
Metoprolol Inhibits Developmental Brain Sterol Biosynthesis in Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Line Cultures
2.3. Primary Neuronal Cultures
2.4. Primary Astroglia Cultures
2.5. Mouse Studies
2.6. Cell Sterol Measurements
2.7. Mouse Sterol Measurements
2.8. Drug Extraction
2.9. Drug Measurements
2.10. Statistics
3. Results
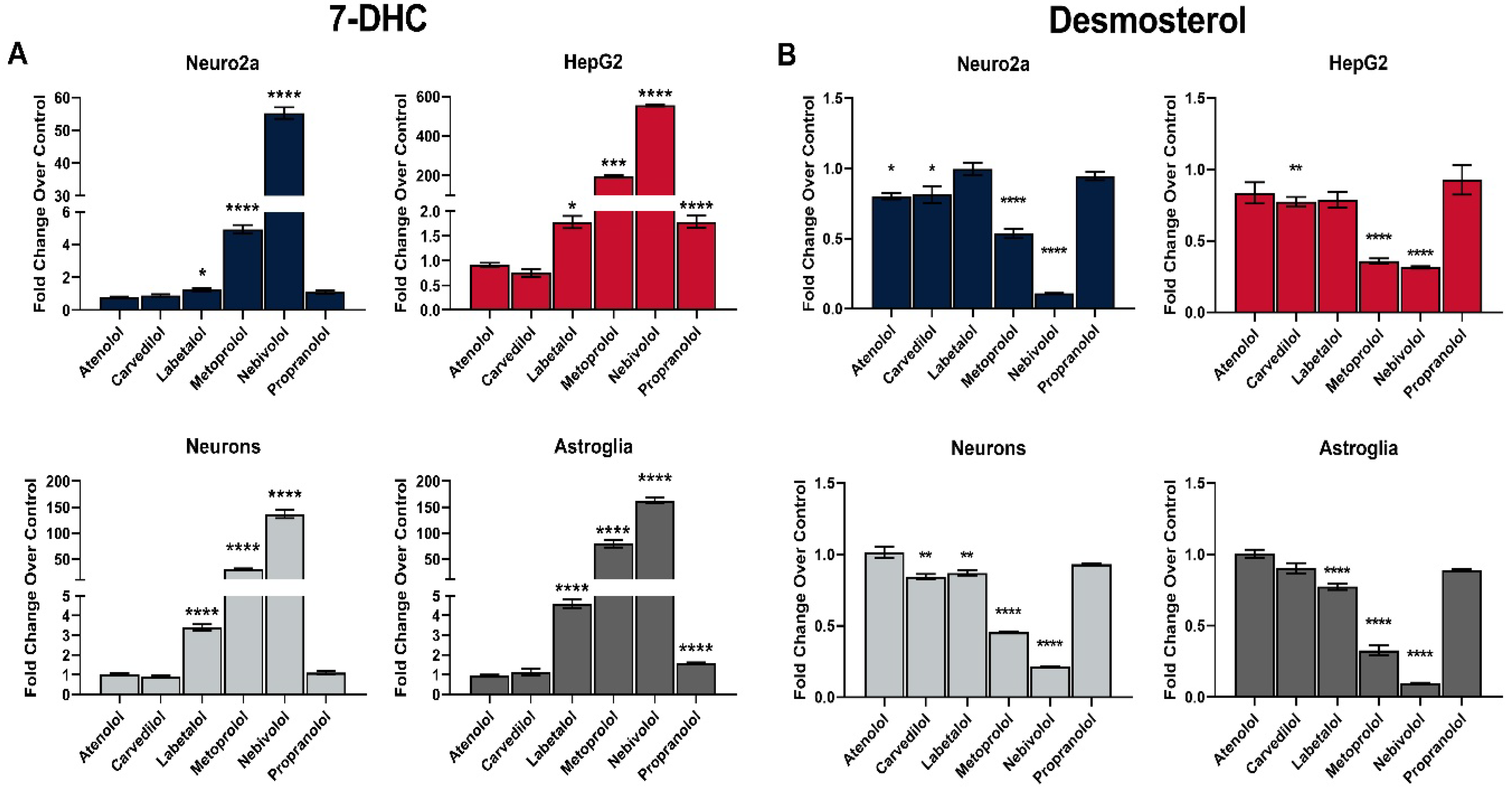
3.1. Multiple Beta-Blockers Inhibit Sterol Biosynthesis In Vitro
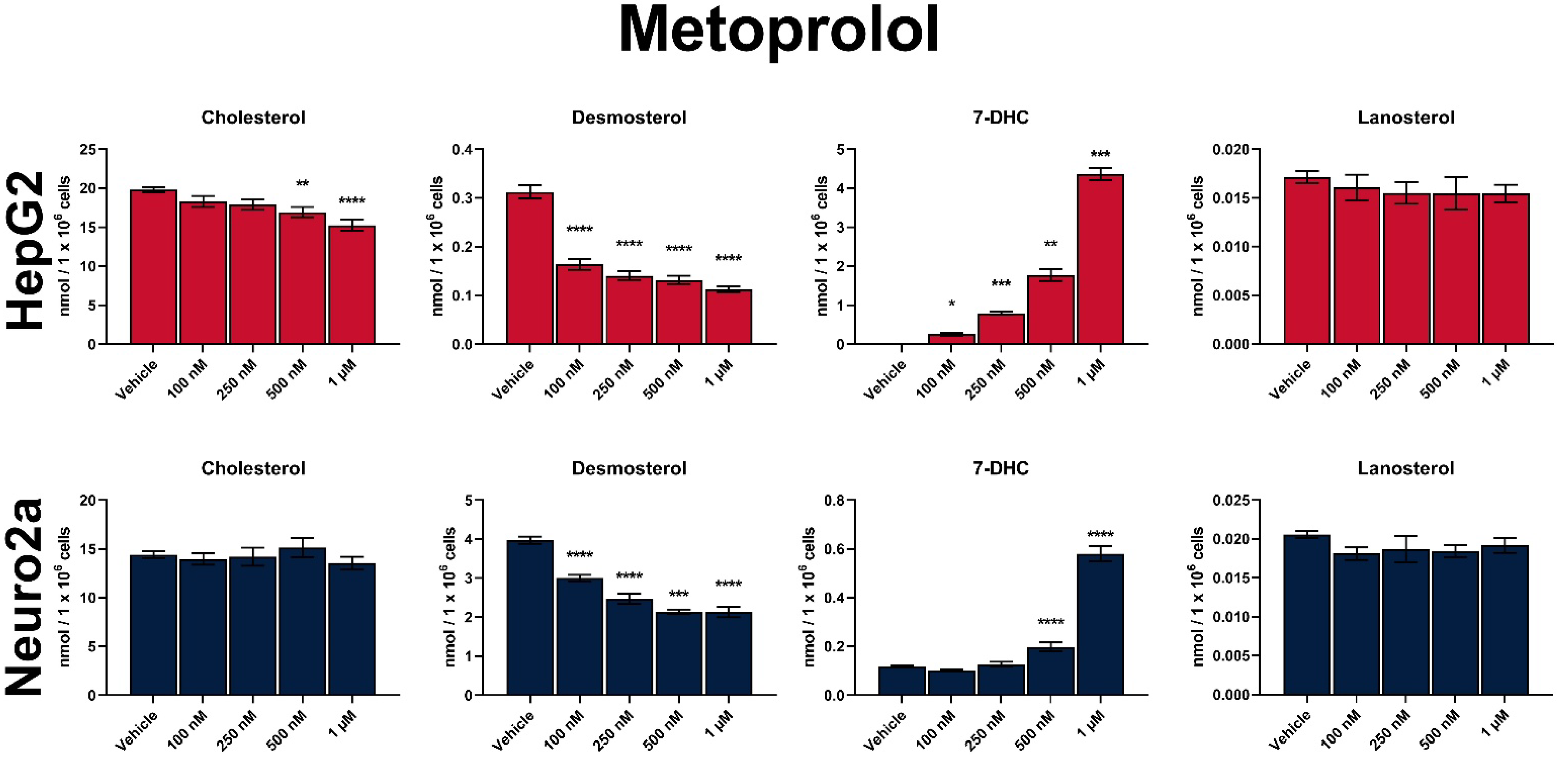
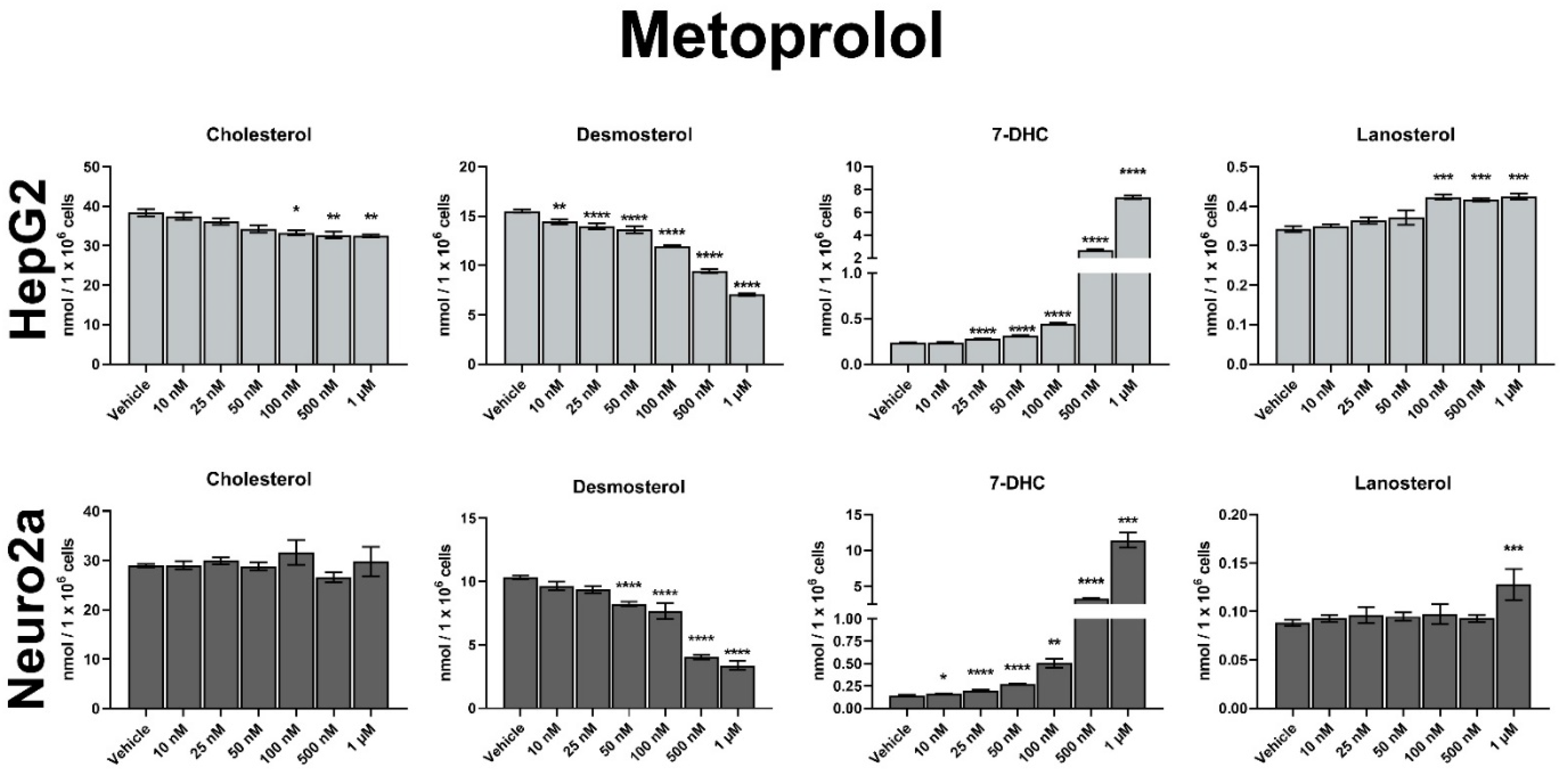
3.2. MTP Is a Strong Inhibitor of Sterol Biosynthesis In Vitro
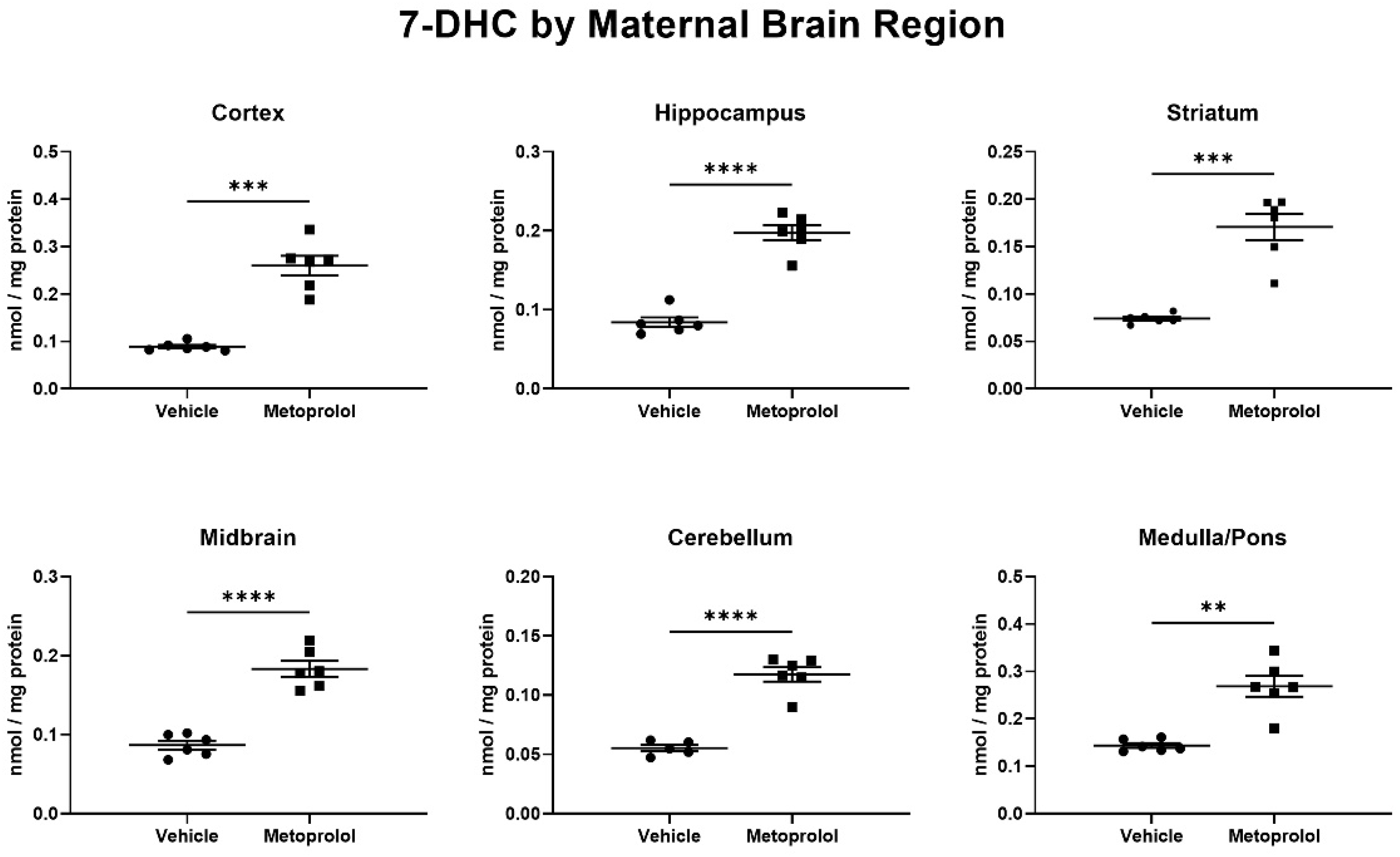
3.3. MTP Inhibits Sterol Biosynthesis in Maternal Mouse Tissues
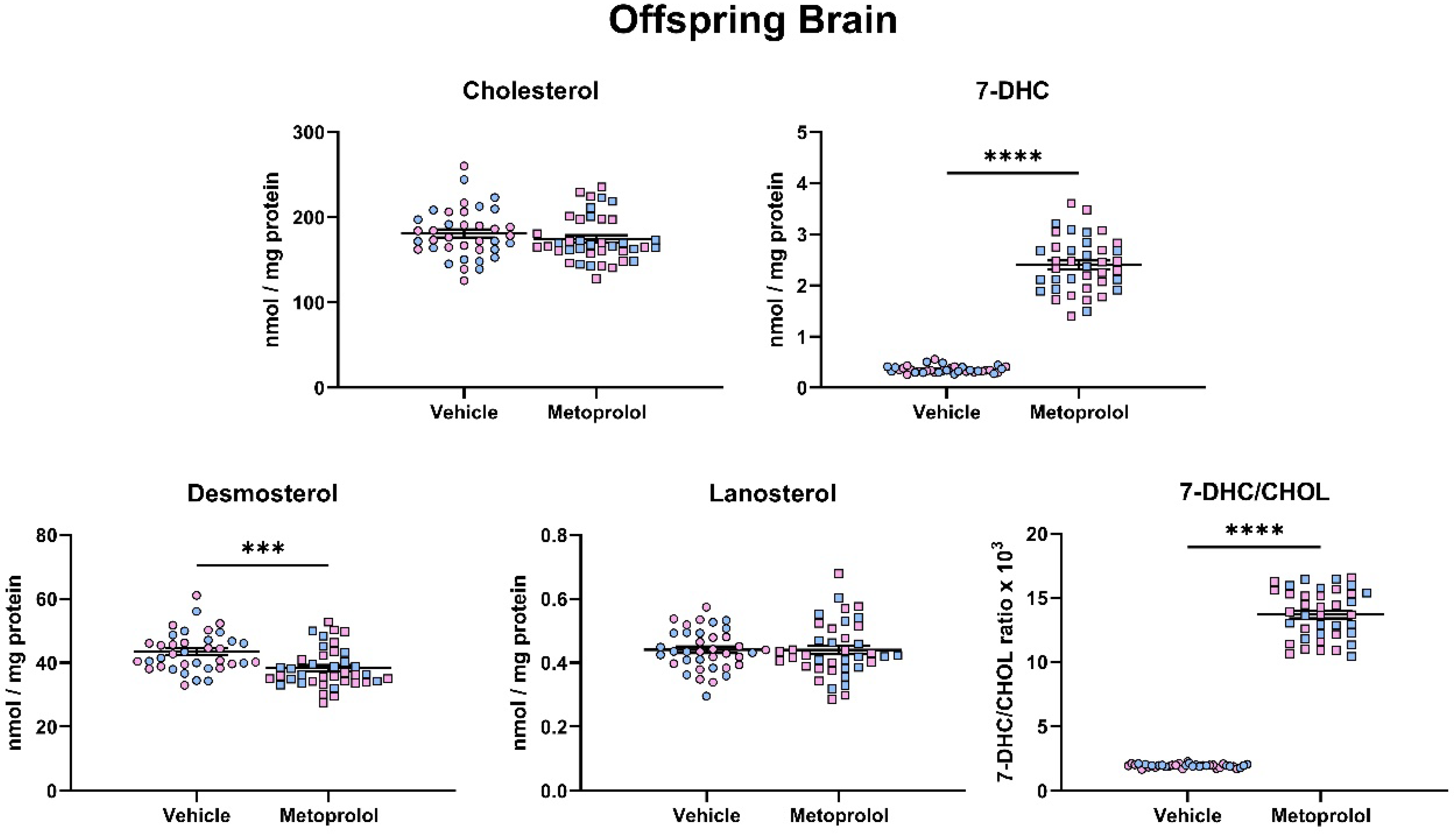
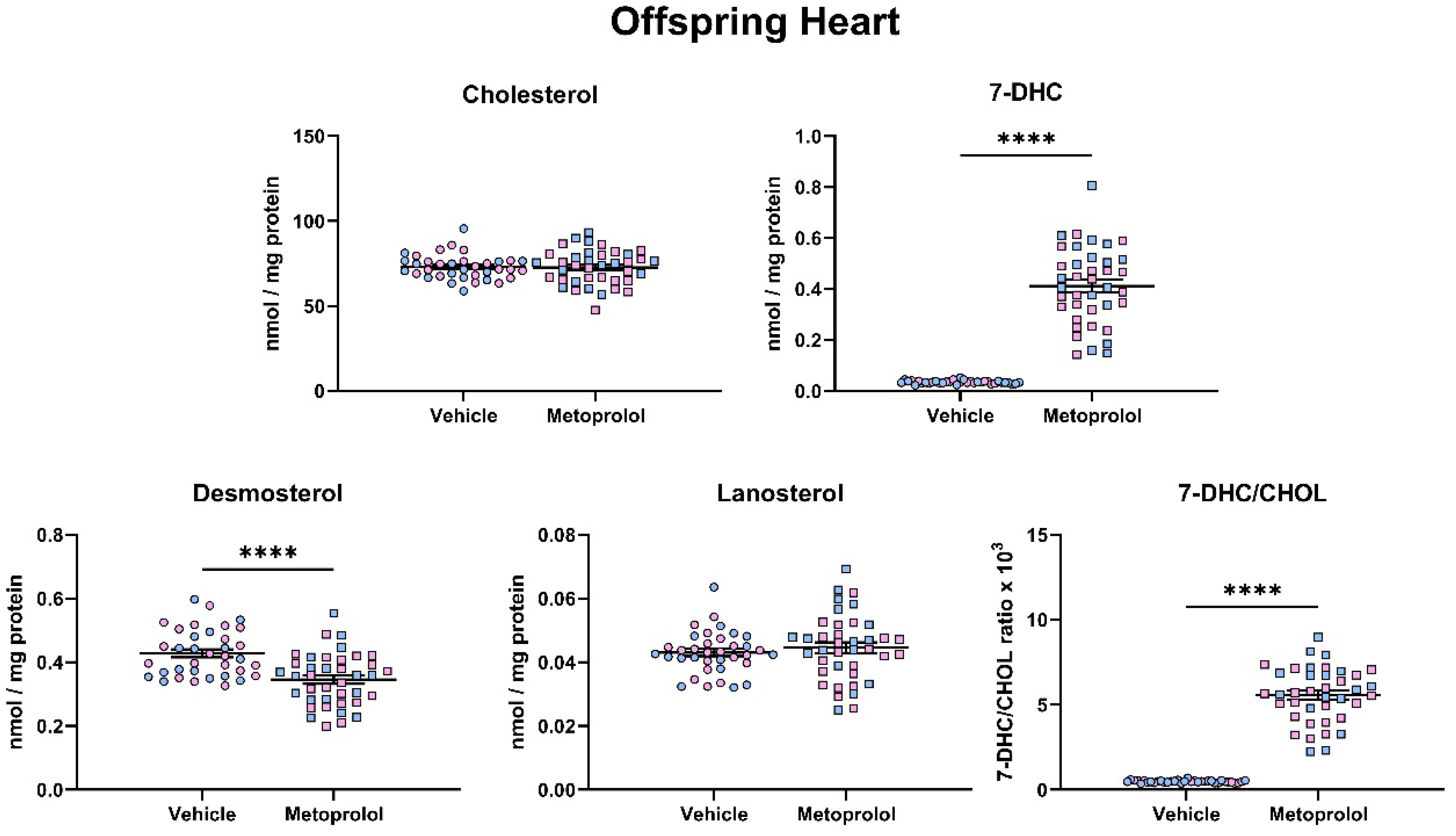
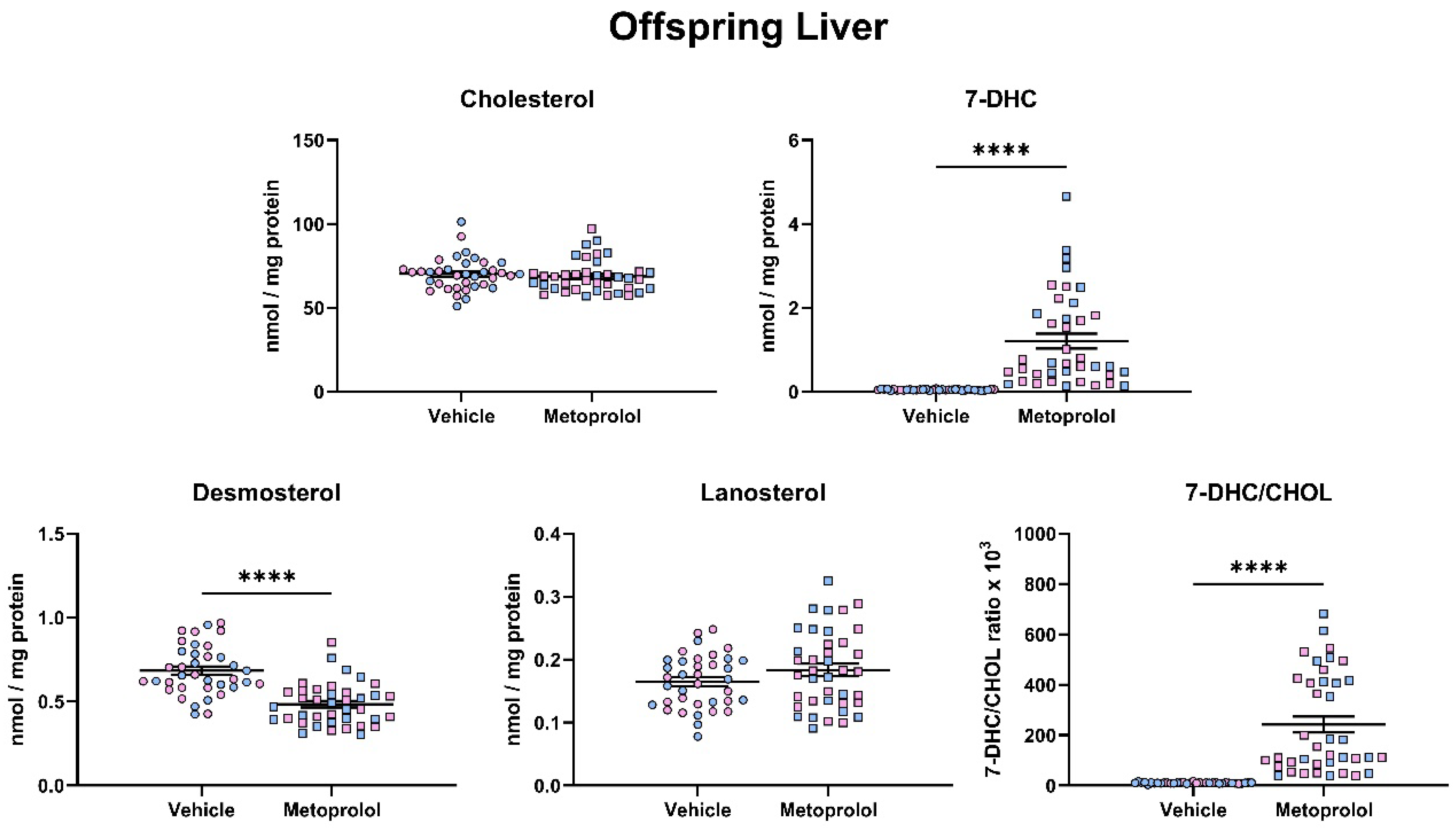
3.4. Maternal Exposure to MTP Alters the Sterol Composition in the Tissue of Offspring
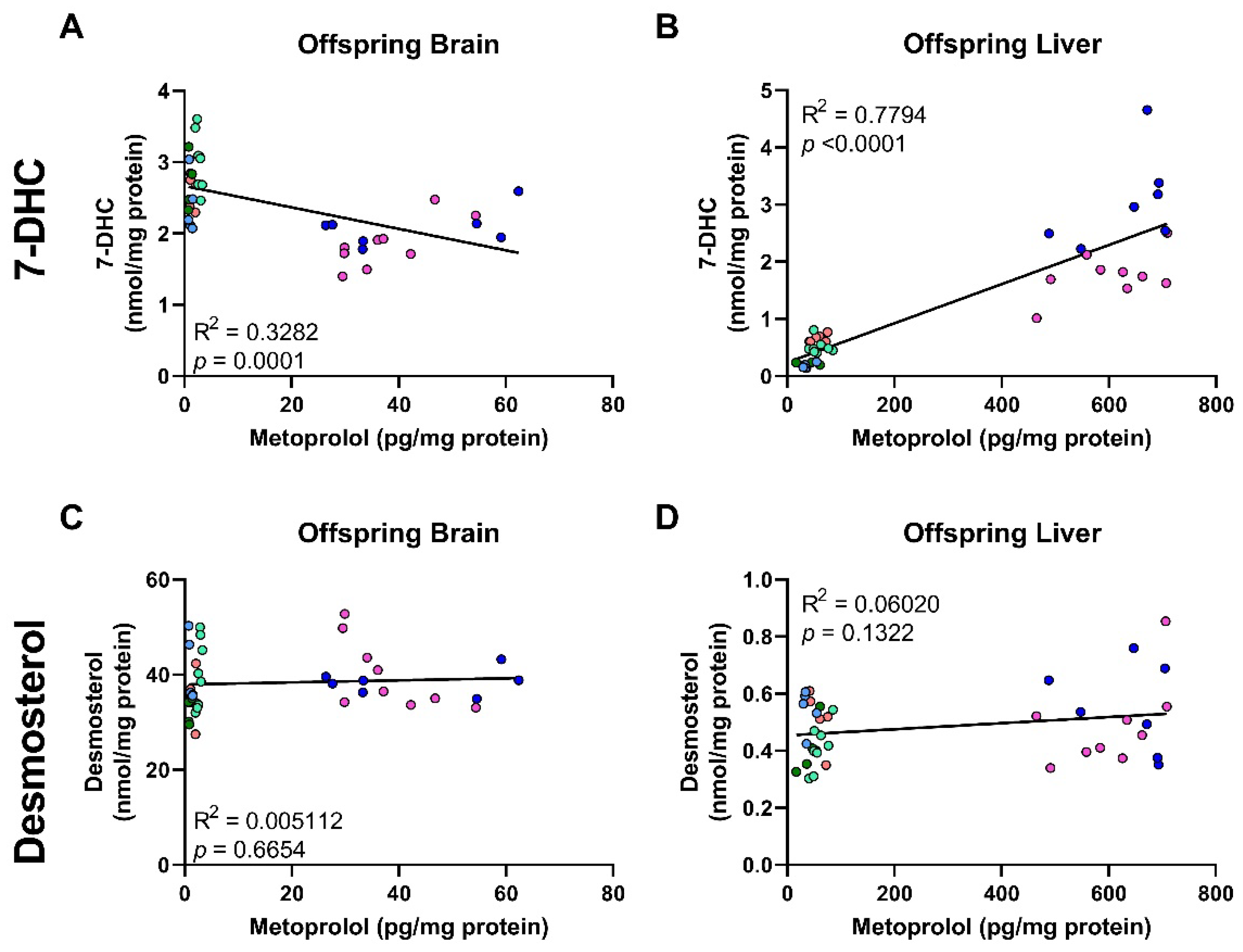
3.5. Correlation of MTP Levels with Sterol Precursors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Korade, Z.; Heffer, M.; Mirnics, K. Medication effects on developmental sterol biosynthesis. Mol. Psychiatry 2022, 27, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J.M. Central nervous system: Cholesterol turnover, brain development and neurodegeneration. Biol. Chem. 2009, 390, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J.M.; Turley, S.D. Cholesterol metabolism in the brain. Curr. Opin. Lipidol. 2001, 12, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J.M.; Turley, S.D. Thematic review series: Brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.D. Biosynthesis of cholesterol and other sterols. Chem. Rev. 2011, 111, 6423–6451. [Google Scholar] [CrossRef]
- Tint, G.S.; Yu, H.; Shang, Q.; Xu, G.; Patel, S.B. The use of the Dhcr7 knockout mouse to accurately determine the origin of fetal sterols. J. Lipid Res. 2006, 47, 1535–1541. [Google Scholar] [CrossRef]
- Pfrieger, F.W. Role of cholesterol in synapse formation and function. Biochim. Biophys. Acta 2003, 1610, 271–280. [Google Scholar] [CrossRef]
