Total Fatty Acid Analysis of Human Blood Samples in One Minute by High-Resolution Mass Spectrometry

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Standards
2.2. Human Plasma Samples
2.3. Sample Preparation
2.4. Mass Spectrometric Analysis
2.5. Lipid Identification and Quantification
2.6. FAME Analysis
3. Results and Discussion
3.1. Total Fatty Acyl Analysis by Direct Infusion FTMS Analysis
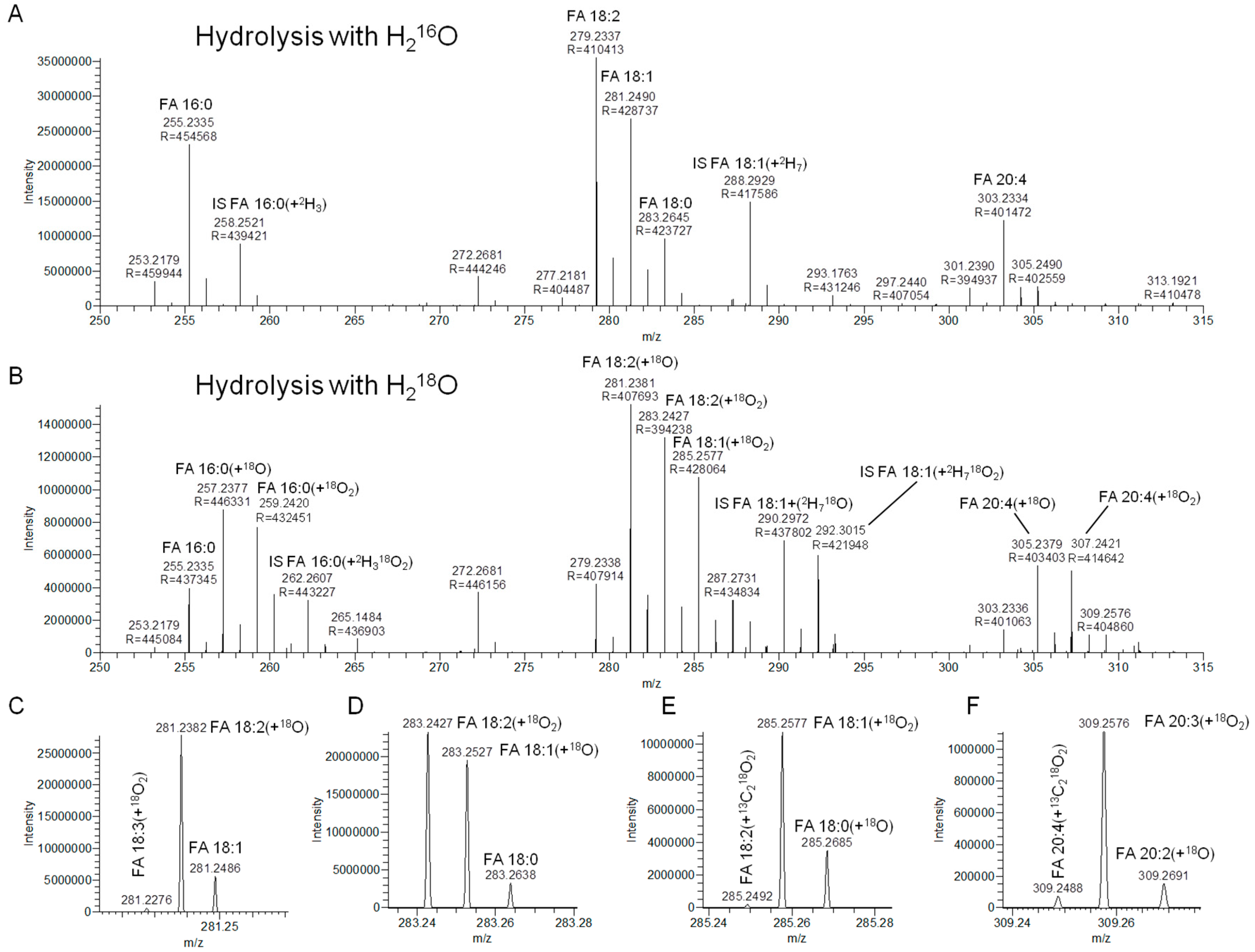
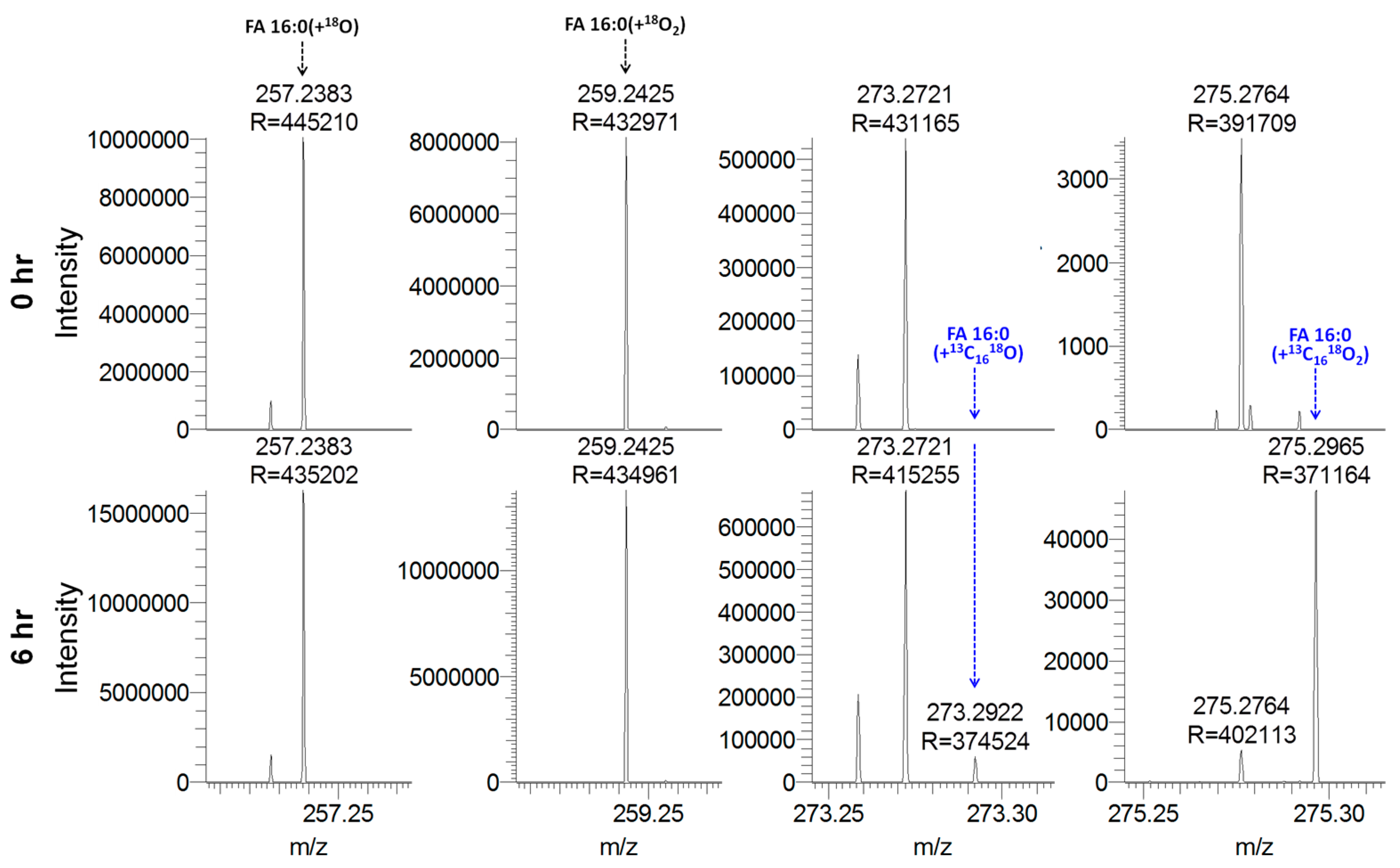
3.2. Hydrolysis with H218O Improves Signal-to-Background
3.3. Instrument Response Is Independent of Fatty Acyl Chain Length and Double Bond Number
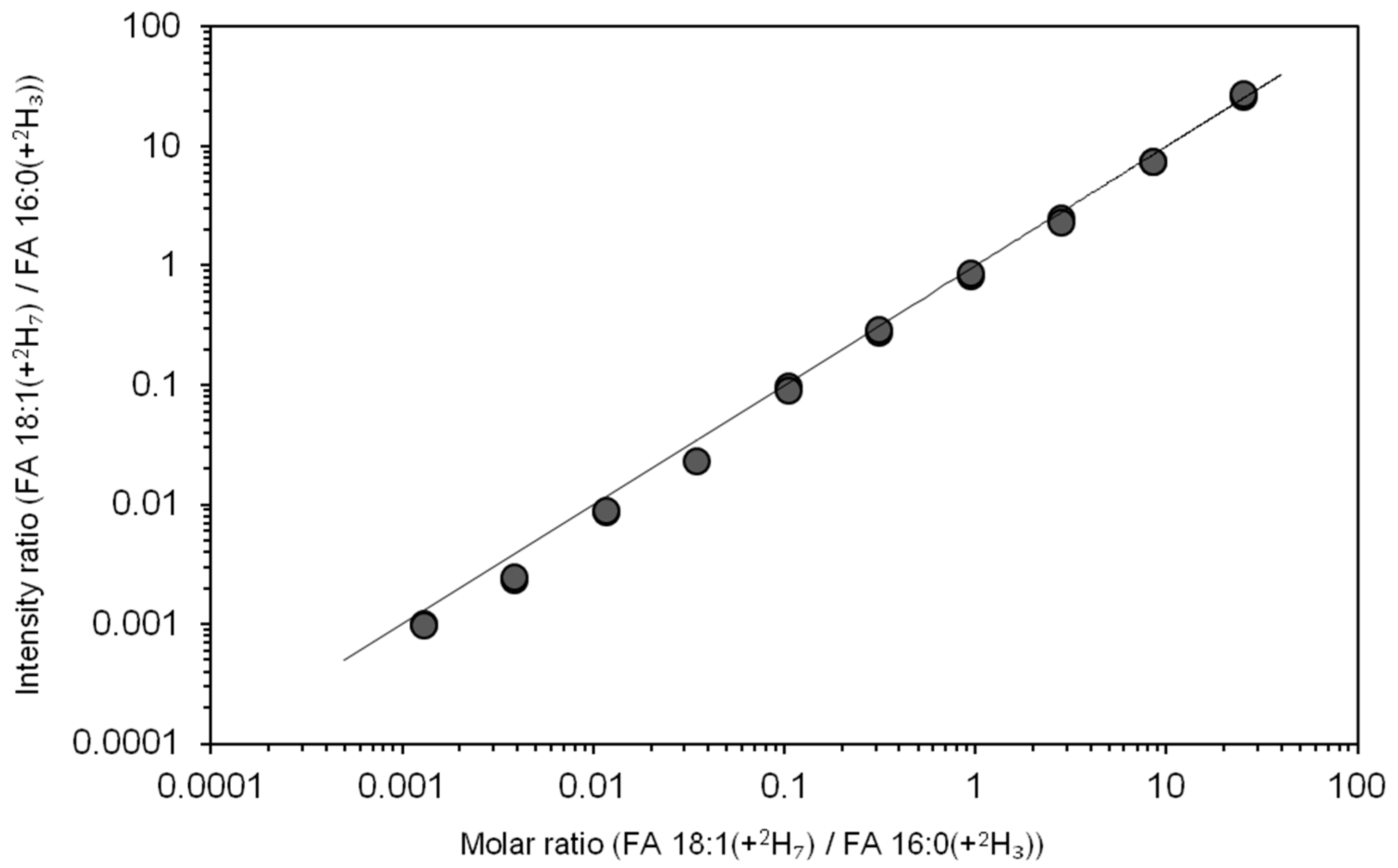
3.4. Dynamic Quantification Range of Total Fatty Acyl Analysis by FTMS
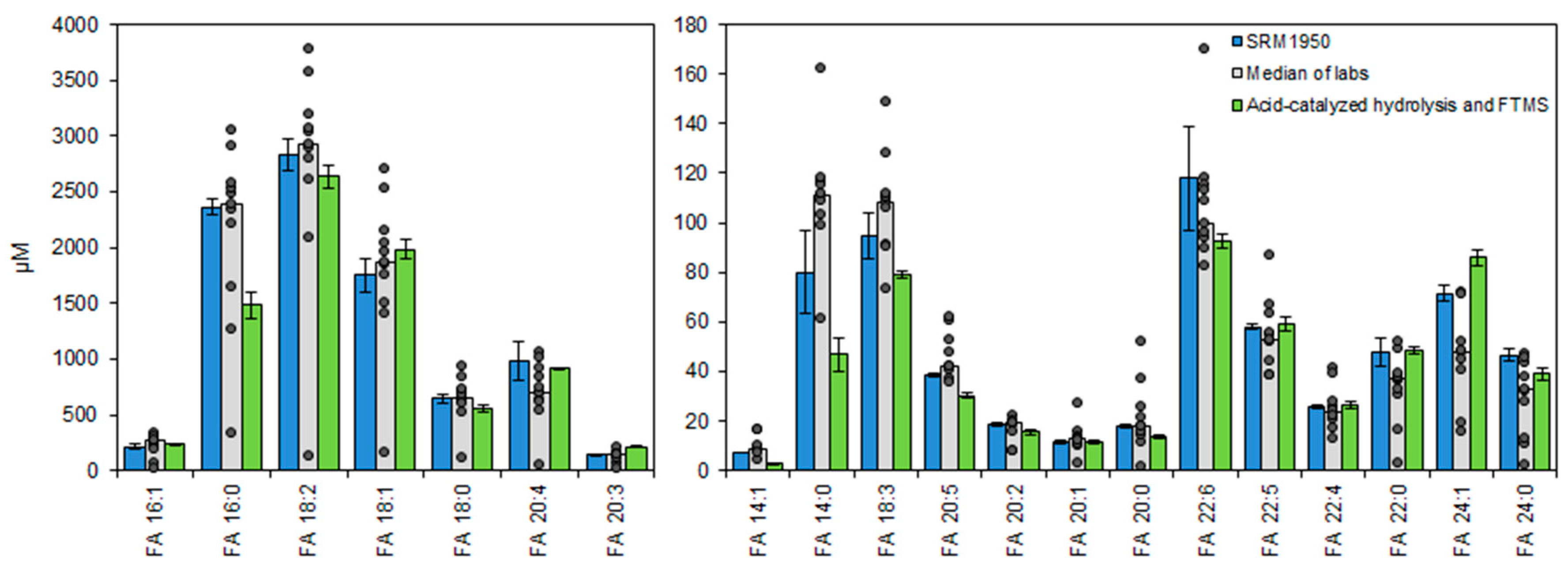
3.5. Accurate Fatty Acyl Quantification by High Resolution FTMS Analysis
3.6. Total Fatty Acyl Analysis by FTMS Is Precise and Applicable for Routine Analysis
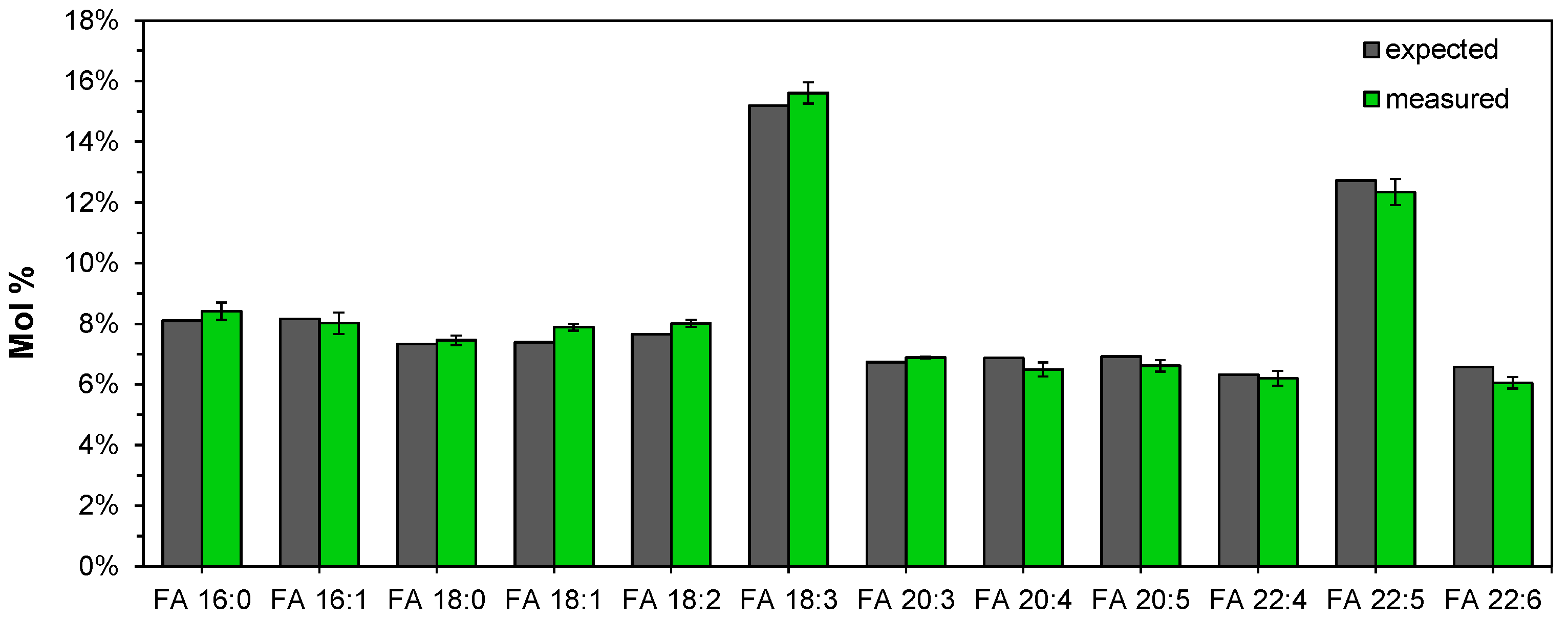
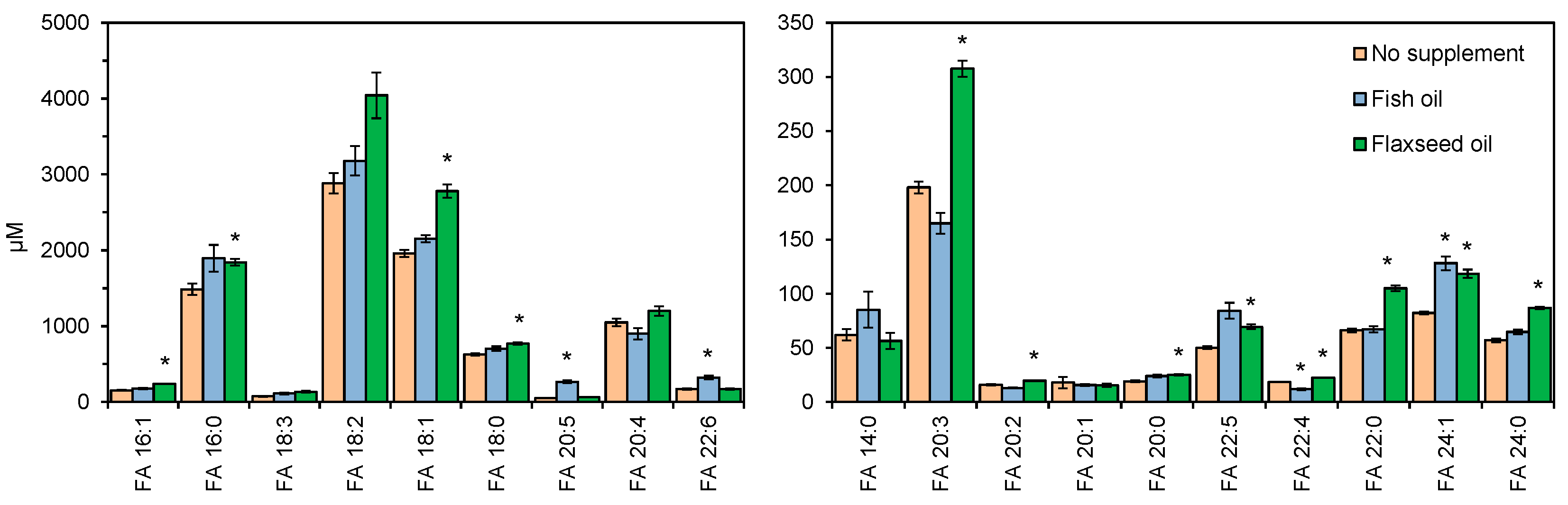
3.7. Lipotyping of Human Serum Samples by Total Fatty Acyl Analysis
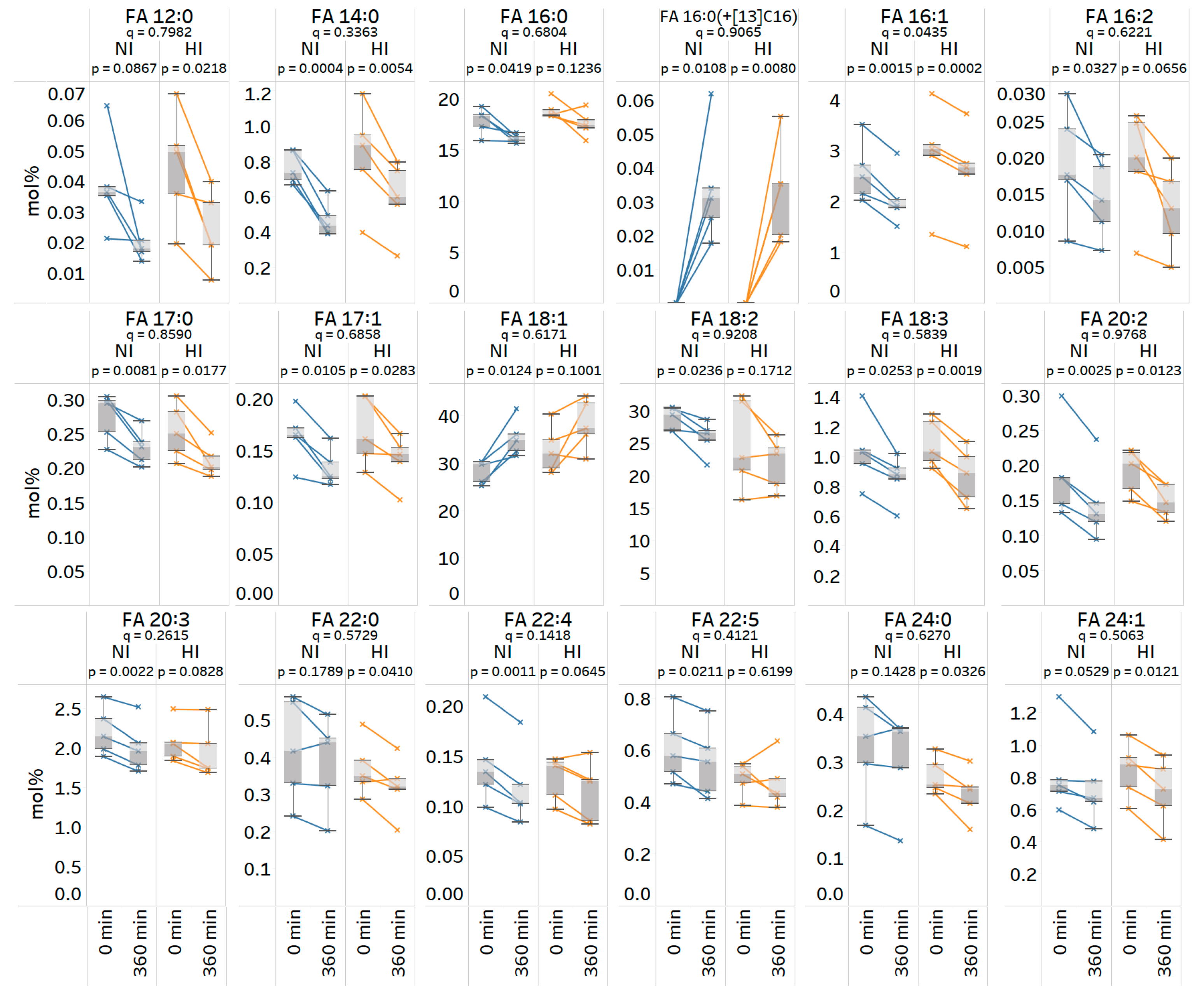
3.8. Quantitative Analysis of Stable Isotope-Labeled Fatty Acyls in Human Plasma
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siguel, E. Diagnosing essential fatty acid deficiency. Circulation 1998, 97, 2580–2583. [Google Scholar] [CrossRef] [PubMed]
- Moser, A.B.; Kreiter, N.; Bezman, L.; Lu, S.; Raymond, G.V.; Naidu, S.; Moser, H.W. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann. Neurol. 1999, 45, 100–110. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Katan, M.B.; Ascherio, A.; Stampfer, M.J.; Willett, W.C. Trans fatty acids and cardiovascular disease. N. Engl. J. Med. 2006, 354, 1601–1613. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.T.; Rimm, E.B.; Spiegelman, D.; Rifai, N.; Tofler, G.H.; Willett, W.C.; Hu, F.B. Association between dietary patterns and plasma biomarkers of obesity and cardiovascular disease risk. Am. J. Clin. Nutr. 2001, 73, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Forouhi, N.G.; Koulman, A.; Sharp, S.J.; Imamura, F.; Kroger, J.; Schulze, M.B.; Crowe, F.L.; Huerta, J.M.; Guevara, M.; Beulens, J.W.; et al. Differences in the prospective association between individual plasma phospholipid saturated fatty acids and incident type 2 diabetes: The EPIC-InterAct case-cohort study. Lancet Diabetes Endocrinol. 2014, 2, 810–818. [Google Scholar] [CrossRef]
- Loomba, R.; Quehenberger, O.; Armando, A.; Dennis, E.A. Polyunsaturated fatty acid metabolites as novel lipidomic biomarkers for noninvasive diagnosis of nonalcoholic steatohepatitis. J. Lipid Res. 2015, 56, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Moghaddam, L.; Brinin, A.; Williams, B.; Mundree, S.; Haritos, V.S. Functional assessment of plant and microalgal lipid pathway genes in yeast to enhance microbial industrial oil production. Biotechnol. Appl. Biochem. 2018, 65, 138–144. [Google Scholar] [CrossRef]
- Liang, M.H.; Jiang, J.G. Advancing oleaginous microorganisms to produce lipid via metabolic engineering technology. Prog. Lipid Res. 2013, 52, 395–408. [Google Scholar] [CrossRef]
- Mansson, H.L. Fatty acids in bovine milk fat. Food Nutr. Res. 2008, 52. [Google Scholar] [CrossRef]
- Roberts, L.D.; McCombie, G.; Titman, C.M.; Griffin, J.L. A matter of fat: An introduction to lipidomic profiling methods. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 871, 174–181. [Google Scholar] [CrossRef]
