Nuclear Ubiquitin-Proteasome Pathways in Proteostasis Maintenance

Abstract
1. Introduction
1.1. Ubiquitin-Proteasome System
1.2. The Nucleus and the Nuclear Envelope at a Glance
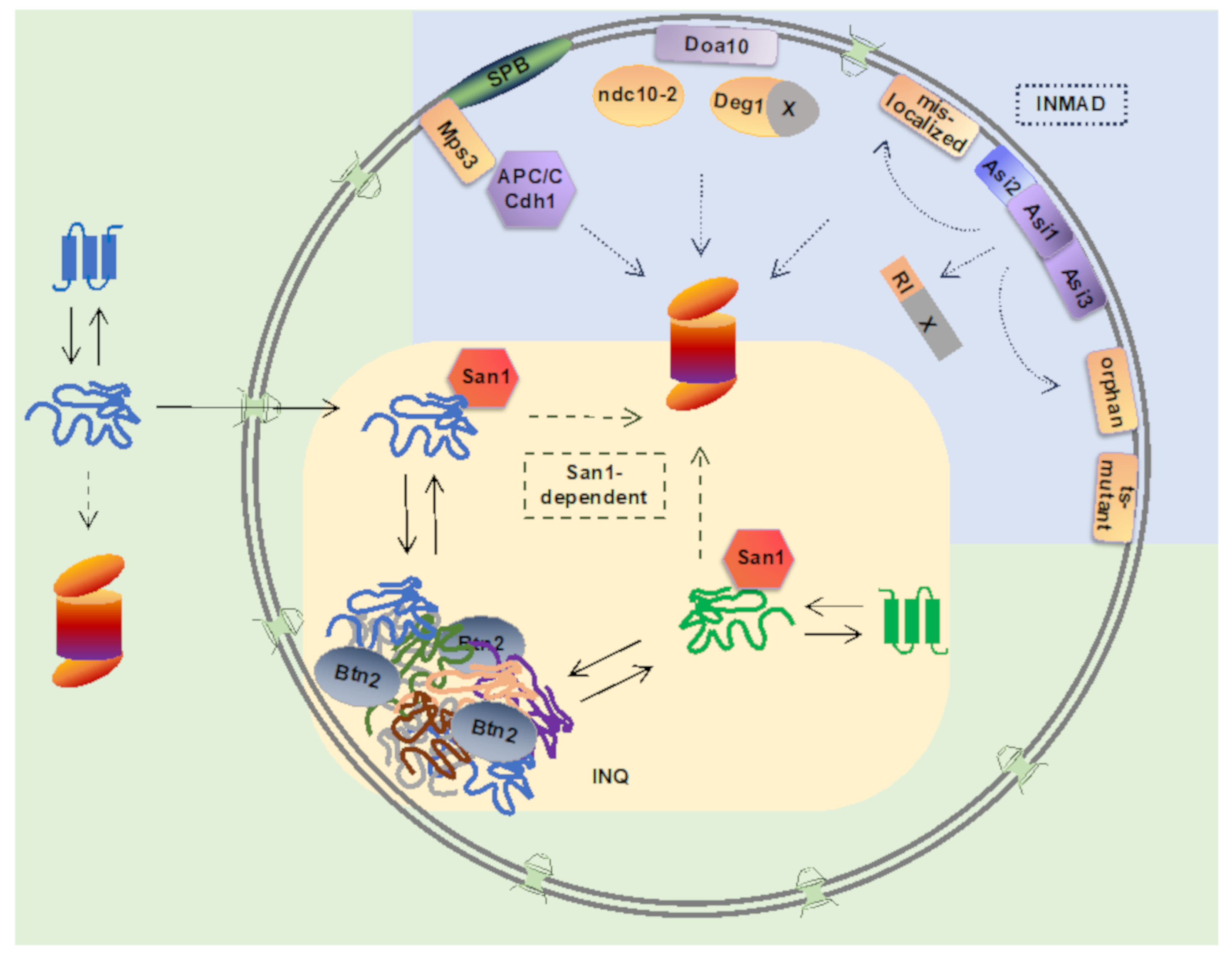
2. Inner Nuclear Membrane-Associated Degradation (INMAD)
2.1. Asi1-3 Complex—An Integral Membrane E3 Ubiquitin Ligase at the INM
2.2. Degradation of Nuclear Proteins by the Integral Membrane E3 Ligase Doa10
2.3. Regulation of INM SUN-Domain Protein Mps3 Levels via E3 Ligase APC/C-Dependent Pathway
3. Nuclear Pathways for Managing Misfolded Proteins
3.1. Proteasomal Degradation of Misfolded Proteins via Nuclear San1-Dependent Ubiquitination Pathway
3.2. Managing Ubiquitin-Proteasome System Overload by Sequestration of Misfolded Proteins into Nuclear Inclusions
4. SUMO-Targeted Ubiquitin Ligases (STUbLs) in Nuclear Protein Quality Control
5. Concluding Remarks

{kind=link}
| E3 Ubiquitin Ligase | E2 Involved | Nuclear PQC Substrates | Degradation Signal | ||
|---|---|---|---|---|---|
| INMAD | |||||
| Asi1-Asi3 | Ubc6 and Ubc7 [31] Ubc4 and Ubc7 [30,34] | nuclear proteins: | Stp1 and Stp2 [31,37] | nuclear RI region (amphipathic helix) [31,37] | |
| integral INM 1 proteins: | INM-mislocalized [31,40] | transmembrane domains [34] | |||
| orphan subunits and lone proteins [34] | |||||
| ts2 mutants [34] | |||||
| other [30,31,40] | |||||
| Doa10 | Ubc6 and Ubc7 [41] | nuclear proteins: | Matalpha2 [41] | amphipathic helix [50] | |
| Ndc10-2 (ts) [43] | amphipathic helix and hydrophobic region [53] | ||||
| integral INM 1 proteins: | Asi2 [47] | not determined | |||
| Mps2-1 (ts) [43] | not determined | ||||
| APC/C Cdh1 | Ubc7 [58] | integral INM 1 protein: | Mps3 [58] | nucleoplasmic KEN-box and D-box-like sequences [58] | |
| Nuclear | |||||
| San1 | Ubc1 or Ubc3/ Cdc34 [62,63] | nuclear proteins | ts 2 mutants of nuclear proteins [62] | exposed hydrophobicity [66] | |
| truncated proteins [66,70,76] | |||||
| artificial substrates and other mutants [66,67,70,71,72,73,75,76,77] | |||||
Author Contributions
Funding
Conflicts of Interest
References
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, J.S.; Lindquist, S. Mechanisms of protein-folding diseases at a glance. Dis. Model. Mech. 2014, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792806. [Google Scholar] [CrossRef] [PubMed]
- Buchberger, A.; Bukau, B.; Sommer, T. Protein Quality Control in the Cytosol and the Endoplasmic Reticulum: Brothers in Arms. Mol. Cell 2010, 40, 238–252. [Google Scholar] [CrossRef]
- Nielsen, S.V.; Poulsen, E.G.; Rebula, C.A.; Hartmann-Petersen, R. Protein quality control in the nucleus. Biomolecules 2014, 4, 646–661. [Google Scholar] [CrossRef]
- Lafarga, M.; Fernández, R.; Mayo, I.; Berciano, M.T.; Castaño, J.G. Proteasome dynamics during cell cycle in rat Schwann cells. Glia 2002, 38, 313–328. [Google Scholar] [CrossRef]
- Rivett, A.J.; Palmer, A.; Knecht, E. Electron Microscopic Localization of the Multicatalytic Proteinase Complex in Rat Liver and in Cultured Cells. J. Histochem. Cytochem. 1992, 40, 1165–1172. [Google Scholar] [CrossRef]
