
Enthalpy–Entropy Compensation in the Promiscuous Interaction of an Intrinsically Disordered Protein with Homologous Protein Partners

 and
and 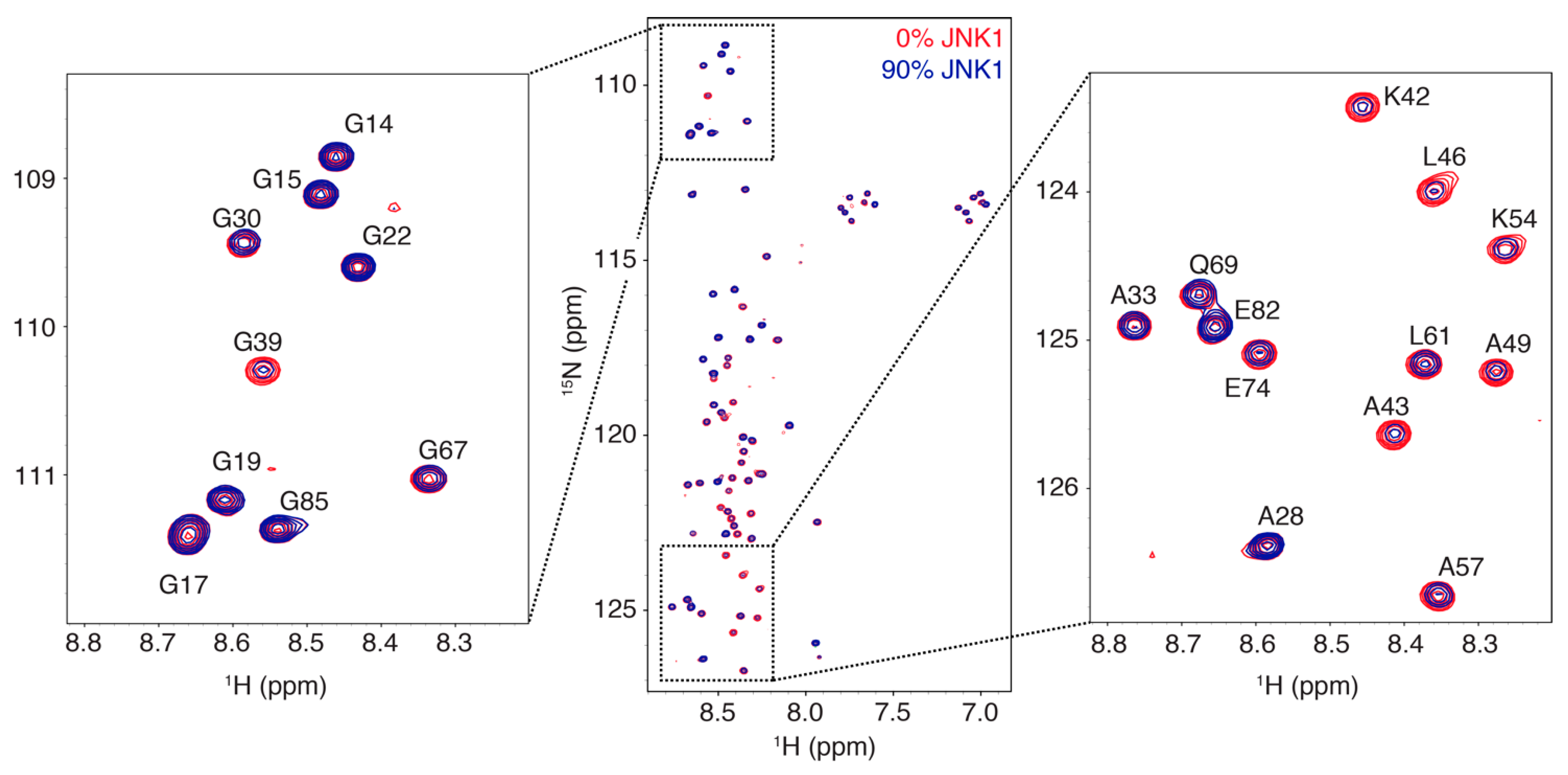{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. ITC Experiments
2.3. NMR Titrations
2.4. NMR Exchange Experiments
3. Results
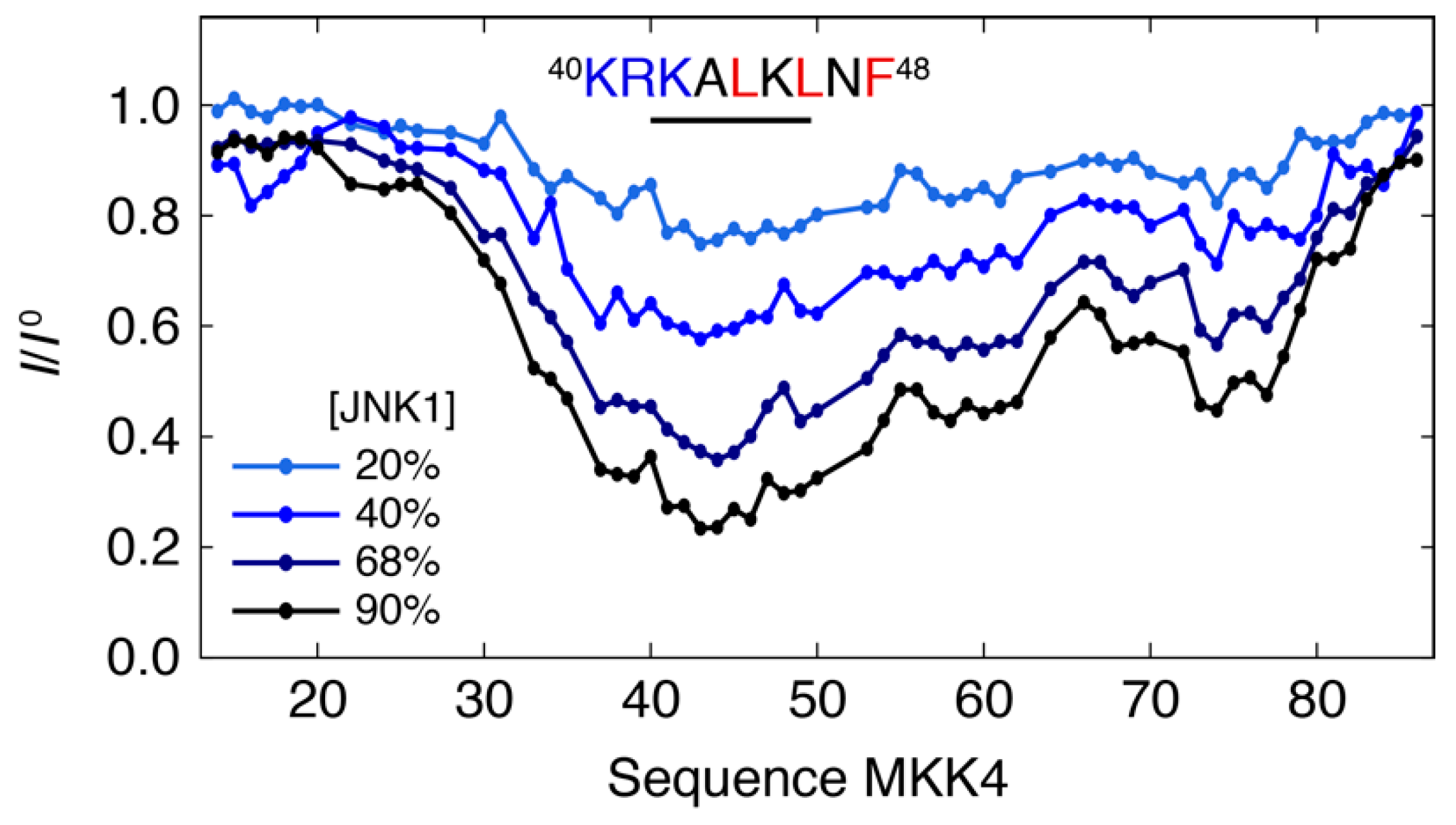
3.1. Interaction Profile of the Regulatory Domain of MKK4 with the JNK1 Kinase
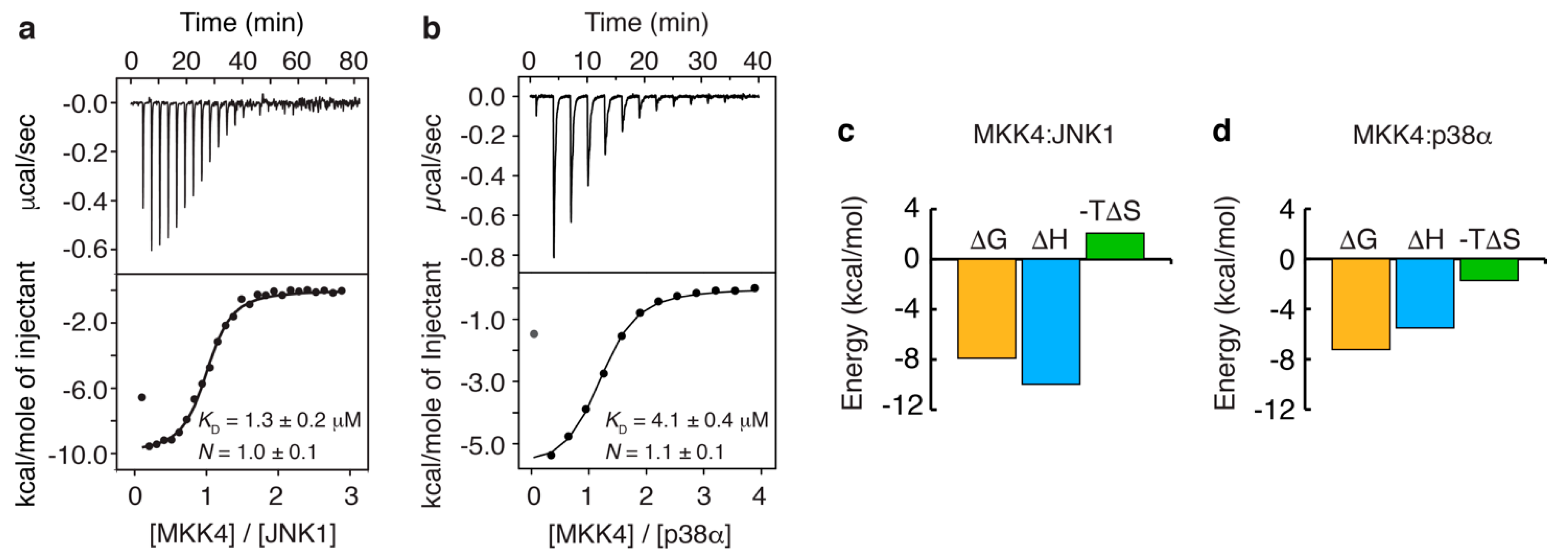
3.2. Thermodynamics of the Interaction of MKK4 with the Two MAPK Paralogs
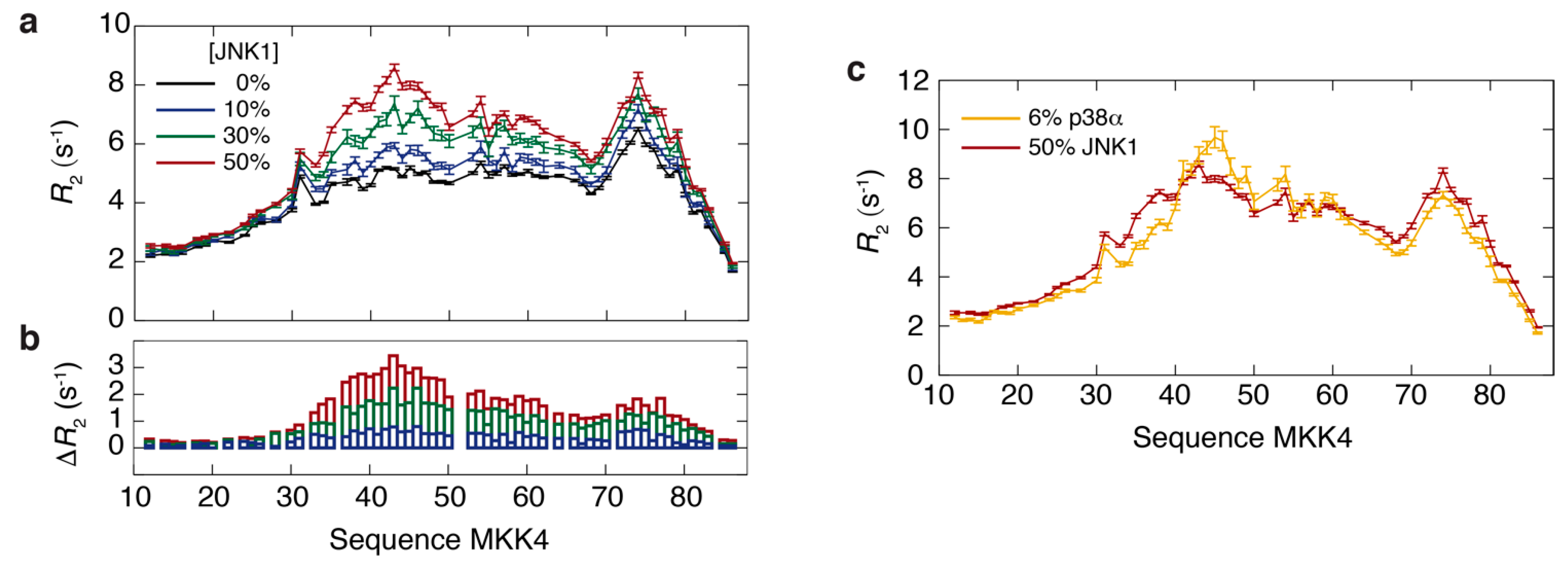
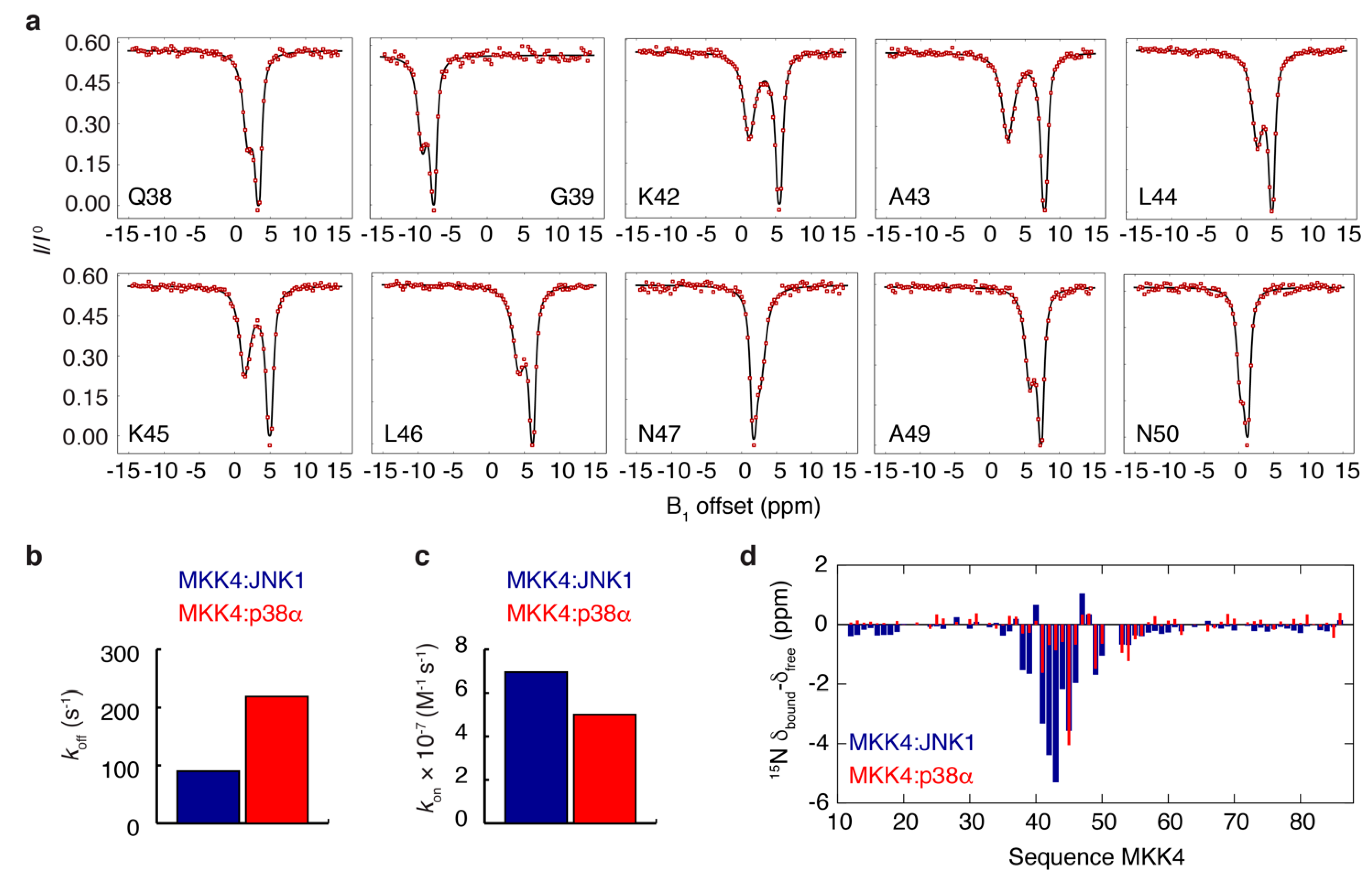
3.3. NMR Exchange Experiments of the MKK4:JNK1 and MKK4:p38α Complexes
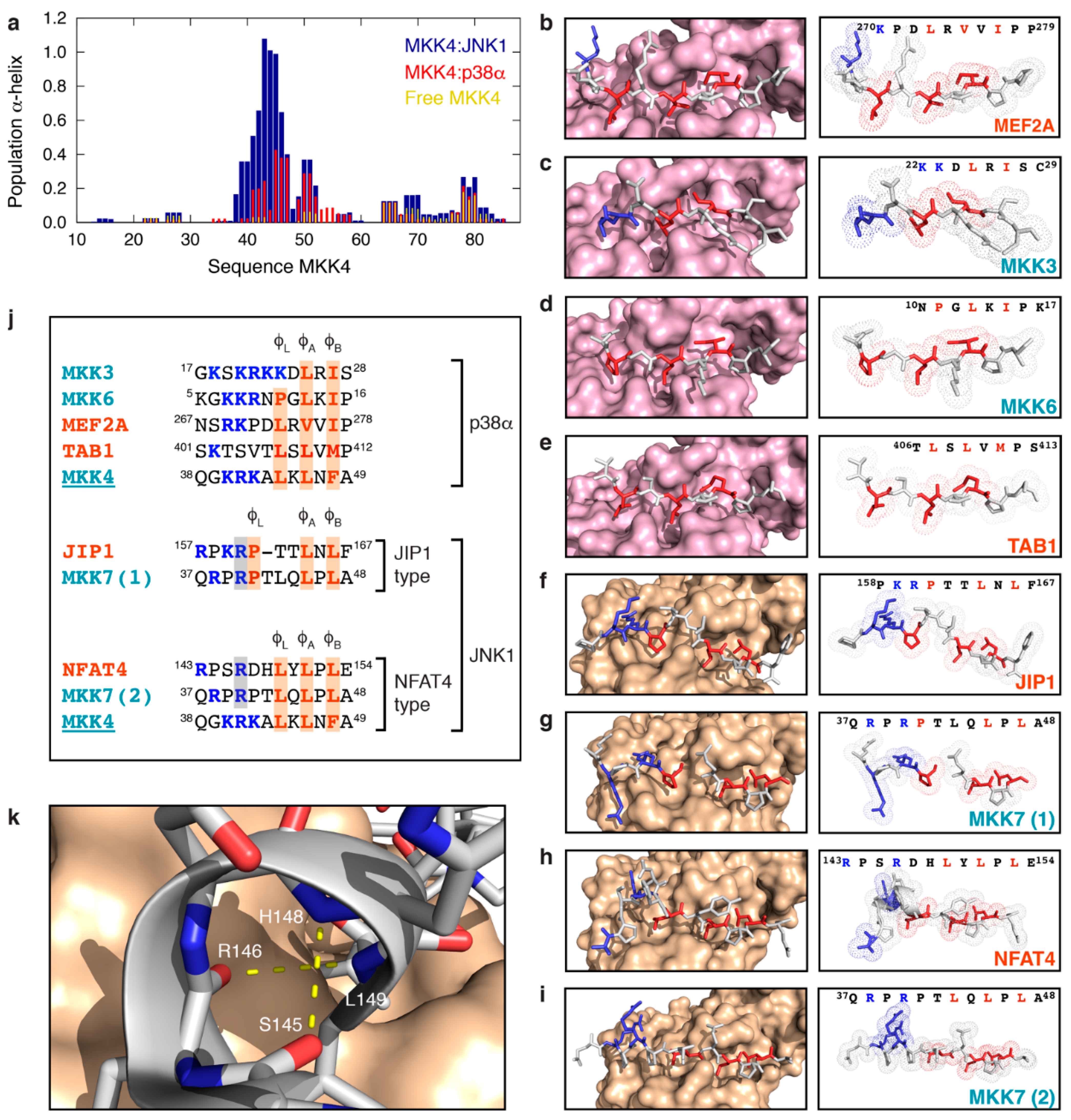
3.4. MKK4 Folds into Two Different Conformations upon Binding to JNK1 and p38α
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wright, P.E.; Dyson, H.J. Intrinsically Unstructured Proteins: Re-Assessing the Protein Structure-Function Paradigm. J. Mol. Biol 1999, 293, 321–331. [Google Scholar] [CrossRef]
- Uversky, V.N. Natively Unfolded Proteins: A Point Where Biology Waits for Physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef]
- Tompa, P. Unstructural Biology Coming of Age. Curr. Opin. Struct. Biol. 2011, 21, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing Protein Intrinsic Disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef]
