
Structural View of Cryo-Electron Microscopy-Determined ATP-Binding Cassette Transporters in Human Multidrug Resistance


Abstract
1. Introduction
2. ABC Transporters and Their Roles in MDR
3. Cryo-EM Study of Drug Recognition, Translocation, and Inhibition of MDR-Related ABC Transporters
3.1. ABCB1
3.1.1. Drug Binding and Translocation of ABCB1
3.1.2. Inhibition of ABCB1
3.2. ABCG2
3.2.1. Drug Binding and Translocation of ABCG2
3.2.2. Inhibition of ABCG2
3.3. ABCC Family: ABCC1, ABCC3, and ABCC4
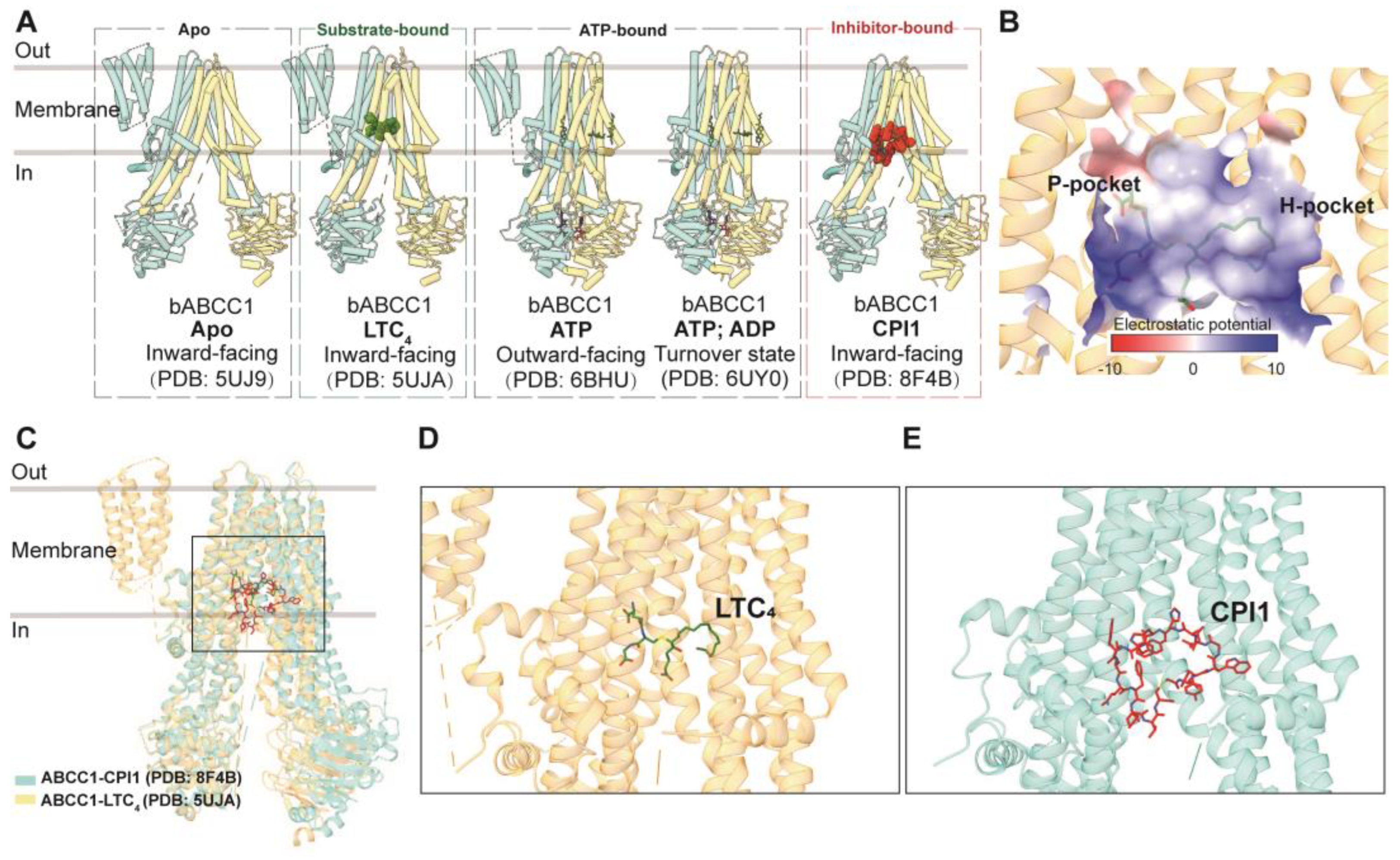
3.3.1. ABCC1
Drug Binding and Translocation of ABCC1
Inhibition of ABCC1
GSH and GSH-Conjugated Molecule Transport of ABCC1
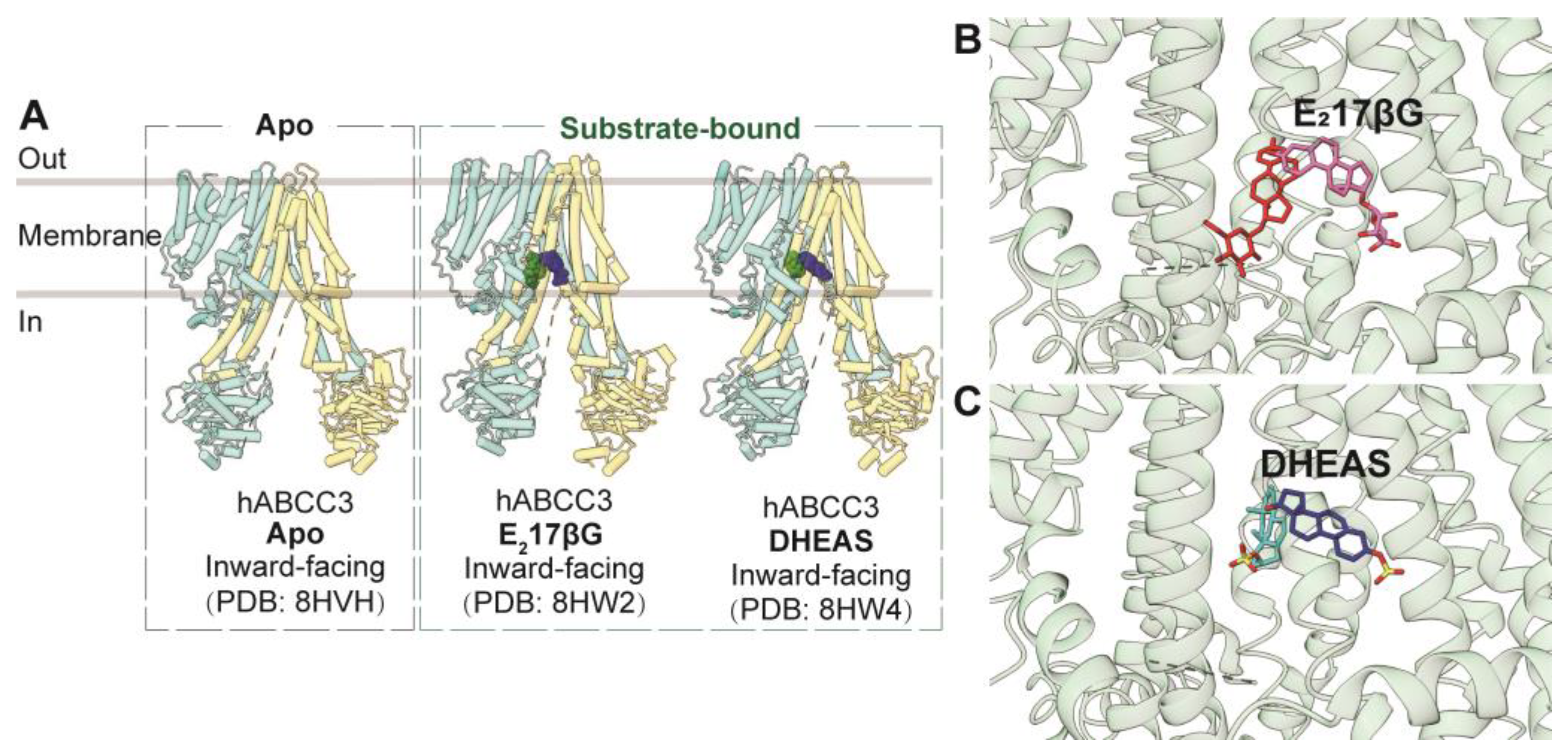
3.3.2. ABCC3
3.3.3. ABCC4
4. The Mechanisms of Drug Recognition, Translocation, and Inhibition in Multidrug Resistance
4.1. Putative Mechanism of Multidrug Recognition
4.2. Mechanism of Drug Translocation
4.3. Inhibition Mechanisms of MDR-Related ABC Transporters
5. The Unknowns Pending Future Studies
5.1. Central Linker with Unknown Structures and Functions
5.2. TMD0 with Less Clear Functions in the Context of ABC Transporters
5.3. Structure-Based Drug Design
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rueff, J.; Rodrigues, A.S. Cancer Drug Resistance: A Brief Overview from a Genetic Viewpoint. Methods Mol. Biol. 2016, 1395, 1–18. [Google Scholar] [PubMed]
- Kaye, S.B. The multidrug resistance phenotype. Br. J. Cancer 1988, 58, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Fojo, A.; Hamilton, T.C.; Young, R.C.; Ozols, R.F. Multidrug resistance in ovarian cancer. Cancer 1987, 60, 2075–2080. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Shukla, N.; Singh, S.S.; Kushwaha, S.; Shrivastava, R. Mechanism of interaction between autophagy and apoptosis in cancer. Apoptosis 2021, 26, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Mayer, L.D. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur. J. Pharm. Sci. 2000, 11, 265–283. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesman, M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. [Google Scholar] [CrossRef]
- Xiao, H.; Zheng, Y.; Ma, L.; Tian, L.; Sun, Q. Clinically-Relevant ABC Transporter for Anti-Cancer Drug Resistance. Front. Pharmacol. 2021, 12, 648407. [Google Scholar] [CrossRef]
- Alam, A.; Locher, K.P. Structure and Mechanism of Human ABC Transporters. Annu. Rev. Biophys. 2023, 52, 275–300. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC transporters: The power to change. Nat. Rev. Mol. Cell. Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F. ABC transporters: From microorganisms to man. Annu. Rev. Cell. Biol. 1992, 8, 67–113. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; de Lourdes Bastos, M.; Remiao, F. Modulation of P-glycoprotein efflux pump: Induction and activation as a therapeutic strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2009, 3, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Haber, M.; Smith, J.; Bordow, S.B.; Flemming, C.; Cohn, S.L.; London, W.B.; Marshall, G.M.; Norris, M.D. Association of high-level MRP1 expression with poor clinical outcome in a large prospective study of primary neuroblastoma. J. Clin. Oncol. 2006, 24, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Sarkadi, B.; Homolya, L.; Szakacs, G.; Varadi, A. Human multidrug resistance ABCB and ABCG transporters: Participation in a chemoimmunity defense system. Physiol. Rev. 2006, 86, 1179–1236. [Google Scholar] [CrossRef]
- Leslie, E.M.; Deeley, R.G.; Cole, S.P. Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol. 2005, 204, 216–237. [Google Scholar] [CrossRef]
- Choi, Y.H.; Yu, A.M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.P.; Bhardwaj, G.; Gerlach, J.H.; Mackie, J.E.; Grant, C.E.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef]
- Wang, J.Q.; Yang, Y.; Cai, C.Y.; Teng, Q.X.; Cui, Q.; Lin, J.; Assaraf, Y.G.; Chen, Z.S. Multidrug resistance proteins (MRPs): Structure, function and the overcoming of cancer multidrug resistance. Drug Resist. Updates 2021, 54, 100743. [Google Scholar] [CrossRef]
- Kool, M.; de Haas, M.; Scheffer, G.L.; Scheper, R.J.; van Eijk, M.J.; Juijn, J.A.; Baas, F.; Borst, P. Analysis of expression of cMOAT (MRP2), MRP3, MRP4, and MRP5, homologues of the multidrug resistance-associated protein gene (MRP1), in human cancer cell lines. Cancer Res. 1997, 57, 3537–3547. [Google Scholar]