- Dodds, E.D.; McCoy, M.R.; Rea, L.D.; Kennish, J.M. Gas chromatographic quantification of fatty acid methyl esters: Flame ionization detection vs. electron impact mass spectrometry. Lipids 2005, 40, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Ruttkay-Nedecky, B.; Subik, J. The OGD1 gene, affecting 2-oxoglutarate dehydrogenase in S. cerevisiae, is closely linked to HIS5 on chromosome IX. Curr. Genet. 1990, 17, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, H.C.; Wei, N.; McGunigale, S.L.; Vesper, H.W. Quantitation of trans-fatty acids in human blood via isotope dilution-gas chromatography-negative chemical ionization-mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1076, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, O.; Armando, A.M.; Dennis, E.A. High sensitivity quantitative lipidomics analysis of fatty acids in biological samples by gas chromatography-mass spectrometry. Biochim. Biophys. Acta 2011, 1811, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Pettinella, C.; Lee, S.H.; Cipollone, F.; Blair, I.A. Targeted quantitative analysis of fatty acids in atherosclerotic plaques by high sensitivity liquid chromatography/tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 850, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Fan, J.; Lu, W.; White, E.; Rabinowitz, J.D. Liquid chromatography-high resolution mass spectrometry analysis of fatty acid metabolism. Anal. Chem. 2011, 83, 9114–9122. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Choi, A.A.; Wu, R. systematic analysis of fatty acids in human cells with a multiplexed isobaric tag (TMT)-based method. J. Proteome Res. 2018, 17, 1606–1614. [Google Scholar] [CrossRef]