- Shrivastava, S.; Paila, Y.D.; Kombrabail, M.; Krishnamoorthy, G.; Chattopadhyay, A. Role of Cholesterol and Its Immediate Biosynthetic Precursors in Membrane Dynamics and Heterogeneity: Implications for Health and Disease. J. Phys. Chem. B 2020, 124, 6312–6320. [Google Scholar] [CrossRef]
- Korade, Z.; Kenworthy, A.K. Lipid rafts, cholesterol, and the brain. Neuropharmacology 2008, 55, 1265–1273. [Google Scholar] [CrossRef]
- Fitzky, B.U.; Witsch-Baumgartner, M.; Erdel, M.; Lee, J.N.; Paik, Y.K.; Glossmann, H.; Utermann, G.; Moebius, F.F. Mutations in the Delta7-sterol reductase gene in patients with the Smith-Lemli-Opitz syndrome. Proc. Natl. Acad. Sci. USA 1998, 95, 8181–8186. [Google Scholar] [CrossRef] [Green Version]
- Lamberson, C.R.; Muchalski, H.; McDuffee, K.B.; Tallman, K.A.; Xu, L.; Porter, N.A. Propagation rate constants for the peroxidation of sterols on the biosynthetic pathway to cholesterol. Chem. Phys. Lipids 2017, 207, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Porter, F.D. RSH/Smith-Lemli-Opitz syndrome: A multiple congenital anomaly/mental retardation syndrome due to an inborn error of cholesterol biosynthesis. Mol. Genet. Metab. 2000, 71, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Porter, F.D. Smith-Lemli-Opitz syndrome: Pathogenesis, diagnosis and management. Eur. J. Hum. Genet. 2008, 16, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Porter, F.D.; Herman, G.E. Malformation syndromes caused by disorders of cholesterol synthesis. J. Lipid Res. 2011, 52, 6–34. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Davis, T.A.; Porter, N.A. Rate constants for peroxidation of polyunsaturated fatty acids and sterols in solution and in liposomes. J. Am. Chem. Soc. 2009, 131, 13037–13044. [Google Scholar] [CrossRef]
- Xu, L.; Korade, Z.; Porter, N.A. Oxysterols from free radical chain oxidation of 7-dehydrocholesterol: Product and mechanistic studies. J. Am. Chem. Soc. 2010, 132, 2222–2232. [Google Scholar] [CrossRef]