- Reits, E.A.J.; Benham, A.M.; Plougastel, B.; Neefjes, J.; Trowsdale, J. Dynamics of proteasome distribution in living cells. EMBO J. 1997, 16, 6087–6094. [Google Scholar] [CrossRef]
- Ádori, C.; Low, P.; Moszkovkin, G.; Bagdy, G.; László, L.; Kovács, G.G. Subcellular distribution of components of the ubiquitin-proteasome system in non-diseased human and rat brain. J. Histochem. Cytochem. 2006, 54, 263–267. [Google Scholar] [CrossRef]
- Russell, S.J.; Steger, K.A.; Johnston, S.A. Subcellular localization, stoichiometry, and protein levels of 26 S proteasome subunits in yeast. J. Biol. Chem. 1999, 274, 21943–21952. [Google Scholar] [CrossRef]
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Best Pr. Res. Clin. Haematol. 2017, 30, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Finley, D.; Chen, X.; Walters, K.J. Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem. Sci. 2016, 41, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Boban, M.; Foisner, R. Degradation-mediated protein quality control at the inner nuclear membrane. Nucleus 2016, 7, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. Proteolysis: From the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 2005, 6, 79–86. [Google Scholar] [CrossRef]
- Varshavsky, A. Letter to the Editor Naming a Targeting Signal. Cell 1991, 64, 13–15. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Rapoport, T.; Bodnar, N. Toward an understanding of the Cdc48/p97 ATPase. F1000Research 2017, 6, 1–10. [Google Scholar] [CrossRef]
- Hänzelmann, P.; Schindelin, H. The interplay of cofactor interactions and post-translational modifications in the regulation of the AAA+ ATPase p97. Front. Mol. Biosci. 2017, 4, 21. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Ohtake, F.; Arai, N.; Kaiho, A.; Yasuda, S.; Tanaka, K.; Saeki, Y. In Vivo Ubiquitin Linkage-type Analysis Reveals that the Cdc48-Rad23/Dsk2 Axis Contributes to K48-Linked Chain Specificity of the Proteasome. Mol. Cell 2017, 66, 488–502.e7. [Google Scholar] [CrossRef]
- Su, V.; Lau, A.F. Ubiquitin-like and ubiquitin-associated domain proteins: Significance in proteasomal degradation. Cell. Mol. Life Sci. 2009, 66, 2819–2833. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Clague, M.J.; Urbé, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Suresh, H.G.; Pascoe, N.; Andrews, B. The structure and function of deubiquitinases: Lessons from budding yeast. Open Biol. 2020, 10, 200279. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ng, D.T.W.; Cheong, I.; Matsudaira, P. The degradation-promoting roles of deubiquitinases Ubp6 and Ubp3 in cytosolic and ER protein quality control. PLoS ONE 2020, 15, e0232755. [Google Scholar] [CrossRef]
- Wilson, K.L.; Berk, J.M. The nuclear envelope at a glance. J. Cell Sci. 2010, 123, 1973–1978. [Google Scholar] [CrossRef]