- Whitlock, B.D.; Leslie, E.M. Efflux transporters in anti-cancer drug resistance: Molecular and functional identification and characterization of multidrug resistance proteins (MRPs/ABCCs). In Drug Efflux Pumps in Cancer Resistance Pathways: From Molecular Recognition and Characterization to Possible Inhibition Strategies in Chemotherapy; Elsevier: Amsterdam, The Netherlands, 2020; pp. 31–65. [Google Scholar]
- Biemans-Oldehinkel, E.; Doeven, M.K.; Poolman, B. ABC transporter architecture and regulatory roles of accessory domains. FEBS Lett. 2006, 580, 1023–1035. [Google Scholar] [CrossRef]
- Hollenstein, K.; Dawson, R.J.; Locher, K.P. Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 2007, 17, 412–418. [Google Scholar] [CrossRef]
- Locher, K.P.; Lee, A.T.; Rees, D.C. The E. coli BtuCD structure: A framework for ABC transporter architecture and mechanism. Science 2002, 296, 1091–1098. [Google Scholar] [CrossRef]
- Choudhuri, S.; Klaassen, C.D. Structure, function, expression, genomic organization, and single nucleotide polymorphisms of human ABCB1 (MDR1), ABCC (MRP), and ABCG2 (BCRP) efflux transporters. Int. J. Toxicol. 2006, 25, 231–259. [Google Scholar] [CrossRef]
- Fung, K.L.; Gottesman, M.M. A synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein function. Biochim. Biophys. Acta 2009, 1794, 860–871. [Google Scholar] [CrossRef]
- Roninson, I.B.; Chin, J.E.; Choi, K.G.; Gros, P.; Housman, D.E.; Fojo, A.; Shen, D.W.; Gottesman, M.M.; Pastan, I. Isolation of human mdr DNA sequences amplified in multidrug-resistant KB carcinoma cells. Proc. Natl. Acad. Sci. USA 1986, 83, 4538–4542. [Google Scholar] [CrossRef]
- Riordan, J.R.; Deuchars, K.; Kartner, N.; Alon, N.; Trent, J.; Ling, V. Amplification of P-glycoprotein genes in multidrug-resistant mammalian cell lines. Nature 1985, 316, 817–819. [Google Scholar] [CrossRef]
- Roninson, I.B.; Abelson, H.T.; Housman, D.E.; Howell, N.; Varshavsky, A. Amplification of specific DNA sequences correlates with multi-drug resistance in Chinese hamster cells. Nature 1984, 309, 626–628. [Google Scholar] [CrossRef]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef]
- Sun, J.; He, Z.G.; Cheng, G.; Wang, S.J.; Hao, X.H.; Zou, M.J. Multidrug resistance P-glycoprotein: Crucial significance in drug disposition and interaction. Med. Sci. Monit. 2004, 10, RA5-14. [Google Scholar] [PubMed]
- Schinkel, A.H.; Smit, J.J.; van Tellingen, O.; Beijnen, J.H.; Wagenaar, E.; van Deemter, L.; Mol, C.A.; van der Valk, M.A.; Robanus-Maandag, E.C.; te Riele, H.P.; et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 1994, 77, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef]
- Sharom, F.J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. [Google Scholar] [CrossRef]
- Chan, H.S.; Haddad, G.; Thorner, P.S.; DeBoer, G.; Lin, Y.P.; Ondrusek, N.; Yeger, H.; Ling, V. P-glycoprotein expression as a predictor of the outcome of therapy for neuroblastoma. N. Engl. J. Med. 1991, 325, 1608–1614. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.S.; Thorner, P.S.; Haddad, G.; Ling, V. Immunohistochemical detection of P-glycoprotein: Prognostic correlation in soft tissue sarcoma of childhood. J. Clin. Oncol. 1990, 8, 689–704. [Google Scholar] [CrossRef]
- Shaffer, B.C.; Gillet, J.P.; Patel, C.; Baer, M.R.; Bates, S.E.; Gottesman, M.M. Drug resistance: Still a daunting challenge to the successful treatment of AML. Drug Resist. Updates 2012, 15, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Amiri-Kordestani, L.; Basseville, A.; Kurdziel, K.; Fojo, A.T.; Bates, S.E. Targeting MDR in breast and lung cancer: Discriminating its potential importance from the failure of drug resistance reversal studies. Drug Resist. Updates 2012, 15, 50–61. [Google Scholar] [CrossRef]
- Silverberg, G.D.; Miller, M.C.; Messier, A.A.; Majmudar, S.; Machan, J.T.; Donahue, J.E.; Stopa, E.G.; Johanson, C.E. Amyloid deposition and influx transporter expression at the blood-brain barrier increase in normal aging. J. Neuropathol. Exp. Neurol. 2010, 69, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Kung, R.; Kowal, J.; McLeod, R.A.; Tremp, N.; Broude, E.V.; Roninson, I.B.; Stahlberg, H.; Locher, K.P. Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc. Natl. Acad. Sci. USA 2018, 115, E1973–E1982. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.I.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B.; et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. [Google Scholar] [CrossRef]