- Xu, L.; Korade, Z.; Rosado, D.A., Jr.; Mirnics, K.; Porter, N.A. Metabolism of oxysterols derived from nonenzymatic oxidation of 7-dehydrocholesterol in cells. J. Lipid Res. 2013, 54, 1135–1143. [Google Scholar] [CrossRef]
- Pfeffer, B.A.; Xu, L.; Porter, N.A.; Rao, S.R.; Fliesler, S.J. Differential cytotoxic effects of 7-dehydrocholesterol-derived oxysterols on cultured retina-derived cells: Dependence on sterol structure, cell type, and density. Exp. Eye Res. 2016, 145, 297–316. [Google Scholar] [CrossRef]
- Xu, L.; Mirnics, K.; Bowman, A.B.; Liu, W.; Da, J.; Porter, N.A.; Korade, Z. DHCEO accumulation is a critical mediator of pathophysiology in a Smith-Lemli-Opitz syndrome model. Neurobiol. Dis. 2012, 45, 923–929. [Google Scholar] [CrossRef]
- Korade, Z.; Xu, L.; Shelton, R.; Porter, N.A. Biological activities of 7-dehydrocholesterol-derived oxysterols: Implications for Smith-Lemli-Opitz syndrome. J. Lipid Res. 2010, 51, 3259–3269. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Li, W.; Zheng, X.; Qi, L.; Wang, H.; Zhang, C.; Wan, X.; Zheng, Y.; Zhong, R.; Zhou, X.; et al. Targeting 7-Dehydrocholesterol Reductase Integrates Cholesterol Metabolism and IRF3 Activation to Eliminate Infection. Immunity 2020, 52, 109–122.e106. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.R.; Tatonetti, N.P. Investigation of 7-dehydrocholesterol reductase pathway to elucidate off-target prenatal effects of pharmaceuticals: A systematic review. Pharmacogenomics J. 2016, 16, 411–429. [Google Scholar] [CrossRef] [PubMed]
- DeBarber, A.E.; Eroglu, Y.; Merkens, L.S.; Pappu, A.S.; Steiner, R.D. Smith-Lemli-Opitz syndrome. Expert Rev. Mol. Med. 2011, 13, e24. [Google Scholar] [CrossRef]
- Kelley, R.I.; Hennekam, R.C. The Smith-Lemli-Opitz syndrome. J. Med. Genet. 2000, 37, 321–335. [Google Scholar] [CrossRef]
- Smith, D.W.; Lemli, L.; Opitz, J.M. A Newly Recognized Syndrome of Multiple Congenital Anomalies. J. Pediatr. 1964, 64, 210–217. [Google Scholar] [CrossRef]
