hayes, P.M.; et al. The USDA Barley Core Collection: Genetic Diversity, Population Structure, and Potential for Genome-Wide Association Studies. PLoS ONE 2014, 9, e94688. [Google Scholar] [CrossRef]
- Bengtsson, T.; The PPP Barley Consortium; Manninen, O.; Jahoor, A.; Orabi, J. Genetic diversity, population structure and linkage disequilibrium in Nordic spring barley (Hordeum vulgare L. subsp. vulgare). Genet. Resour. Crop. Evol. 2017, 64, 2021–2033. [Google Scholar] [CrossRef]
- Elakhdar, A.; Kumamaru, T.; Qualset, C.O.; Brueggeman, R.S.; Amer, K.; Capo-chichi, L. Assessment of genetic diversity in Egyptian barley (Hordeum vulgare L.) genotypes using SSR and SNP markers. Genet. Resour. Crop. Evol. 2018, 65, 1937–1951. [Google Scholar] [CrossRef]
- Hill, C.B.; Angessa, T.T.; Zhang, X.Q.; Chen, K.; Zhou, G.; Tan, C.; Wang, P.; Westcott, S.; Li, C. A global barley panel revealing genomic signatures of breeding in modern Australian cultivars. Plant J. 2021, 106, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Sato, K. History and future perspectives of barley genomics. DNA Res. 2020, 27, dsaa023. [Google Scholar] [CrossRef] [PubMed]
- Jayakodi, M.; Padmarasu, S.; Haberer, G.; Bonthala, V.S.; Gundlach, H.; Monat, C.; Lux, T.; Kamal, N.; Lang, D.; Himmelbach, A.; et al. The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 2020, 588, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Jayakodi, M.; Schreiber, M.N.; Stein, N.; Mascher, M. Building pan-genome infrastructures for crop plants and their use in association genetics. DNA Res. 2021, 28, dsaa030. [Google Scholar] [CrossRef] [PubMed]
- Lister, D.L.; Jones, H.; Oliveira, H.R.; Petrie, C.A.; Liu, X.; Cockram, J.; Kneale, C.J.; Kovaleva, O.; Jones, M.K. Barley heads east: Genetic analyses reveal routes of spread through diverse Eurasian landscapes. PLoS ONE 2018, 13, e0196652. [Google Scholar] [CrossRef] [PubMed]
- Turuspekov, Y.; Abugalieva, S.; Ermekbayev, K.; Sato, K. Genetic characterization of wild barley populations (Hordeum vulgare ssp. spontaneum) from Kazakhstan based on genome wide SNP analysis. Breed. Sci. 2014, 64, 399–403. [Google Scholar] [CrossRef]
- Germinate Barley SNP Platforms. Available online: https://ics.hutton.ac.uk (accessed on 16 March 2021).
- Zohary, D.; Hopf, M. Domestication of Plants in the Old World, 2nd ed.; Oxford University Press: Oxford, UK, 1993. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; von Holdt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Thorndike, R.L. Who belongs in the family? Psychometrika 1953, 18, 267–276. [Google Scholar] [CrossRef]
- Alqudah, A.M.; Koppolu, R.; Wolde, G.M.; Graner, A.; Schnurbusch, T. The genetic architecture of barley plant stature. Front. Genet. 2016, 7, 117. [Google Scholar] [CrossRef]
- Alqudah, A.M.; Youssef, H.M.; Graner, A.; Schnurbusch, T. Natural variation and genetic make-up of leaf blade area in spring barley. Theor. Appl. Genet. 2018, 131, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Amezrou, R.; Gyawali, S.; Belqadi, L.; Chao, S.; Arbaoui, M.; Mamidi, S.; Rehman, S.; Sreedasyam, A.; Verma, R.P.S. Molecular and phenotypic diversity of ICARDA spring barley (Hordeum vulgare L.) collection. Genet. Resour. Crop. Evol. 2018, 65, 255–269. [Google Scholar] [CrossRef]