- Manolaridis, I.; Jackson, S.M.; Taylor, N.M.I.; Kowal, J.; Stahlberg, H.; Locher, K.P. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 2018, 563, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Orlando, B.J.; Liao, M. ABCG2 transports anticancer drugs via a closed-to-open switch. Nat. Commun. 2020, 11, 2264. [Google Scholar] [CrossRef]
- Kowal, J.; Ni, D.; Jackson, S.M.; Manolaridis, I.; Stahlberg, H.; Locher, K.P. Structural Basis of Drug Recognition by the Multidrug Transporter ABCG2. J. Mol. Biol. 2021, 433, 166980. [Google Scholar] [CrossRef]
- Johnson, Z.L.; Chen, J. Structural Basis of Substrate Recognition by the Multidrug Resistance Protein MRP1. Cell 2017, 168, 1075–1085.e9. [Google Scholar] [CrossRef]
- Pietz, H.L.; Abbas, A.; Johnson, Z.L.; Oldham, M.L.; Suga, H.; Chen, J. A macrocyclic peptide inhibitor traps MRP1 in a catalytically incompetent conformation. Proc. Natl. Acad. Sci. USA 2023, 120, e2220012120. [Google Scholar] [CrossRef]
- Wang, J.; Li, X.; Wang, F.F.; Cheng, M.T.; Mao, Y.X.; Fang, S.C.; Wang, L.; Zhou, C.Z.; Hou, W.T.; Chen, Y. Placing steroid hormones within the human ABCC3 transporter reveals a compatible amphiphilic substrate-binding pocket. EMBO J. 2023, 42, e113415. [Google Scholar] [CrossRef]
- Bloch, M.; Raj, I.; Pape, T.; Taylor, N.M.I. Structural and mechanistic basis of substrate transport by the multidrug transporter MRP4. Structure 2023, 31, 1407–1418.e6. [Google Scholar] [CrossRef]
- Huang, Y.; Xue, C.; Wang, L.; Bu, R.; Mu, J.; Wang, Y.; Liu, Z. Structural basis for substrate and inhibitor recognition of human multidrug transporter MRP4. Commun. Biol. 2023, 6, 549. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.B.; Szewczyk, P.; Grimard, V.; Lee, C.W.; Martinez, L.; Doshi, R.; Caya, A.; Villaluz, M.; Pardon, E.; Cregger, C.; et al. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. USA 2013, 110, 13386–13391. [Google Scholar] [CrossRef] [PubMed]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. [Google Scholar] [CrossRef] [PubMed]
- Vahedi, S.; Lusvarghi, S.; Pluchino, K.; Shafrir, Y.; Durell, S.R.; Gottesman, M.M.; Ambudkar, S.V. Mapping discontinuous epitopes for MRK-16, UIC2 and 4E3 antibodies to extracellular loops 1 and 4 of human P-glycoprotein. Sci. Rep. 2018, 8, 12716. [Google Scholar] [CrossRef]
- Dash, R.P.; Jayachandra Babu, R.; Srinivas, N.R. Therapeutic Potential and Utility of Elacridar with Respect to P-glycoprotein Inhibition: An Insight from the Published In Vitro, Preclinical and Clinical Studies. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 915–933. [Google Scholar] [CrossRef]
- Varma, M.V.; Ashokraj, Y.; Dey, C.S.; Panchagnula, R. P-glycoprotein inhibitors and their screening: A perspective from bioavailability enhancement. Pharmacol. Res. 2003, 48, 347–359. [Google Scholar] [CrossRef]
- Thomas, H.; Coley, H.M. Overcoming multidrug resistance in cancer: An update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control 2003, 10, 159–165. [Google Scholar] [CrossRef]
- Cole, S.P. Targeting multidrug resistance protein 1 (MRP1, ABCC1): Past, present, and future. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 95–117. [Google Scholar] [CrossRef]
- Binkhathlan, Z.; Lavasanifar, A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. [Google Scholar] [CrossRef]
- Patel, K.J.; Tannock, I.F. The influence of P-glycoprotein expression and its inhibitors on the distribution of doxorubicin in breast tumors. BMC Cancer 2009, 9, 356. [Google Scholar] [CrossRef]
- Urgaonkar, S.; Nosol, K.; Said, A.M.; Nasief, N.N.; Bu, Y.; Locher, K.P.; Lau, J.Y.N.; Smolinski, M.P. Discovery and Characterization of Potent Dual P-Glycoprotein and CYP3A4 Inhibitors: Design, Synthesis, Cryo-EM Analysis, and Biological Evaluations. J. Med. Chem. 2022, 65, 191–216. [Google Scholar] [CrossRef]
- Sajid, A.; Lusvarghi, S.; Chufan, E.E.; Ambudkar, S.V. Evidence for the critical role of transmembrane helices 1 and 7 in substrate transport by human P-glycoprotein (ABCB1). PLoS ONE 2018, 13, e0204693. [Google Scholar] [CrossRef]