- Kadoguchi, M.; Arakawa, H.; Honda, R.; Hotta, K.; Shirasaka, Y.; Deguchi, Y.; Tamai, I. Characterization of Aripiprazole Uptake Transporter in the Blood-Brain Barrier Model hCMEC/D3 Cells by Targeted siRNA Screening. Pharm. Res. 2022, 39, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Toth, M.; Varrone, A.; Steiger, C.; Laszlovszky, I.; Horvath, A.; Kiss, B.; Gyertyan, I.; Adham, N.; Halldin, C.; Gulyas, B. Brain uptake and distribution of the dopamine D3/D2 receptor partial agonist [11 C]cariprazine: An in vivo positron emission tomography study in nonhuman primates. Synapse 2013, 67, 258–264. [Google Scholar] [CrossRef]
- Banks, W.A. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009, 9 (Suppl. 1), S3. [Google Scholar] [CrossRef]
- Saija, A.; Princi, P.; Imperatore, C.; De Pasquale, R.; Costa, G. Ageing influences haloperidol-induced changes in the permeability of the blood-brain barrier in the rat. J. Pharm. Pharmacol. 1992, 44, 450–452. [Google Scholar] [CrossRef]
- Genaro-Mattos, T.C.; Allen, L.B.; Anderson, A.; Tallman, K.A.; Porter, N.A.; Korade, Z.; Mirnics, K. Maternal aripiprazole exposure interacts with 7-dehydrocholesterol reductase mutations and alters embryonic neurodevelopment. Mol. Psychiatry 2019, 24, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Genaro-Mattos, T.C.; Anderson, A.; Allen, L.B.; Tallman, K.A.; Porter, N.A.; Korade, Z.; Mirnics, K. Maternal cariprazine exposure inhibits embryonic and postnatal brain cholesterol biosynthesis. Mol. Psychiatry 2020, 25, 2685–2694. [Google Scholar] [CrossRef]
- Korade, Z.; Allen, L.B.; Anderson, A.; Tallman, K.A.; Genaro-Mattos, T.C.; Porter, N.A.; Mirnics, K. Trazodone effects on developing brain. Transl. Psychiatry 2021, 11, 85. [Google Scholar] [CrossRef] [PubMed]
- Korade, Z.; Liu, W.; Warren, E.B.; Armstrong, K.; Porter, N.A.; Konradi, C. Effect of psychotropic drug treatment on sterol metabolism. Schizophr. Res. 2017, 187, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Wages, P.A.; Kim, H.H.; Korade, Z.; Porter, N.A. Identification and characterization of prescription drugs that change levels of 7-dehydrocholesterol and desmosterol. J. Lipid Res. 2018, 59, 1916–1926. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Korade, Z.; Tallman, K.A.; Liu, W.; Weaver, C.D.; Mirnics, K.; Porter, N.A. Inhibitors of 7-Dehydrocholesterol Reductase: Screening of a Collection of Pharmacologically Active Compounds in Neuro2a Cells. Chem. Res. Toxicol. 2016, 29, 892–900. [Google Scholar] [CrossRef]
- Korade, Z.; Kim, H.Y.; Tallman, K.A.; Liu, W.; Koczok, K.; Balogh, I.; Xu, L.; Mirnics, K.; Porter, N.A. The Effect of Small Molecules on Sterol Homeostasis: Measuring 7-Dehydrocholesterol in Dhcr7-Deficient Neuro2a Cells and Human Fibroblasts. J. Med. Chem. 2016, 59, 1102–1115. [Google Scholar] [CrossRef]
- Podymow, T.; August, P. Update on the use of antihypertensive drugs in pregnancy. Hypertension 2008, 51, 960–969. [Google Scholar] [CrossRef]
- Bateman, B.T.; Huybrechts, K.F.; Fischer, M.A.; Seely, E.W.; Ecker, J.L.; Oberg, A.S.; Franklin, J.M.; Mogun, H.; Hernandez-Diaz, S. Chronic hypertension in pregnancy and the risk of congenital malformations: A cohort study. Am. J. Obstet. Gynecol. 2015, 212, 337.e1–337.e14. [Google Scholar] [CrossRef]
- Bateman, B.T.; Heide-Jorgensen, U.; Einarsdottir, K.; Engeland, A.; Furu, K.; Gissler, M.; Hernandez-Diaz, S.; Kieler, H.; Lahesmaa-Korpinen, A.M.; Mogun, H.; et al. beta-Blocker Use in Pregnancy and the Risk for Congenital Malformations: An International Cohort Study. Ann. Intern. Med. 2018, 169, 665–673. [Google Scholar] [CrossRef]
- Podymow, T.; August, P. Antihypertensive drugs in pregnancy. Semin. Nephrol. 2011, 31, 70–85. [Google Scholar] [CrossRef]
- Kayser, A.; Beck, E.; Hoeltzenbein, M.; Zinke, S.; Meister, R.; Weber-Schoendorfer, C.; Schaefer, C. Neonatal effects of intrauterine metoprolol/bisoprolol exposure during the second and third trimester: A cohort study with two comparison groups. J. Hypertens. 