- Stahlman, M.; Ejsing, C.S.; Tarasov, K.; Perman, J.; Boren, J.; Ekroos, K. High-throughput shotgun lipidomics by quadrupole time-of-flight mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 2664–2672. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, X.; Li, J.; Zhang, W.; Cheng, J.X.; Ouyang, Z.; Xia, Y. Photochemical tagging for quantitation of unsaturated fatty acids by mass spectrometry. Anal. Chem. 2016, 88, 8931–8935. [Google Scholar] [CrossRef]
- Wang, M.; Han, R.H.; Han, X. Fatty acidomics: Global analysis of lipid species containing a carboxyl group with a charge-remote fragmentation-assisted approach. Anal. Chem. 2013, 85, 9312–9320. [Google Scholar] [CrossRef]
- Gallego, S.F.; Sprenger, R.R.; Neess, D.; Pauling, J.K.; Faergeman, N.J.; Ejsing, C.S. Quantitative lipidomics reveals age-dependent perturbations of whole-body lipid metabolism in ACBP deficient mice. Biochim. Biophys. Acta 2016, 1862, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Pramfalk, C.; Pavlides, M.; Banerjee, R.; McNeil, C.A.; Neubauer, S.; Karpe, F.; Hodson, L. fasting plasma insulin concentrations are associated with changes in hepatic fatty acid synthesis and partitioning prior to changes in liver fat content in healthy adults. Diabetes 2016, 65, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Ellis, S.R.; Paine, M.R.L.; Eijkel, G.B.; Pauling, J.K.; Husen, P.; Jervelund, M.W.; Hermansson, M.; Ejsing, C.S.; Heeren, R.M.A. Automated, parallel mass spectrometry imaging and structural identification of lipids. Nat. Methods 2018, 15, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Ecker, J.; Scherer, M.; Schmitz, G.; Liebisch, G. A rapid GC-MS method for quantification of positional and geometric isomers of fatty acid methyl esters. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 897, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Gallego, S.F.; Hojlund, K.; Ejsing, C.S. Easy, fast, and reproducible quantification of cholesterol and other lipids in human plasma by combined high resolution MSX and FTMS analysis. J. Am. Soc. Mass Spectrom. 