- Sajid, A.; Lusvarghi, S.; Murakami, M.; Chufan, E.E.; Abel, B.; Gottesman, M.M.; Durell, S.R.; Ambudkar, S.V. Reversing the direction of drug transport mediated by the human multidrug transporter P-glycoprotein. Proc. Natl. Acad. Sci. USA 2020, 117, 29609–29617. [Google Scholar] [CrossRef]
- Robey, R.W.; To, K.K.; Polgar, O.; Dohse, M.; Fetsch, P.; Dean, M.; Bates, S.E. ABCG2: A perspective. Adv. Drug Deliv. Rev. 2009, 61, 3–13. [Google Scholar] [CrossRef]
- Stiburkova, B.; Pavelcova, K.; Pavlikova, M.; Jesina, P.; Pavelka, K. The impact of dysfunctional variants of ABCG2 on hyperuricemia and gout in pediatric-onset patients. Arthritis Res. Ther. 2019, 21, 77. [Google Scholar] [CrossRef]
- Toyoda, Y.; Takada, T.; Suzuki, H. Inhibitors of Human ABCG2: From Technical Background to Recent Updates with Clinical Implications. Front. Pharmacol. 2019, 10, 208. [Google Scholar] [CrossRef]
- Houghton, P.J.; Germain, G.S.; Harwood, F.C.; Schuetz, J.D.; Stewart, C.F.; Buchdunger, E.; Traxler, P. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res. 2004, 64, 2333–2337. [Google Scholar] [CrossRef]
- Candeil, L.; Gourdier, I.; Peyron, D.; Vezzio, N.; Copois, V.; Bibeau, F.; Orsetti, B.; Scheffer, G.L.; Ychou, M.; Khan, Q.A.; et al. ABCG2 overexpression in colon cancer cells resistant to SN38 and in irinotecan-treated metastases. Int. J. Cancer 2004, 109, 848–854. [Google Scholar] [CrossRef]
- Xiong, H.; Callaghan, D.; Jones, A.; Bai, J.; Rasquinha, I.; Smith, C.; Pei, K.; Walker, D.; Lue, L.F.; Stanimirovic, D.; et al. ABCG2 is upregulated in Alzheimer’s brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood-brain barrier for Abeta(1-40) peptides. J. Neurosci. 2009, 29, 5463–5475. [Google Scholar] [CrossRef]
- Taylor, N.M.I.; Manolaridis, I.; Jackson, S.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Structure of the human multidrug transporter ABCG2. Nature 2017, 546, 504–509. [Google Scholar] [CrossRef]
- Rasouli, A.; Yu, Q.; Dehghani-Ghahnaviyeh, S.; Wen, P.C.; Kowal, J.; Locher, K.P.; Tajkhorshid, E. Differential dynamics and direct interaction of bound ligands with lipids in multidrug transporter ABCG2. Proc. Natl. Acad. Sci. USA 2023, 120, e2213437120. [Google Scholar] [CrossRef]
- Yu, Q.; Ni, D.; Kowal, J.; Manolaridis, I.; Jackson, S.M.; Stahlberg, H.; Locher, K.P. Structures of ABCG2 under turnover conditions reveal a key step in the drug transport mechanism. Nat. Commun. 2021, 12, 4376. [Google Scholar] [CrossRef]
- Honjo, Y.; Hrycyna, C.A.; Yan, Q.W.; Medina-Perez, W.Y.; Robey, R.W.; van de Laar, A.; Litman, T.; Dean, M.; Bates, S.E. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res. 2001, 61, 6635–6639. [Google Scholar]
- Ozvegy-Laczka, C.; Koblos, G.; Sarkadi, B.; Varadi, A. Single amino acid (482) variants of the ABCG2 multidrug transporter: Major differences in transport capacity and substrate recognition. Biochim. Biophys. Acta 2005, 1668, 53–63. [Google Scholar] [CrossRef][Green Version]
- Woodward, O.M.; Kottgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Kottgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef]
- Zambo, B.; Mozner, O.; Bartos, Z.; Torok, G.; Varady, G.; Telbisz, A.; Homolya, L.; Orban, T.I.; Sarkadi, B. Cellular expression and function of naturally occurring variants of the human ABCG2 multidrug transporter. Cell. Mol. Life Sci. 2020, 77, 365–378. [Google Scholar] [CrossRef]
- Rabindran, S.K.; He, H.; Singh, M.; Brown, E.; Collins, K.I.; Annable, T.; Greenberger, L.M. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res. 1998, 58, 5850–5858. [Google Scholar]
- Allen, J.D.; van Loevezijn, A.; Lakhai, J.M.; van der Valk, M.; van Tellingen, O.; Reid, G.; Schellens, J.H.; Koomen, G.J.; Schinkel, A.H. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer Ther. 2002, 1, 417–425. [Google Scholar]
- Weidner, L.D.; Zoghbi, S.S.; Lu, S.; Shukla, S.; Ambudkar, S.V.; Pike, V.W.; Mulder, J.; Gottesman, M.M.; Innis, R.B.; Hall, M.D. The Inhibitor Ko143 Is Not Specific for ABCG2. J. Pharmacol. Exp. Ther. 2015, 354, 384–393. [Google Scholar] [CrossRef]
- Irobalieva, R.N.; Manolaridis, I.; Jackson, S.M.; Ni, D.; Pardon, E.; Stahlberg, H.; Steyaert, J.; Locher, K.P. Structural Basis of the Allosteric Inhibition of Human ABCG2 by Nanobodies. J. Mol. Biol. 2023, 435, 168234. [Google Scholar] [CrossRef]
- Zhang, Y.K.; Wang, Y.J.; Gupta, P.; Chen, Z.S. Multidrug Resistance Proteins (MRPs) and Cancer Therapy. AAPS J. 2015, 17, 802–812. [Google Scholar] [CrossRef]