- Schirmer, E.C.; Foisner, R. Proteins that associate with lamins: Many faces, many functions. Exp. Cell Res. 2007, 313, 2167–2179. [Google Scholar] [CrossRef]
- Gruenbaum, Y.; Foisner, R. Lamins: Nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 2015, 84, 131–164. [Google Scholar] [CrossRef]
- Katta, S.S.; Smoyer, C.J.; Jaspersen, S.L. Destination: Inner nuclear membrane. Trends Cell Biol. 2014, 24, 221–229. [Google Scholar] [CrossRef]
- King, M.C.; Lusk, C.P.; Blobel, G. Karyopherin-mediated import of integral inner nuclear membrane proteins. Nature 2006, 442, 1003–1007. [Google Scholar] [CrossRef]
- Foresti, O.; Rodriguez-Vaello, V.; Funaya, C.; Carvalho, P. Quality control of inner nuclear membrane proteins by the Asi complex. Science 2014, 346, 751–755. [Google Scholar] [CrossRef]
- Khmelinskii, A.; Blaszczak, E.; Pantazopoulou, M.; Fischer, B.; Omnus, D.J.; le Dez, G.; Brossard, A.; Gunnarsson, A.; Barry, J.D.; Meurer, M.; et al. Protein quality control at the inner nuclear membrane. Nature 2014, 516, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Boban, M.; Zargari, A.; Andreasson, C.; Heessen, S.; Thyberg, J.; Ljungdahl, P.O. Asi1 is an inner nuclear membrane protein that restricts promoter access of two latent transcription factors. J. Cell Biol. 2006, 173, 695–707. [Google Scholar] [CrossRef] [PubMed]
- Zargari, A.; Boban, M.; Heessen, S.; Andreasson, C.; Thyberg, J.; Ljungdahl, P.O. Inner nuclear membrane proteins Asi1, Asi2, and Asi3 function in concert to maintain the latent properties of transcription factors Stp1 and Stp2. J. Biol. Chem. 2007, 282, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, N.; Foresti, O.; Wendrich, K.; Stein, A.; Carvalho, P. Quality Control of Protein Complex Assembly by a Transmembrane Recognition Factor. Mol. Cell 2020, 77, 108–119.e9. [Google Scholar] [CrossRef]
- Biederer, T.; Volkwein, C.; Sommer, T. Role of Cue1p in Ubiquitination and Degradation at the ER Surface. Science 1997, 278, 1806–1809. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Cohen, I.; Popp, O.; Dittmar, G.; Reiss, Y.; Sommer, T.; Ravid, T.; Jarosch, E. Sequential Poly-ubiquitylation by Specialized Conjugating Enzymes Expands the Versatility of a Quality Control Ubiquitin Ligase. Mol. Cell 2016, 63, 827–839. [Google Scholar] [CrossRef]
- Omnus, D.J.; Ljungdahl, P.O. Latency of transcription factor Stp1 depends on a modular regulatory motif that functions as cytoplasmic retention determinant and nuclear degron. Mol. Biol. Cell 2014, 25, 3823–3833. [Google Scholar] [CrossRef]
- Andréasson, C.; Ljungdahl, P.O. Receptor-mediated endoproteolytic activation of two transcription factors in yeast. Genes Dev. 2002, 16, 3158–3172. [Google Scholar] [CrossRef]
- Andréasson, C.; Ljungdahl, P.O. The N-Terminal Regulatory Domain of Stp1p Is Modular and, Fused to an Artificial Transcription Factor, Confers Full Ssy1p-Ptr3p-Ssy5p Sensor Control. Mol. Cell. Biol. 2004, 24, 7503–7513. [Google Scholar] [CrossRef][Green Version]