2018, 29, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, A.; Sprenger, R.R.; Tarasov, K.; Ruckerbauer, D.E.; Hannibal-Bach, H.K.; Zanghellini, J.; Jensen, O.N.; Ejsing, C.S. Quantitative analysis of proteome and lipidome dynamics reveals functional regulation of global lipid metabolism. Chem. Biol. 2015, 22, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Graessler, J.; Schwudke, D.; Schwarz, P.E.; Herzog, R.; Shevchenko, A.; Bornstein, S.R. Top-down lipidomics reveals ether lipid deficiency in blood plasma of hypertensive patients. PLoS ONE 2009, 4, e6261. [Google Scholar] [CrossRef]
- Yao, C.H.; Liu, G.Y.; Yang, K.; Gross, R.W.; Patti, G.J. Inaccurate quantitation of palmitate in metabolomics and isotope tracer studies due to plastics. Metabolomics 2016, 12. [Google Scholar] [CrossRef]
- Schantz, M.M.; Powers, C.D.; Schleicher, R.L.; Betz, J.M.; Wise, S.A. Interlaboratory analytical comparison of fatty acid concentrations in serum or plasma. Clin. Chim. Acta 2016, 462, 148–152. [Google Scholar] [CrossRef]
- Liebisch, G.; Lieser, B.; Rathenberg, J.; Drobnik, W.; Schmitz, G. High-throughput quantification of phosphatidylcholine and sphingomyelin by electrospray ionization tandem mass spectrometry coupled with isotope correction algorithm. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2004, 1686, 108–117. [Google Scholar] [CrossRef]
- Liebisch, G.; Binder, M.; Schifferer, R.; Langmann, T.; Schulz, B.; Schmitz, G. High throughput quantification of cholesterol and cholesteryl ester by electrospray ionization tandem mass spectrometry (ESI-MS/MS). Biochim. Biophys. Acta 2006, 176, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Benner, B.A., Jr.; Schantz, M.M.; Powers, C.D.; Schleicher, R.L.; Camara, J.E.; Sharpless, K.E.; Yen, J.H.; Sniegoski, L.T. Standard