- Dallas, S.; Miller, D.S.; Bendayan, R. Multidrug resistance-associated proteins: Expression and function in the central nervous system. Pharmacol. Rev. 2006, 58, 140–161. [Google Scholar] [CrossRef]
- Sugiyama, D.; Kusuhara, H.; Lee, Y.J.; Sugiyama, Y. Involvement of multidrug resistance associated protein 1 (Mrp1) in the efflux transport of 17beta estradiol-D-17beta-glucuronide (E217betaG) across the blood-brain barrier. Pharm. Res. 2003, 20, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Krohn, M.; Lange, C.; Hofrichter, J.; Scheffler, K.; Stenzel, J.; Steffen, J.; Schumacher, T.; Bruning, T.; Plath, A.S.; Alfen, F.; et al. Cerebral amyloid-beta proteostasis is regulated by the membrane transport protein ABCC1 in mice. J. Clin. Investig. 2011, 121, 3924–3931. [Google Scholar] [CrossRef]
- Slot, A.J.; Molinski, S.V.; Cole, S.P. Mammalian multidrug-resistance proteins (MRPs). Essays Biochem. 2011, 50, 179–207. [Google Scholar]
- Zhang, Z.; Chen, J. Atomic Structure of the Cystic Fibrosis Transmembrane Conductance Regulator. Cell 2016, 167, 1586–1597.e9. [Google Scholar] [CrossRef]
- Westlake, C.J.; Qian, Y.M.; Gao, M.; Vasa, M.; Cole, S.P.; Deeley, R.G. Identification of the structural and functional boundaries of the multidrug resistance protein 1 cytoplasmic loop 3. Biochemistry 2003, 42, 14099–14113. [Google Scholar] [CrossRef]
- Koch, J.; Guntrum, R.; Heintke, S.; Kyritsis, C.; Tampe, R. Functional dissection of the transmembrane domains of the transporter associated with antigen processing (TAP). J. Biol. Chem. 2004, 279, 10142–10147. [Google Scholar] [CrossRef]
- Chan, K.W.; Zhang, H.; Logothetis, D.E. N-terminal transmembrane domain of the SUR controls trafficking and gating of Kir6 channel subunits. EMBO J. 2003, 22, 3833–3843. [Google Scholar] [CrossRef]
- Al-Shawi, M.K. Catalytic and transport cycles of ABC exporters. Essays Biochem. 2011, 50, 63–83. [Google Scholar]
- Lu, J.F.; Pokharel, D.; Bebawy, M. MRP1 and its role in anticancer drug resistance. Drug. Metab. Rev. 2015, 47, 406–419. [Google Scholar] [CrossRef]
- Zhou, S.F.; Wang, L.L.; Di, Y.M.; Xue, C.C.; Duan, W.; Li, C.G.; Li, Y. Substrates and inhibitors of human multidrug resistance associated proteins and the implications in drug development. Curr. Med. Chem. 2008, 15, 1981–2039. [Google Scholar] [CrossRef]
- Hlavac, V.; Brynychova, V.; Vaclavikova, R.; Ehrlichova, M.; Vrana, D.; Pecha, V.; Kozevnikovova, R.; Trnkova, M.; Gatek, J.; Kopperova, D.; et al. The expression profile of ATP-binding cassette transporter genes in breast carcinoma. Pharmacogenomics 2013, 14, 515–529. [Google Scholar] [CrossRef]
- Norris, M.D.; Bordow, S.B.; Marshall, G.M.; Haber, P.S.; Cohn, S.L.; Haber, M. Expression of the gene for multidrug-resistance-associated protein and outcome in patients with neuroblastoma. N. Engl. J. Med. 1996, 334, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Johnson, Z.L.; Wasserman, M.R.; Levring, J.; Chen, J.; Liu, S. Characterization of the kinetic cycle of an ABC transporter by single-molecule and cryo-EM analyses. Elife 2020, 9, e56451. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Z.L.; Chen, J. ATP Binding Enables Substrate Release from Multidrug Resistance Protein 1. Cell 2018, 172, 81–89.e10. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, A.C.; Al Saig, F.; Cloos, J.; Jansen, G.; Peters, G.J. How to overcome ATP-binding cassette drug efflux transporter-mediated drug resistance. Cancer Drug Resist. 2018, 1, 6–29. [Google Scholar] [CrossRef]
- de Groot, D.J.; van der Deen, M.; Le, T.K.; Regeling, A.; de Jong, S.; de Vries, E.G. Indomethacin induces apoptosis via a MRP1-dependent mechanism in doxorubicin-resistant small-cell lung cancer cells overexpressing MRP1. Br. J. Cancer 2007, 97, 1077–1083. [Google Scholar] [CrossRef]
- Trompier, D.; Chang, X.B.; Barattin, R.; du Moulinet D’Hardemare, A.; Di Pietro, A.; Baubichon-Cortay, H. Verapamil and its derivative trigger apoptosis through glutathione extrusion by multidrug resistance protein MRP1. Cancer Res. 2004, 64, 4950–4956. [Google Scholar] [CrossRef]
- Wijnholds, J.; Evers, R.; van Leusden, M.R.; Mol, C.A.; Zaman, G.J.; Mayer, U.; Beijnen, J.H.; van der Valk, M.; Krimpenfort, P.; Borst, P. Increased sensitivity to anticancer drugs and decreased inflammatory response in mice lacking the multidrug resistance-associated protein. Nat. Med. 1997, 3, 1275–1279. [Google Scholar] [CrossRef]