- Pancsa, R.; Fuxreiter, M. Interactions via Intrinsically Disordered Regions: What Kind of Motifs? IUBMB Life 2012, 64, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Davey, N.E.; Gibson, T.J.; Babu, M.M. A Million Peptide Motifs for the Molecular Biologist. Mol. Cell 2014, 55, 161–169. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Coupling of Folding and Binding for Unstructured Proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Romero, P.; Uversky, V.N.; Dunker, A.K. Coupled Folding and Binding with Alpha-Helix-Forming Molecular Recognition Elements. Biochemistry 2005, 44, 12454–12470. [Google Scholar] [CrossRef]
- Sugase, K.; Dyson, H.J.; Wright, P.E. Mechanism of Coupled Folding and Binding of an Intrinsically Disordered Protein. Nature 2007, 447, 1021–1025. [Google Scholar] [CrossRef]
- Gianni, S.; Dogan, J.; Jemth, P. Coupled Binding and Folding of Intrinsically Disordered Proteins: What Can We Learn from Kinetics? Curr. Opin. Struct. Biol. 2015, 36, 18–24. [Google Scholar] [CrossRef]
- Schneider, R.; Maurin, D.; Communie, G.; Kragelj, J.; Hansen, D.F.; Ruigrok, R.W.H.; Jensen, M.R.; Blackledge, M. Visualizing the Molecular Recognition Trajectory of an Intrinsically Disordered Protein Using Multinuclear Relaxation Dispersion NMR. J. Am. Chem. Soc. 2015, 137, 1220–1229. [Google Scholar] [CrossRef]
- Tompa, P.; Schad, E.; Tantos, A.; Kalmar, L. Intrinsically Disordered Proteins: Emerging Interaction Specialists. Curr. Opin. Struct. Biol. 2015, 35, 49–59. [Google Scholar] [CrossRef]