Reference Material (SRM) 2378 fatty acids in frozen human serum. Certification of a clinical SRM based on endogenous supplementation of polyunsaturated fatty acids. Anal. Bioanal. Chem. 2018, 410, 2321–2329. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, A.S.; Roberts, R.; Fielding, B.A.; Hodson, L.; Blaak, E.E.; Wagenmakers, A.J.; Gilbert, M.; Karpe, F.; Frayn, K.N. Preferential uptake of dietary fatty acids in adipose tissue and muscle in the postprandial period. Diabetes 2007, 56, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Fernandez, L.; Laiglesia, L.M.; Huerta, A.E.; Martinez, J.A.; Moreno-Aliaga, M.J. Omega-3 fatty acids and adipose tissue function in obesity and metabolic syndrome. Prostag. Other Lipid Mediat. 2015, 121, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, K.; Ekroos, K.; Suoniemi, M.; Kauhanen, D.; Sylvanne, T.; Hurme, R.; Gouni-Berthold, I.; Berthold, H.K.; Kleber, M.E.; Laaksonen, R.; et al. Molecular lipids identify cardiovascular risk and are efficiently lowered by simvastatin and PCSK9 deficiency. J. Clin. Endocrinol. Metab. 2014, 99, E45–E52. [Google Scholar] [CrossRef] [PubMed]
- Laaksonen, R.; Ekroos, K.; Sysi-Aho, M.; Hilvo, M.; Vihervaara, T.; Kauhanen, D.; Suoniemi, M.; Hurme, R.; Marz, W.; Scharnagl, H.; et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur. Heart J. 2016, 37, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Pauling, J.K.; Hermansson, M.; Hartler, J.; Christiansen, K.; Gallego, S.F.; Peng, B.; Ahrends, R.; Ejsing, C.S. Proposal for a common nomenclature for fragment ions in mass spectra of lipids. PLoS ONE 2017, 12, e0188394. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.W.; Pham, H.; Thomas, M.C.; Blanksby, S.J. Identification of double bond position in lipids: From GC to OzID. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 2722–2735. [Google Scholar] [CrossRef]