- Ebner, J.; Schmoellerl, J.; Piontek, M.; Manhart, G.; Troester, S.; Carter, B.Z.; Neubauer, H.; Moriggl, R.; Szakacs, G.; Zuber, J.; et al. ABCC1 and glutathione metabolism limit the efficacy of BCL-2 inhibitors in acute myeloid leukemia. Nat. Commun. 2023, 14, 5709. [Google Scholar] [CrossRef]
- Cole, S.P.; Deeley, R.G. Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol. Sci. 2006, 27, 438–446. [Google Scholar] [CrossRef]
- Hanssen, K.M.; Wheatley, M.S.; Yu, D.M.T.; Conseil, G.; Norris, M.D.; Haber, M.; Cole, S.P.C.; Fletcher, J.I. GSH facilitates the binding and inhibitory activity of novel multidrug resistance protein 1 (MRP1) modulators. FEBS J. 2022, 289, 3854–3875. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, S.S.; Park, J.G.; Kim, J.W.; Ju, S.; Choi, S.H.; Kim, S.; Kim, N.J.; Hong, S.; Kang, J.Y.; et al. Structural Insights into Porphyrin Recognition by the Human ATP-Binding Cassette Transporter ABCB6. Mol. Cells 2022, 45, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.; Pierik, A.J.; Lill, R. Crystal structures of nucleotide-free and glutathione-bound mitochondrial ABC transporter Atm1. Science 2014, 343, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Bodo, A.; Bakos, E.; Szeri, F.; Varadi, A.; Sarkadi, B. Differential modulation of the human liver conjugate transporters MRP2 and MRP3 by bile acids and organic anions. J. Biol. Chem. 2003, 278, 23529–23537. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Bain, L.J.; Belinsky, M.G.; Kruh, G.D. Expression of multidrug resistance protein-3 (multispecific organic anion transporter-D) in human embryonic kidney 293 cells confers resistance to anticancer agents. Cancer Res. 1999, 59, 5964–5967. [Google Scholar]
- Kool, M.; van der Linden, M.; de Haas, M.; Scheffer, G.L.; de Vree, J.M.; Smith, A.J.; Jansen, G.; Peters, G.J.; Ponne, N.; Scheper, R.J.; et al. MRP3, an organic anion transporter able to transport anti-cancer drugs. Proc. Natl. Acad. Sci. USA 1999, 96, 6914–6919. [Google Scholar] [CrossRef]
- Zhao, Y.; Lu, H.; Yan, A.; Yang, Y.; Meng, Q.; Sun, L.; Pang, H.; Li, C.; Dong, X.; Cai, L. ABCC3 as a marker for multidrug resistance in non-small cell lung cancer. Sci. Rep. 2013, 3, 3120. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; Konig, J.; Pfannschmidt, M.; Klar, E.; Hofmann, W.J.; Keppler, D. Expression of the multidrug resistance proteins MRP2 and MRP3 in human hepatocellular carcinoma. Int. J. Cancer 2001, 94, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Sodani, K.; Patel, A.; Kathawala, R.J.; Chen, Z.S. Multidrug resistance associated proteins in multidrug resistance. Chin. J. Cancer 2012, 31, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.; Wielinga, P.; Zelcer, N.; van der Heijden, I.; Kuil, A.; de Haas, M.; Wijnholds, J.; Borst, P. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. USA 2003, 100, 9244–9249. [Google Scholar] [CrossRef] [PubMed]
- Russel, F.G.; Koenderink, J.B.; Masereeuw, R. Multidrug resistance protein 4 (MRP4/ABCC4): A versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol. Sci. 2008, 29, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Guo, Y.; Yue, W.; Zhang, L.; Gu, M.; Wang, Y. ABCC4 is required for cell proliferation and tumorigenesis in non-small cell lung cancer. Oncol. Targets Ther. 2014, 7, 343–351. [Google Scholar]
- Borel, F.; Han, R.; Visser, A.; Petry, H.; van Deventer, S.J.; Jansen, P.L.; Konstantinova, P. Adenosine triphosphate-binding cassette transporter genes up-regulation in untreated hepatocellular carcinoma is mediated by cellular microRNAs. Hepatology 2012, 55, 821–832. [Google Scholar] [CrossRef]
- Ho, L.L.; Kench, J.G.; Handelsman, D.J.; Scheffer, G.L.; Stricker, P.D.; Grygiel, J.G.; Sutherland, R.L.; Henshall, S.M.; Allen, J.D.; Horvath, L.G. Androgen regulation of multidrug resistance-associated protein 4 (MRP4/ABCC4) in prostate cancer. Prostate 2008, 68, 1421–1429. [Google Scholar] [CrossRef]
- Norris, M.D.; Smith, J.; Tanabe, K.; Tobin, P.; Flemming, C.; Scheffer, G.L.; Wielinga, P.; Cohn, S.L.; London, W.B.; Marshall, G.M.; et al. Expression of multidrug transporter MRP4/ABCC4 is a marker of poor prognosis in neuroblastoma and confers resistance to irinotecan in vitro. Mol. Cancer Ther. 2005, 4, 547–553. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, L.; Hou, W.-T.; Zha, Z.; Xu, K.; Zhou, C.-Z.; Li, Q.; Chen, Y. Structural insights into human ABCC4-mediated transport of platelet agonist and antagonist. Nat. Cardiovasc. Res. 2023, 2, 693–701. [Google Scholar] [CrossRef]