- Charlier, C.; Bouvignies, G.; Pelupessy, P.; Walrant, A.; Marquant, R.; Kozlov, M.; De Ioannes, P.; Bolik-Coulon, N.; Sagan, S.; Cortes, P.; et al. Structure and Dynamics of an Intrinsically Disordered Protein Region That Partially Folds upon Binding by Chemical-Exchange NMR. J. Am. Chem. Soc. 2017, 139, 12219–12227. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically Unstructured Proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically Unstructured Proteins and Their Functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically Disordered Proteins in Cellular Signalling and Regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Demarest, S.J.; Martinez-Yamout, M.; Chung, J.; Chen, H.; Xu, W.; Dyson, H.J.; Evans, R.M.; Wright, P.E. Mutual Synergistic Folding in Recruitment of CBP/P300 by P160 Nuclear Receptor Coactivators. Nature 2002, 415, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, Z. Do Intrinsically Disordered Proteins Possess High Specificity in Protein-Protein Interactions? Chemistry 2013, 19, 4462–4467. [Google Scholar] [CrossRef]
- Teilum, K.; Olsen, J.G.; Kragelund, B.B. Globular and Disordered-the Non-Identical Twins in Protein-Protein Interactions. Front. Mol. Biosci. 2015, 2, 40. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Simon, I.; Friedrich, P.; Tompa, P. Preformed Structural Elements Feature in Partner Recognition by Intrinsically Unstructured Proteins. J. Mol. Biol. 2004, 338, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Gely, S.; Lowry, D.F.; Bernard, C.; Jensen, M.R.; Blackledge, M.; Costanzo, S.; Bourhis, J.-M.; Darbon, H.; Daughdrill, G.; Longhi, S. Solution Structure of the C-Terminal X Domain of the Measles Virus Phosphoprotein and Interaction with the Intrinsically Disordered C-Terminal Domain of the Nucleoprotein. J. Mol. Recognit. 