- Surma, M.A.; Herzog, R.; Vasilj, A.; Klose, C.; Christinat, N.; Morin-Rivron, D.; Simons, K.; Masoodi, M.; Sampaio, J.L. An automated shotgun lipidomics platform for high throughput, comprehensive, and quantitative analysis of blood plasma intact lipids. Eur. J. Lipid Sci. Technol. 2015, 117, 1540–1549. [Google Scholar] [CrossRef]
- Sigruener, A.; Kleber, M.E.; Heimerl, S.; Liebisch, G.; Schmitz, G.; Maerz, W. Glycerophospholipid and sphingolipid species and mortality: The Ludwigshafen Risk and Cardiovascular Health (LURIC) study. PLoS ONE 2014, 9, e85724. [Google Scholar] [CrossRef]
- Sales, S.; Graessler, J.; Ciucci, S.; Al-Atrib, R.; Vihervaara, T.; Schuhmann, K.; Kauhanen, D.; Sysi-Aho, M.; Bornstein, S.R.; Bickle, M.; et al. Gender, Contraceptives and individual metabolic predisposition shape a healthy plasma lipidome. Sci. Rep. 2016, 6, 27710. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intra-Day | Inter-Day | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Day 1 (n = 5) | Day 2 (n = 5) | Day 3 (n = 5) | (n= 3 × 5) | ||||||
| Analyte | Insulin | µM ± SD | CV (%) | µM ± SD | CV (%) | µM ± SD | CV (%) | µM ± SD | CV (%) |
| FA 16:0 | Normal | 1852 ± 103 | 5.5 | 2085 ± 179 | 8.6 | 2028 ± 105 | 5.2 | 1988 ± 161 | 8.1 |
| High | 4239 ± 294 | 6.9 | 3798 ± 288 | 7.6 | 4373 ± 218 | 5.0 | 4137 ± 356 | 8.6 | |
| FA 18:1 | Normal | 2898 ± 54 | 1.9 | 3132 ± 156 | 5.0 | 3130 ± 183 | 5.8 | 3053 ± 174 | 5.7 |
| High | 10859 ± 534 | 4.9 | 9618 ± 279 | 2.9 | 11114 ± 508 | 4.6 | 10531 ± 797 | 7.6 | |
| FA 18:2 | Normal | 3404 ± 133 | 3.9 | 3734 ± 192 | 5.1 | 3765 ± 162 | 4.3 | 3634 ± 227 | 6.2 |