2020, 38, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Welzel, T.; Donner, B.; van den Anker, J.N. Intrauterine Growth Retardation in Pregnant Women with Long QT Syndrome Treated with Beta-Receptor Blockers. Neonatology 2021, 118, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Orbach, H.; Matok, I.; Gorodischer, R.; Sheiner, E.; Daniel, S.; Wiznitzer, A.; Koren, G.; Levy, A. Hypertension and antihypertensive drugs in pregnancy and perinatal outcomes. Am. J. Obstet. Gynecol. 2013, 208, 301.e1–301.e6. [Google Scholar] [CrossRef] [PubMed]
- Genaro-Mattos, T.C.; Anderson, A.; Allen, L.B.; Korade, Z.; Mirnics, K. Cholesterol Biosynthesis and Uptake in Developing Neurons. ACS Chem. Neurosci. 2019, 10, 3671–3681. [Google Scholar] [CrossRef]
- Tallman, K.A.; Allen, L.B.; Klingelsmith, K.; Anderson, A.; Genaro-Mattos, T.C.; Mirnics, K.; Porter, N.A.; Korade, Z. Prescription Medications Alter Neuronal and Glial Cholesterol Synthesis. ACS Chem. Neurosci. 2021. [Google Scholar] [CrossRef]
- Goudriaan, A.; Camargo, N.; Carney, K.E.; Oliet, S.H.; Smit, A.B.; Verheijen, M.H. Novel cell separation method for molecular analysis of neuron-astrocyte co-cultures. Front. Cell. Neurosci. 2014, 8, 12. [Google Scholar] [CrossRef]
- Liu, W.; Xu, L.; Lamberson, C.; Haas, D.; Korade, Z.; Porter, N.A. A highly sensitive method for analysis of 7-dehydrocholesterol for the study of Smith-Lemli-Opitz syndrome. J. Lipid Res. 2014, 55, 329–337. [Google Scholar] [CrossRef]
- Genaro-Mattos, T.C.; Klingelsmith, K.B.; Allen, L.B.; Anderson, A.; Tallman, K.A.; Porter, N.A.; Korade, Z.; Mirnics, K. Sterol Biosynthesis Inhibition in Pregnant Women Taking Prescription Medications. ACS Pharmacol. Transl. Sci. 2021, 4, 848–857. [Google Scholar] [CrossRef]
- Allen, L.B.; Genaro-Mattos, T.C.; Anderson, A.; Porter, N.A.; Mirnics, K.; Korade, Z. Amiodarone Alters Cholesterol Biosynthesis through Tissue-Dependent Inhibition of Emopamil Binding Protein and Dehydrocholesterol Reductase 24. ACS Chem. Neurosci. 2020, 11, 1413–1423. [Google Scholar] [CrossRef]
- Kane, S.P. Evidence-Based Clinical Decision Support Tools and Calculators for Medical Professionals. Available online: Clincalc.com (accessed on 19 July 2022).