2010, 23, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Tompa, P.; Simon, I.; Dosztányi, Z. Molecular Principles of the Interactions of Disordered Proteins. J. Mol. Biol. 2007, 372, 549–561. [Google Scholar] [CrossRef]
- Dogan, J.; Gianni, S.; Jemth, P. The Binding Mechanisms of Intrinsically Disordered Proteins. Phys. Chem. Chem. Phys. 2014, 16, 6323–6331. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Szász, C.; Buday, L. Structural Disorder Throws New Light on Moonlighting. Trends Biochem. Sci. 2005, 30, 484–489. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible Nets: Disorder and Induced Fit in the Associations of P53 and 14–3-3 with Their Partners. BMC Genomics 2008, 9 (Suppl. 1), S1. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK Signal Transduction Pathways Activated by Stress and Inflammation: A 10-Year Update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and P38 Protein Kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Tanoue, T.; Nishida, E. Docking Interactions in the Mitogen-Activated Protein Kinase Cascades. Pharmacol. Ther. 2002, 93, 193–202. [Google Scholar] [CrossRef]
- Bardwell, L. Mechanisms of MAPK Signalling Specificity. Biochem. Soc. Trans. 2006, 34, 837–841. [Google Scholar] [CrossRef]
- Bardwell, A.J.; Frankson, E.; Bardwell, L. Selectivity of Docking Sites in MAPK Kinases. J. Biol. Chem. 2009, 284, 13165–13173. [Google Scholar] [CrossRef]