- Bretani, G.; Rossini, L.; Ferrandi, C.; Russell, J.; Waugh, R.; Kilian, B.; Bagnaresi, P.; Cattivelli, L.; Fricano, A. Segmental duplications are hot spots of copy number variants affecting barley gene content. Plant J. 2020, 103, 1073–1088. [Google Scholar] [CrossRef]
- Takahashi, R. The origin and evolution of cultivated barley. Adv. Genet. 1955, 7, 227–266. [Google Scholar] [CrossRef]
- Turuspekov, Y.; Plieske, J.; Ganal, M.; Akhunov, E.; Abugalieva, S. Phylogenetic analysis of wheat cultivars in Kazakhstan based on the wheat 90 K single nucleotide polymorphism array. Plant Genet. Resour. 2017, 15, 29–35. [Google Scholar] [CrossRef]
- Malysheva-Otto, L.; Ganal, M.W.; Law, J.R.; Reeves, J.C.; Röder, M.S. Temporal trends of genetic diversity in European barley cultivars (Hordeum vulgare L.). Mol. Breed. 2007, 20, 309–322. [Google Scholar] [CrossRef]
- Orabi, J.; Jahoor, A.; Backes, G. Genetic diversity and population structure of wild and cultivated barley from West Asia and North Africa. Plant Breed. 2009, 128, 606–614. [Google Scholar] [CrossRef]
- Tondelli, A.; Xu, X.; Moragues, M.; Sharma, R.; Schnaithmann, F.; Ingvardsen, C.; Manninen, O.; Comadran, J.; Russell, J.; Waugh, R.; et al. Structural and Temporal Variation in Genetic Diversity of European Spring Two-Row Barley Cultivars and Association Mapping of Quantitative Traits. The Plant Genome 2013, 6. [Google Scholar] [CrossRef]
- Gryaznov, A.A. Karabalyk Barley (Forage, Groats, Beer); Kustanay Printing House: Kustanay, Kazakhstan, 1996; p. 448. (In Russian) [Google Scholar]
- Griffiths, S.; Dunford, R.P.; Coupland, G.; Laurie, D.A. The evolution of CONSTANS-like gene families in barley, rice, and Arabidopsis. Plant Physiol. 2003, 131, 1855–1867. [Google Scholar] [CrossRef]
- Cockram, J.; Thiel, T.; Steuernagel, B.; Stein, N.; Taudien, S.; Bailey, P.C.; O′Sullivan, D.M. Genome Dynamics Explain the Evolution of Flowering Time CCT Domain Gene Families in the Poaceae. PLoS ONE 2012, 7, e45307. [Google Scholar] [CrossRef] [PubMed]
- Herzig, P.; Maurer, A.; Draba, V.; Sharma, R.; Draicchio, F.; Bull, H.; Milne, L.; Thomas, W.T.B.; Flavell, A.J.; Pillen, K. Contrasting genetic regulation of plant development in wild barley grown in two European environments revealed by nested association mapping. J. Exp. Bot. 2018, 69, 1517–1531. [Google Scholar] [CrossRef]
- Zikhali, M.; Wingen, L.U.; Griffiths, S. Delimitation of the Earliness per se D1 (Eps-D1) flowering gene to a subtelomeric chromosomal deletion in bread wheat (Triticum aestivum). J. Exp. Bot. 2016, 67, 287–299. [Google Scholar] [CrossRef]
- Chary, S.N.; Hicks, G.R.; Choi, Y.G.; Carter, D.; Raikhel, N.V. Trehalose-6-phosphate synthase/phosphatase regulates cell shape and plant architecture in Arabidopsis. Plant Physiol. 2008, 146, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.J.; Watson, A.; Griffiths, C.A. Trehalose 6-phosphate signalling and impact on crop yield. Biochem. Soc. Trans. 2020, 48, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria; Available online: http://www.R-project.org/ (accessed on 30 March 2021).