- Bateman, B.T.; Huybrechts, K.F.; Hernandez-Diaz, S.; Kieler, H.; Zoega, H. beta-Blocker Use in Pregnancy and the Risk for Congenital Malformations. Ann. Intern. Med. 2019, 170, 909–910. [Google Scholar] [CrossRef]
- Hogstedt, S.; Lindberg, B.; Rane, A. Increased oral clearance of metoprolol in pregnancy. Eur. J. Clin. Pharmacol. 1983, 24, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Meidahl Petersen, K.; Jimenez-Solem, E.; Andersen, J.T.; Petersen, M.; Brodbaek, K.; Kober, L.; Torp-Pedersen, C.; Poulsen, H.E. beta-Blocker treatment during pregnancy and adverse pregnancy outcomes: A nationwide population-based cohort study. BMJ Open 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Ryu, R.J.; Eyal, S.; Easterling, T.R.; Caritis, S.N.; Venkataraman, R.; Hankins, G.; Rytting, E.; Thummel, K.; Kelly, E.J.; Risler, L.; et al. Pharmacokinetics of metoprolol during pregnancy and lactation. J. Clin. Pharmacol. 2016, 56, 581–589. [Google Scholar] [CrossRef] [PubMed]
- ClinCalc. Available online: https://clincalc.com/DrugStats/Drugs/Metoprolol (accessed on 21 July 2022).
- Burkauskas, J.; Noreikaite, A.; Bunevicius, A.; Brozaitiene, J.; Neverauskas, J.; Mickuviene, N.; Bunevicius, R. Beta-1-Selective Beta-Blockers and Cognitive Functions in Patients With Coronary Artery Disease: A Cross-Sectional Study. J. Neuropsychiatry Clin. Neurosci. 2016, 28, 143–146. [Google Scholar] [CrossRef]
- Burris, J.F. Central nervous system side effects with beta-blockers. Arch. Intern. Med. 1988, 148, 980. [Google Scholar] [CrossRef]
- Cove-Smith, J.R.; Kirk, C.A. CNS-related side-effects with metoprolol and atenolol. Eur. J. Clin. Pharmacol. 1985, 28, 69–72. [Google Scholar] [CrossRef]
- Gliebus, G.; Lippa, C.F. The influence of beta-blockers on delayed memory function in people with cognitive impairment. Am. J. Alzheimers Dis. Other Demen. 2007, 22, 57–61. [Google Scholar] [CrossRef]
- Goldner, J.A. Metoprolol-induced visual hallucinations: A case series. J. Med. Case Rep. 2012, 6, 65. [Google Scholar] [CrossRef]
- Liu, X.; Lou, X.; Cheng, X.; Meng, Y. Impact of metoprolol treatment on mental status of chronic heart failure patients with neuropsychiatric disorders. Drug Des. Devel. Ther. 2017, 11, 305–312. [Google Scholar] [CrossRef]
- McAinsh, J.; Cruickshank, J.M. Beta-blockers and central nervous system side effects. Pharmacol. Ther. 1990, 46, 163–197. [Google Scholar] [CrossRef]
- Rietveld, L.; van der Hoek, T.; van Beek, M.H.; Schellekens, A.F. Familial liability for metoprolol-induced psychosis. Gen. Hosp. Psychiatry 2015, 37, e625–e626. [Google Scholar] [CrossRef] [PubMed]
- Sirois, F.J. Visual hallucinations and metoprolol. Psychosomatics 2006, 47, 537–538. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.I.; van Mierlo, P.J.; van Waarde, J.A.; Jansen, P.A. [Hallucinations and vivid dreams by use of metoprolol]. Tijdschr. Psychiatr. 2010, 52, 117–121. [Google Scholar] [PubMed]
- Westerlund, A. Central nervous system side-effects with hydrophilic and lipophilic beta-blockers. Eur. J. Clin. Pharmacol. 1985, 28, 73–76. [Google Scholar] [CrossRef]
- Neil-Dwyer, G. The clinical importance of lipid solubility in beta blockers. Aviat. Space Environ. Med. 1981, 52, S19–S22. [Google Scholar]
- Evans, A.K.; Ardestani, P.M.; Yi, B.; Park, H.H.; Lam, R.K.; Shamloo, M. Beta-adrenergic receptor antagonism is proinflammatory and exacerbates neuroinflammation in a mouse model of Alzheimer’s Disease. Neurobiol. Dis. 2020, 146, 105089. [Google Scholar] [CrossRef]
- Laurens, C.; Abot, A.; Delarue, A.; Knauf, C. Central Effects of Beta-Blockers May Be Due to Nitric Oxide and Hydrogen Peroxide Release Independently of Their Ability to Cross the Blood-Brain Barrier. Front. Neurosci. 2019, 13, 33. [Google Scholar] [CrossRef]
- Coyle, C.H.; Martinez, L.J.; Coleman, M.C.; Spitz, D.R.; Weintraub, N.L.; Kader, K.N. Mechanisms of H2O2-induced oxidative stress in endothelial cells. Free Radic. Biol. Med. 2006, 40, 2206–2213. [Google Scholar] [CrossRef]
- Xu, L.; Korade, Z.; Rosado, J.D.A.; Liu, W.; Lamberson, C.R.; Porter, N.A. An oxysterol biomarker for 7-dehydrocholesterol oxidation in cell/mouse models for Smith-Lemli-Opitz syndrome. J. Lipid Res. 2011, 52, 1222–1233. [Google Scholar] [CrossRef]
- Duc, D.; Vigne, S.; Pot, C. Oxysterols in Autoimmunity. Int. J. Mol. Sci. 2019, 20, 4522. [Google Scholar] [CrossRef]
- Koella, W.P. CNS-related (side-)effects of beta-blockers with special reference to mechanisms of action. Eur. J. Clin. Pharmacol. 1985, 28, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Kostis, J.B.; Rosen, R.C. Central nervous system effects of beta-adrenergic-blocking drugs: The role of ancillary properties. Circulation 1987, 75, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Muller, G.; Lubow, C.; Weindl, G. Lysosomotropic beta blockers induce oxidative stress and IL23A production in Langerhans cells. Autophagy 2020, 16, 1380–1395. [Google Scholar] [CrossRef] [PubMed]
- Oliver, E.; Mayor, F., Jr.; D’Ocon, P. Beta-blockers: Historical Perspective and Mechanisms of Action. Rev. Esp. Cardiol. (Engl. Ed.) 2019, 72, 853–862. [Google Scholar] [CrossRef]
- Yoshida, H.; Suzukawa, M.; Ishikawa, T.; Shige, H.; Nishio, E.; Hosoai, H.; Ayaori, M.; Nakamura, H. Effects of beta-blockers on HMG CoA reductase and LDL receptor activity in cultured human skin fibroblasts. Cardiovasc. Drugs Ther. 1996, 10, 67–74. [Google Scholar] [CrossRef]