- Jardetzky, O. Simple allosteric model for membrane pumps. Nature 1966, 211, 969–970. [Google Scholar] [CrossRef]
- Jones, P.M.; George, A.M. The Switch and Reciprocating Models for the Function of ABC Multidrug Exporters: Perspectives on Recent Research. Int. J. Mol. Sci. 2023, 24, 2624. [Google Scholar] [CrossRef]
- Wilmes, M.; Pinto Espinoza, C.; Ludewig, P.; Stabernack, J.; Liesz, A.; Nicke, A.; Gelderblom, M.; Gerloff, C.; Falzoni, S.; Tolosa, E.; et al. Blocking P2X7 by intracerebroventricular injection of P2X7-specific nanobodies reduces stroke lesions. J. Neuroinflammation 2022, 19, 256. [Google Scholar] [CrossRef]
- Thomas, C.; Tampe, R. Multifaceted structures and mechanisms of ABC transport systems in health and disease. Curr. Opin. Struct. Biol. 2018, 51, 116–128. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320-4. [Google Scholar] [CrossRef] [PubMed]
- Hrycyna, C.A.; Airan, L.E.; Germann, U.A.; Ambudkar, S.V.; Pastan, I.; Gottesman, M.M. Structural flexibility of the linker region of human P-glycoprotein permits ATP hydrolysis and drug transport. Biochemistry 1998, 37, 13660–13673. [Google Scholar] [CrossRef]
- Georges, E. The P-glycoprotein (ABCB1) linker domain encodes high-affinity binding sequences to alpha- and beta-tubulins. Biochemistry 2007, 46, 7337–7342. [Google Scholar] [CrossRef]
- Sato, T.; Kodan, A.; Kimura, Y.; Ueda, K.; Nakatsu, T.; Kato, H. Functional role of the linker region in purified human P-glycoprotein. FEBS J. 2009, 276, 3504–3516. [Google Scholar] [CrossRef]
- Esser, L.; Zhou, F.; Pluchino, K.M.; Shiloach, J.; Ma, J.; Tang, W.K.; Gutierrez, C.; Zhang, A.; Shukla, S.; Madigan, J.P.; et al. Structures of the Multidrug Transporter P-glycoprotein Reveal Asymmetric ATP Binding and the Mechanism of Polyspecificity. J. Biol. Chem. 2017, 292, 446–461. [Google Scholar] [CrossRef]
- Ambadipudi, R.; Georges, E. Sequences in Linker-1 domain of the multidrug resistance associated protein (MRP1 or ABCC1) bind to tubulin and their binding is modulated by phosphorylation. Biochem. Biophys. Res. Commun. 2017, 482, 1001–1006. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Z.; Mo, W.; Ambadipudi, R.; Arnold, R.J.; Hrncirova, P.; Novotny, M.V.; Georges, E.; Zhang, J.T. Human ABCC1 interacts and colocalizes with ATP synthase alpha, revealed by interactive proteomics analysis. J. Proteome Res. 2012, 11, 1364–1372. [Google Scholar] [CrossRef]
- Ford, R.C.; Marshall-Sabey, D.; Schuetz, J. Linker Domains: Why ABC Transporters ‘Live in Fragments no Longer’. Trends Biochem. Sci. 2020, 45, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Kroll, T.; Prescher, M.; Smits, S.H.J.; Schmitt, L. Structure and Function of Hepatobiliary ATP Binding Cassette Transporters. Chem. Rev. 2021, 121, 5240–5288. [Google Scholar] [CrossRef] [PubMed]
- Demirel, O.; Bangert, I.; Tampe, R.; Abele, R. Tuning the cellular trafficking of the lysosomal peptide transporter TAPL by its N-terminal domain. Traffic 2010, 11, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Kiss, K.; Kucsma, N.; Brozik, A.; Tusnady, G.E.; Bergam, P.; van Niel, G.; Szakacs, G. Role of the N-terminal transmembrane domain in the endo-lysosomal targeting and function of the human ABCB6 protein. Biochem. J. 2015, 467, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Bandler, P.E.; Westlake, C.J.; Grant, C.E.; Cole, S.P.; Deeley, R.G. Identification of regions required for apical membrane localization of human multidrug resistance protein 2. Mol. Pharmacol. 2008, 74, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Bakos, E.; Evers, R.; Szakacs, G.; Tusnady, G.E.; Welker, E.; Szabo, K.; de Haas, M.; van Deemter, L.; Borst, P.; Varadi, A.; et al. Functional multidrug resistance protein (MRP1) lacking the N-terminal transmembrane domain. J. Biol. Chem. 1998, 273, 32167–32175. [Google Scholar] [CrossRef] [PubMed]
- Goebel, J.; Chmielewski, J.; Hrycyna, C.A. The roles of the human ATP-binding cassette transporters P-glycoprotein and ABCG2 in multidrug resistance in cancer and at endogenous sites: Future opportunities for structure-based drug design of inhibitors. Cancer Drug Resist 2021, 4, 784–804. [Google Scholar] [CrossRef] [PubMed]