| High | 3442 ± 275 | 8.0 | * 3031 ± 102 | 3.4 | 3640 ± 412 | 11.3 | 3396 ± 378 | 11.1 | |
| FA 20:4 | Normal | 920 ± 24 | 2.6 | 948 ± 26 | 2.8 | 998 ± 80 | 8.0 | 955 ± 58 | 6.0 |
| High | 1728 ± 95 | 5.5 | 1504 ± 56 | 3.7 | 1750 ± 111 | 6.4 | 1661 ± 142 | 8.6 | |
| FA 20:5 | Normal | 108 ± 3.6 | 3.3 | 115 ± 3.1 | 2.7 | 121 ± 10 | 8.6 | 115 ± 8.2 | 7.1 |
| High | 188 ± 8.9 | 4.7 | 165 ± 6.5 | 3.9 | 194 ± 14 | 7.0 | 182 ± 16 | 8.8 | |
| FA 22:5 | Normal | 73 ± 3.0 | 4.1 | 76 ± 1.8 | 2.4 | 82 ± 5.9 | 7.2 | 77 ± 5.3 | 6.8 |
| High | 146 ± 8.5 | 5.8 | 123 ± 4.4 | 3.6 | 147 ± 9.9 | 6.7 | 139 ± 13 | 9.6 | |
| FA 22:6 | Normal | 197 ± 5.2 | 2.7 | 205 ± 5.3 | 2.6 | 216 ± 16 | 7.4 | 206 ± 12 | 6.0 |
| High | 163 ± 8.5 | 5.2 | 141 ± 6.4 | 4.5 | 167 ± 14 | 8.6 | 157 ± 15 | 9.7 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallego, S.F.; Hermansson, M.; Liebisch, G.; Hodson, L.; Ejsing, C.S. Total Fatty Acid Analysis of Human Blood Samples in One Minute by High-Resolution Mass Spectrometry. Biomolecules 2019, 9, 7. https://doi.org/10.3390/biom9010007
Gallego SF, Hermansson M, Liebisch G, Hodson L, Ejsing CS. Total Fatty Acid Analysis of Human Blood Samples in One Minute by High-Resolution Mass Spectrometry. Biomolecules. 2019; 9(1):7. https://doi.org/10.3390/biom9010007
Chicago/Turabian StyleGallego, Sandra F., Martin Hermansson, Gerhard Liebisch, Leanne Hodson, and Christer S. Ejsing. 2019. "Total Fatty Acid Analysis of Human Blood Samples in One Minute by High-Resolution Mass Spectrometry" Biomolecules 9, no. 1: 7. https://doi.org/10.3390/biom9010007
APA StyleGallego, S. F., Hermansson, M., Liebisch, G., Hodson, L., & Ejsing, C. S. (2019). Total Fatty Acid Analysis of Human Blood Samples in One Minute by High-Resolution Mass Spectrometry. Biomolecules, 9(1), 7. https://doi.org/10.3390/biom9010007