- Derijard, B.; Raingeaud, J.; Barrett, T.; Wu, I.; Han, J.; Ulevitch, R.; Davis, R. Independent Human MAP-Kinase Signal Transduction Pathways Defined by MEK and MKK Isoforms. Science 1995, 267, 682–685. [Google Scholar] [CrossRef]
- Lin, A.; Minden, A.; Martinetto, H.; Claret, F.X.; Lange-Carter, C.; Mercurio, F.; Johnson, G.L.; Karin, M. Identification of a Dual Specificity Kinase That Activates the Jun Kinases and P38-Mpk2. Science 1995, 268, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Garai, Á.; Zeke, A.; Gógl, G.; Törő, I.; Fördős, F.; Blankenburg, H.; Bárkai, T.; Varga, J.; Alexa, A.; Emig, D.; et al. Specificity of Linear Motifs That Bind to a Common Mitogen-Activated Protein Kinase Docking Groove. Sci Signal 2012, 5, ra74. [Google Scholar] [CrossRef] [PubMed]
- Kragelj, J.; Palencia, A.; Nanao, M.H.; Maurin, D.; Bouvignies, G.; Blackledge, M.; Jensen, M.R. Structure and Dynamics of the MKK7-JNK Signaling Complex. Proc. Natl. Acad. Sci. USA 2015, 112, 3409–3414. [Google Scholar] [CrossRef] [PubMed]
- Delaforge, E.; Kragelj, J.; Tengo, L.; Palencia, A.; Milles, S.; Bouvignies, G.; Salvi, N.; Blackledge, M.; Jensen, M.R. Deciphering the Dynamic Interaction Profile of an Intrinsically Disordered Protein by NMR Exchange Spectroscopy. J. Am. Chem. Soc. 2018, 140, 1148–1158. [Google Scholar] [CrossRef]
- Lakomek, N.-A.; Ying, J.; Bax, A. Measurement of 15N Relaxation Rates in Perdeuterated Proteins by TROSY-Based Methods. J. Biomol. NMR 2012, 53, 209–221. [Google Scholar] [CrossRef]
- Akke, M.; Palmer, A.G. Monitoring Macromolecular Motions on Microsecond to Millisecond Time Scales by R1ρ−R1 Constant Relaxation Time NMR Spectroscopy. J. Am. Chem. Soc. 1996, 118, 911–912. [Google Scholar] [CrossRef]