- Pote, M.S.; Gacche, R.N. ATP-binding cassette efflux transporters and MDR in cancer. Drug Discov. Today 2023, 28, 103537. [Google Scholar] [CrossRef] [PubMed]
- Le, M.T.; Hoang, V.N.; Nguyen, D.N.; Bui, T.H.; Phan, T.V.; Huynh, P.N.; Tran, T.D.; Thai, K.M. Structure-Based Discovery of ABCG2 Inhibitors: A Homology Protein-Based Pharmacophore Modeling and Molecular Docking Approach. Molecules 2021, 26, 3115. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Takahashi, K.; Ikeda, N.; Kajimoto, Y.; Hagiya, Y.; Ogura, S.; Miyatake, S.; Kuroiwa, T. Transporter-Mediated Drug Interaction Strategy for 5-Aminolevulinic Acid (ALA)-Based Photodynamic Diagnosis of Malignant Brain Tumor: Molecular Design of ABCG2 Inhibitors. Pharmaceutics 2011, 3, 615–635. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name (Alternate) | Active Sites in Central Cavity | Substrates | Inhibitors | Function and Mechanism |
|---|---|---|---|---|
| ABCB1 (P-gp, MDR1) | Y310 *, I340, F343, S344, Q347, Q725, Q946, Y953, F983, M986, A987, Q990 [46,47] | A variety of chemotherapy drugs (Taxol, vincristine, doxorubicin) | Cyclic peptide (QZ59-RRR, QZ59-SSS, cyclosporine A) Small molecules inhibitor (zosuquidar, elacridar, tariquidar, encequiar, verapamil) Nanobody (Nb592) | MDR; transports poly-specificity substrates; globular hydrophobic pocket primarily suitable for hydrophobic or weakly amphipathic compounds |
| ABCG2 (BCRP) | L405, F432, T435, N436, F439, S440, V442, T542, V546, M549 [48,49,50,51] | Chemotherapy drugs (topotecan, SN-38, mitoxantrone, doxorubicin) Endogenous substrates (sulfate conjugates of steroids, uric acid, and porphyrins, e.g., E1S) Exogenous cytotoxic compounds | FTC, Ko143, MZ29 Tariquidar, MB136 Tyrosine kinase inhibitors (TKIs) Antibody and nanobody (5D3, Nb8, Nb17, Nb96) | MDR; transports a broad spectrum of substrates; the deep/slit-like cavity preference for flat molecules |
| ABCC1 (MRP1) | K332, H335, L381, F385, Y440, T550, W553, F594, M1092, R1196, Y1242, N1244, W1245, R1248 [52,53] | Physiological substrates, xenobiotic compounds (leukotriene C4-LTC4, E217βG) GSH and GSH-conjugated molecules | Indomethacin, verapamil, and its derivatives Macrocyclic peptide inhibitor (CPI1) | MDR; transports a wide range of substrates; employs large bipartite pockets to recognize amphiphilic substrates |
| ABCC3 (MRP3) | K318, Y371, F375, F426, L429, T535, W539, L580, M584, M1089, Y1188, R1193, F1238, N1241, W1242, R1245, M1246 [54] | Endogenous metabolites (bilirubin glucuronides, bile acids, and steroid hormones, like E217βG, DHEAS) Anticancer drugs (etoposide, teniposide, methotrexate, and vincristine) | Cyclosporine A, MK-571 | MDR; transports diverse endogenous metabolites and a limited number of anticancer drugs; the binding pockets comprise a substantial hydrophobic surface and two polar patches |
| ABCC4 (MRP4) | F211, F324, L363, L367, F368, R375, R946, Q994, W995, R998 [55,56] | Physiological substrates (cyclic nucleotides, steroid conjugates, folate, and prostaglandins (PGE1 and PGE2)). Xenobiotics (antibiotics, antiviral agents, anticancer drugs, aspirin, U46619) | Dipyridamole and sulindac | MDR; transports physiological substrates and xenobiotics; possesses a hydrophobic pocket (H-pocket) and positively charged pocket (P-pocket) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, W.; Shao, K.; Luo, M. Structural View of Cryo-Electron Microscopy-Determined ATP-Binding Cassette Transporters in Human Multidrug Resistance. Biomolecules 2024, 14, 231. https://doi.org/10.3390/biom14020231
Fan W, Shao K, Luo M. Structural View of Cryo-Electron Microscopy-Determined ATP-Binding Cassette Transporters in Human Multidrug Resistance. Biomolecules. 2024; 14(2):231. https://doi.org/10.3390/biom14020231
Chicago/Turabian StyleFan, Wenjie, Kai Shao, and Min Luo. 2024. "Structural View of Cryo-Electron Microscopy-Determined ATP-Binding Cassette Transporters in Human Multidrug Resistance" Biomolecules 14, no. 2: 231. https://doi.org/10.3390/biom14020231
APA StyleFan, W., Shao, K., & Luo, M. (2024). Structural View of Cryo-Electron Microscopy-Determined ATP-Binding Cassette Transporters in Human Multidrug Resistance. Biomolecules, 14(2), 231. https://doi.org/10.3390/biom14020231