- Ruys, T.P.; Maggioni, A.; Johnson, M.R.; Sliwa, K.; Tavazzi, L.; Schwerzmann, M.; Nihoyannopoulos, P.; Kozelj, M.; Marelli, A.; Elkayam, U.; et al. Cardiac medication during pregnancy, data from the ROPAC. Int. J. Cardiol. 2014, 177, 124–128. [Google Scholar] [CrossRef]
- Eroli, F.; Johnell, K.; Latorre-Leal, M.; Hilmer, S.; Wastesson, J.; Cedazo-Minguez, A.; Maioli, S. Long-term exposure to polypharmacy impairs cognitive functions in young adult female mice. Aging (Albany NY) 2021, 13, 14729–14744. [Google Scholar] [CrossRef]
- Siwek, M.; Woron, J.; Gorostowicz, A.; Wordliczek, J. Adverse effects of interactions between antipsychotics and medications used in the treatment of cardiovascular disorders. Pharmacol. Rep. 2020, 72, 350–359. [Google Scholar] [CrossRef]
- Pringsheim, T.; Gardner, D.; Addington, D.; Martino, D.; Morgante, F.; Ricciardi, L.; Poole, N.; Remington, G.; Edwards, M.; Carson, A.; et al. The Assessment and Treatment of Antipsychotic-Induced Akathisia. Can. J. Psychiatry 2018, 63, 719–729. [Google Scholar] [CrossRef]
- Salem, H.; Nagpal, C.; Pigott, T.; Teixeira, A.L. Revisiting Antipsychotic-induced Akathisia: Current Issues and Prospective Challenges. Curr. Neuropharmacol. 2017, 15, 789–798. [Google Scholar] [CrossRef]
- Adler, L.A.; Angrist, B.; Rotrosen, J. Metoprolol versus propranolol. Biol. Psychiatry 1990, 27, 673–675. [Google Scholar] [CrossRef]
- Genaro-Mattos, T.C.; Tallman, K.A.; Allen, L.B.; Anderson, A.; Mirnics, K.; Korade, Z.; Porter, N.A. Dichlorophenyl piperazines, including a recently-approved atypical antipsychotic, are potent inhibitors of DHCR7, the last enzyme in cholesterol biosynthesis. Toxicol. Appl. Pharmacol. 2018, 349, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Balog, M.; Anderson, A.; Genaro-Mattos, T.C.; Korade, Z.; Mirnics, K. Individual and simultaneous treatment with antipsychotic aripiprazole and antidepressant trazodone inhibit sterol biosynthesis in the adult brain. J. Lipid Res. 2022, 63, 100249. [Google Scholar] [CrossRef] [PubMed]
- Genaro-Mattos, T.C.; Anderson, A.; Allen, L.B.; Korade, Z.; Mirnics, K. Altered Cholesterol Biosynthesis Affects Drug Metabolism. ACS Omega 2021, 6, 5490–5498. [Google Scholar] [CrossRef] [PubMed]
- Korade, Z.; Genaro-Mattos, T.C.; Tallman, K.A.; Liu, W.; Garbett, K.A.; Koczok, K.; Balogh, I.; Mirnics, K.; Porter, N.A. Vulnerability of DHCR7(+/-) mutation carriers to aripiprazole and trazodone exposure. J. Lipid Res. 2017, 58, 2139–2146. [Google Scholar] [CrossRef] [PubMed]
- Shariq, O.A.; McKenzie, T.J. Obesity-related hypertension: A review of pathophysiology, management, and the role of metabolic surgery. Gland Surg. 2020, 9, 80–93. [Google Scholar] [CrossRef]
- Fantin, F.; Giani, A.; Zoico, E.; Rossi, A.P.; Mazzali, G.; Zamboni, M. Weight Loss and Hypertension in Obese Subjects. Nutrients 2019, 11, 1667. [Google Scholar] [CrossRef]
- Re, R.N. Obesity-related hypertension. Ochsner. J. 2009, 9, 133–136. [Google Scholar]
- Mikhail, N.; Golub, M.S.; Tuck, M.L. Obesity and hypertension. Prog. Cardiovasc. Dis. 1999, 42, 39–58. [Google Scholar] [CrossRef]
- Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Despres, J.P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. Obesity and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2021, 143, e984–e1010. [Google Scholar] [CrossRef]
- Mandviwala, T.; Khalid, U.; Deswal, A. Obesity and Cardiovascular Disease: A Risk Factor or a Risk Marker? Curr. Atheroscler. Rep. 2016, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, D.R.; Morris, M.A.; Gambone, J.C. Obesity pandemic: Causes, consequences, and solutions-but do we have the will? Fertil. Steril. 2017, 107, 833–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swinburn, B.A.; Sacks, G.; Hall, K.D.; McPherson, K.; Finegood, D.T.; Moodie, M.L.; Gortmaker, S.L. The global obesity pandemic: Shaped by global drivers and local environments. Lancet 2011, 378, 804–814. [Google Scholar] [CrossRef]
- Kelley, R.I. Diagnosis of Smith-Lemli-Opitz syndrome by gas chromatography/mass spectrometry of 7-dehydrocholesterol in plasma, amniotic fluid and cultured skin fibroblasts. Clin. Chim. Acta 1995, 236, 45–58. [Google Scholar] [CrossRef]
- Cross, J.L.; Iben, J.; Simpson, C.L.; Thurm, A.; Swedo, S.; Tierney, E.; Bailey-Wilson, J.E.; Biesecker, L.G.; Porter, F.D.; Wassif, C.A. Determination of the allelic frequency in Smith-Lemli-Opitz syndrome by analysis of massively parallel sequencing data sets. Clin. Genet. 2015, 87, 570–575. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allen, L.B.; Mirnics, K. Metoprolol Inhibits Developmental Brain Sterol Biosynthesis in Mice. Biomolecules 2022, 12, 1211. https://doi.org/10.3390/biom12091211
Allen LB, Mirnics K. Metoprolol Inhibits Developmental Brain Sterol Biosynthesis in Mice. Biomolecules. 2022; 12(9):1211. https://doi.org/10.3390/biom12091211
Chicago/Turabian StyleAllen, Luke B., and Károly Mirnics. 2022. "Metoprolol Inhibits Developmental Brain Sterol Biosynthesis in Mice" Biomolecules 12, no. 9: 1211. https://doi.org/10.3390/biom12091211
APA StyleAllen, L. B., & Mirnics, K. (2022). Metoprolol Inhibits Developmental Brain Sterol Biosynthesis in Mice. Biomolecules, 12(9), 1211. https://doi.org/10.3390/biom12091211