- Cheng, N.-S. Formula for the Viscosity of a Glycerol−Water Mixture. Ind. Eng. Chem. Res. 2008, 47, 3285–3288. [Google Scholar] [CrossRef]
- Bae, S.-H.; Dyson, H.J.; Wright, P.E. Prediction of the Rotational Tumbling Time for Proteins with Disordered Segments. J. Am. Chem. Soc. 2009, 131, 6814–6821. [Google Scholar] [CrossRef]
- Vallurupalli, P.; Bouvignies, G.; Kay, L.E. Studying “Invisible” Excited Protein States in Slow Exchange with a Major State Conformation. J. Am. Chem. Soc. 2012, 134, 8148–8161. [Google Scholar] [CrossRef]
- Fersht, A.R.; Shi, J.P.; Knill-Jones, J.; Lowe, D.M.; Wilkinson, A.J.; Blow, D.M.; Brick, P.; Carter, P.; Waye, M.M.; Winter, G. Hydrogen Bonding and Biological Specificity Analysed by Protein Engineering. Nature 1985, 314, 235–238. [Google Scholar] [CrossRef]
- Kragelj, J.; Ozenne, V.; Blackledge, M.; Jensen, M.R. Conformational Propensities of Intrinsically Disordered Proteins from NMR Chemical Shifts. Chemphyschem 2013, 14, 3034–3045. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.; Wider, G.; Wuethrich, K. Sequence-Corrected 15N “Random Coil” Chemical Shifts. J. Am. Chem. Soc. 1994, 116, 8466–8469. [Google Scholar] [CrossRef]
- Tamiola, K.; Acar, B.; Mulder, F.A.A. Sequence-Specific Random Coil Chemical Shifts of Intrinsically Disordered Proteins. J. Am. Chem. Soc. 2010, 132, 18000–18003. [Google Scholar] [CrossRef]