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2012, 6, 288–295. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Origin | n | Na | Ne | I | h | uh | %P |
|---|---|---|---|---|---|---|---|
| Africa | 31 | 1.929 ± 0.006 | 1.515 ± 0.008 | 0.460 ± 0.005 | 0.304 ± 0.004 | 0.314 ± 0.004 | 92.94% |
| East Asia | 9 | 1.747 ± 0.010 | 1.479 ± 0.008 | 0.413 ± 0.006 | 0.278 ± 0.004 | 0.313 ± 0.005 | 74.68% |
| Europe | 123 | 1.966 ± 0.004 | 1.519 ± 0.008 | 0.463 ± 0.005 | 0.306 ± 0.004 | 0.309 ± 0.004 | 96.57% |
| Kazakhstan | 94 | 1.920 ± 0.006 | 1.445 ± 0.008 | 0.410 ± 0.005 | 0.267 ± 0.004 | 0.270 ± 0.004 | 91.97% |
| North America | 279 | 1.925 ± 0.006 | 1.396 ± 0.008 | 0.357 ± 0.006 | 0.233 ± 0.004 | 0.234 ± 0.004 | 92.53% |
| South America | 3 | 1.411 ± 0.011 | 1.329 ± 0.009 | 0.262 ± 0.007 | 0.183 ± 0.005 | 0.274 ± 0.007 | 41.07% |
| Western Asia | 58 | 1.903 ± 0.007 | 1.453 ± 0.008 | 0.414 ± 0.005 | 0.271 ± 0.004 | 0.276 ± 0.004 | 90.28% |
| Africa | East Asia | Europe | Kazakhstan | North America | South America | Western Asia | |
|---|---|---|---|---|---|---|---|
| Africa | |||||||
| East Asia | 0.027 | ||||||
| Europe | 0.014 | 0.031 | |||||
| Kazakhstan | 0.020 | 0.052 | 0.029 | ||||
| North America | 0.068 | 0.082 | 0.056 | 0.072 | |||
| South America | 0.040 | 0.045 | 0.046 | 0.079 | 0.087 | ||
| Western Asia | 0.019 | 0.078 | 0.054 | 0.049 | 0.110 | 0.106 |
| n | Na | Ne | I | h | uh | %P | |
|---|---|---|---|---|---|---|---|
| Kyzylorda | 20 | 1.932 ± 0.003 | 1.583 ± 0.004 | 0.501 ± 0.003 | 0.337 ± 0.002 | 0.355 ± 0.002 | 93.23% |
| Aktobe | 15 | 1.897 ± 0.004 | 1.540 ± 0.004 | 0.474 ± 0.003 | 0.316 ± 0.002 | 0.341 ± 0.002 | 89.72% |
| Almaty | 16 | 1.853 ± 0.005 | 1.515 ± 0.005 | 0.451 ± 0.003 | 0.302 ± 0.002 | 0.322 ± 0.002 | 85.31% |
| Karabalyk | 15 | 1.868 ± 0.004 | 1.508 ± 0.005 | 0.450 ± 0.003 | 0.299 ± 0.002 | 0.320 ± 0.002 | 56.85% |
| Karaganda | 18 | 1.788 ± 0.005 | 1.430 ± 0.005 | 0.388 ± 0.003 | 0.255 ± 0.002 | 0.271 ± 0.003 | 78.82% |
| Krasnovodopad | 10 | 1.600 ± 0.006 | 1.173 ± 0.002 | 0.224 ± 0.003 | 0.131 ± 0.002 | 0.145 ± 0.002 | 60% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almerekova, S.; Genievskaya, Y.; Abugalieva, S.; Sato, K.; Turuspekov, Y. Population Structure and Genetic Diversity of Two-Rowed Barley Accessions from Kazakhstan Based on SNP Genotyping Data. Plants 2021, 10, 2025. https://doi.org/10.3390/plants10102025
Almerekova S, Genievskaya Y, Abugalieva S, Sato K, Turuspekov Y. Population Structure and Genetic Diversity of Two-Rowed Barley Accessions from Kazakhstan Based on SNP Genotyping Data. Plants. 2021; 10(10):2025. https://doi.org/10.3390/plants10102025
Chicago/Turabian StyleAlmerekova, Shyryn, Yuliya Genievskaya, Saule Abugalieva, Kazuhiro Sato, and Yerlan Turuspekov. 2021. "Population Structure and Genetic Diversity of Two-Rowed Barley Accessions from Kazakhstan Based on SNP Genotyping Data" Plants 10, no. 10: 2025. https://doi.org/10.3390/plants10102025
APA StyleAlmerekova, S., Genievskaya, Y., Abugalieva, S., Sato, K., & Turuspekov, Y. (2021). Population Structure and Genetic Diversity of Two-Rowed Barley Accessions from Kazakhstan Based on SNP Genotyping Data. Plants, 10(10), 2025. https://doi.org/10.3390/plants10102025