- Tamiola, K.; Mulder, F.A.A. Using NMR Chemical Shifts to Calculate the Propensity for Structural Order and Disorder in Proteins. Biochem. Soc. Trans. 2012, 40, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.T.; Mulder, F.A.A. POTENCI: Prediction of Temperature, Neighbor and PH-Corrected Chemical Shifts for Intrinsically Disordered Proteins. J. Biomol. NMR 2018, 70, 141–165. [Google Scholar] [CrossRef]
- Zhang, H.; Neal, S.; Wishart, D.S. RefDB: A Database of Uniformly Referenced Protein Chemical Shifts. J. Biomol. NMR 2003, 25, 173–195. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.I.; Xu, B.; Akella, R.; Cobb, M.H.; Goldsmith, E.J. Crystal Structures of MAP Kinase P38 Complexed to the Docking Sites on Its Nuclear Substrate MEF2A and Activator MKK3b. Mol. Cell 2002, 9, 1241–1249. [Google Scholar] [CrossRef]
- Xin, F.; Wu, J. Crystal Structure of the P38α MAP Kinase in Complex with a Docking Peptide from TAB1. Sci. China Life Sci. 2013, 56, 653–660. [Google Scholar] [CrossRef][Green Version]
- Heo, J.; Kim, S.; Seo, C.; Kim, Y.; Sung, B.; Lee, H.; Lee, J.; Park, S.; Kim, J.; Hwang, K.; et al. Structural Basis for the Selective Inhibition of JNK1 by the Scaffolding Protein JIP1 and SP600125. EMBO J. 2004, 23, 2185–2195. [Google Scholar] [CrossRef]
- Bardwell, A.J.; Bardwell, L. Two Hydrophobic Residues Can Determine the Specificity of Mitogen-Activated Protein Kinase Docking Interactions. J. Biol. Chem. 2015, 290, 26661–26674. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Otieno, S.; Waddell, B.; Iconaru, L.; Kriwacki, R.W.; Chen, J. Electrostatically Accelerated Coupled Binding and Folding of Intrinsically Disordered Proteins. J. Mol. Biol. 2012, 422, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.; Blackledge, M.; Jensen, M.R. Elucidating Binding Mechanisms and Dynamics of Intrinsically Disordered Protein Complexes Using NMR Spectroscopy. Curr. Opin. Struct. Biol. 2019, 54, 10–18. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kragelj, J.; Orand, T.; Delaforge, E.; Tengo, L.; Blackledge, M.; Palencia, A.; Jensen, M.R. Enthalpy–Entropy Compensation in the Promiscuous Interaction of an Intrinsically Disordered Protein with Homologous Protein Partners. Biomolecules 2021, 11, 1204. https://doi.org/10.3390/biom11081204
Kragelj J, Orand T, Delaforge E, Tengo L, Blackledge M, Palencia A, Jensen MR. Enthalpy–Entropy Compensation in the Promiscuous Interaction of an Intrinsically Disordered Protein with Homologous Protein Partners. Biomolecules. 2021; 11(8):1204. https://doi.org/10.3390/biom11081204
Chicago/Turabian StyleKragelj, Jaka, Thibault Orand, Elise Delaforge, Laura Tengo, Martin Blackledge, Andrés Palencia, and Malene Ringkjøbing Jensen. 2021. "Enthalpy–Entropy Compensation in the Promiscuous Interaction of an Intrinsically Disordered Protein with Homologous Protein Partners" Biomolecules 11, no. 8: 1204. https://doi.org/10.3390/biom11081204
APA StyleKragelj, J., Orand, T., Delaforge, E., Tengo, L., Blackledge, M., Palencia, A., & Jensen, M. R. (2021). Enthalpy–Entropy Compensation in the Promiscuous Interaction of an Intrinsically Disordered Protein with Homologous Protein Partners. Biomolecules, 11(8), 1204. https://doi.org/10.3390/biom11081204