- Smoyer, C.J.; Smith, S.E.; Gardner, J.M.; McCroskey, S.; Unruh, J.R.; Jaspersen, S.L. Distribution of proteins at the inner nuclear membrane is regulated by the asi1 E3 ligase in saccharomyces cerevisiae. Genetics 2019, 211, 1269–1282. [Google Scholar] [CrossRef]
- Swanson, R.; Locher, M.; Hochstrasser, M. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matα2 repressor degradation. Genes Dev. 2001, 15, 2660–2674. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Hochstrasser, M. Spatially regulated ubiquitin ligation by an ER/nuclear membrane ligase. Nature 2006, 443, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Ravid, T.; Kreft, S.G.; Hochstrasser, M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 2006, 25, 533–543. [Google Scholar] [CrossRef]
- Habeck, G.; Ebner, F.A.; Shimada-Kreft, H.; Kreft, S.G. The yeast ERAD-C ubiquitin ligase Doa10 recognizes an intramembrane degron. J. Cell Biol. 2015, 209, 261–273. [Google Scholar] [CrossRef]
- Mehrtash, A.B.; Hochstrasser, M. Ubiquitin-dependent protein degradation at the endoplasmic reticulum and nuclear envelope. Semin. Cell Dev. Biol. 2019, 93, 111–124. [Google Scholar] [CrossRef]
- Hassink, G.; Kikkert, M.; van Voorden, S.; Lee, S.J.; Spaapen, R.; van Laar, T.; Coleman, C.S.; Bartee, E.; Fruh, K.; Chau, V.; et al. TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem. J. 2005, 388, 647–655. [Google Scholar] [CrossRef]
- Boban, M.; Pantazopoulou, M.; Schick, A.; Ljungdahl, P.O.; Foisner, R. A nuclear ubiquitin-proteasome pathway targets the inner nuclear membrane protein Asi2 for degradation. J. Cell Sci. 2014, 127, 3603–3613. [Google Scholar] [CrossRef]
- Boban, M.; Ljungdahl, P.O.; Foisner, R. Atypical ubiquitylation in yeast targets lysine-less Asi2 for proteasomal degradation. J. Biol. Chem. 2015, 290, 2489–2495. [Google Scholar] [CrossRef]
- Pantazopoulou, M.; Boban, M.; Foisner, R.; Ljungdahl, P.O. Cdc48 and Ubx1 participate in a pathway associated with the inner nuclear membrane that governs Asi1 degradation. J. Cell Sci. 2016, 129, 3770–3780. [Google Scholar] [CrossRef]
- Johnson, P.; Swanson, R.; Rakhilina, L.; Hochstrasser, M. Degradation Signal Masking by Heterodimerization of MAT2 and MATa1 Blocks Their Mutual Destruction by the Ubiquitin-Proteasome Pathway. Cell 1998, 94, 217–227. [Google Scholar] [CrossRef]
- Gilon, T.; Chomsky, O.; Kulka, R.G. Degradation signals for ubiquitin system proteolysis in Saccharomyces cerevisiae. EMBO J. 1998, 17, 2759–2766. [Google Scholar] [CrossRef] [PubMed]
- Gilon, T.; Chomsky, O.; Kulka, R.G. Degradation Signals Recognized by the Ubc6p-Ubc7p Ubiquitin-Conjugating Enzyme Pair. Mol. Cell. Biol. 2000, 20, 7214–7219. [Google Scholar] [CrossRef] [PubMed]
- Furth, N.; Gertman, O.; Shiber, A.; Alfassy, O.S.; Cohen, I.; Rosenberg, M.M.; Doron, N.K.; Friedler, A.; Ravid, T. Exposure of bipartite hydrophobic signal triggers nuclear quality control of Ndc10 at the endoplasmic reticulum/nuclear envelope. Mol. Biol. Cell 2011, 22, 4726–4739. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Johnson, P.; Sommer, T.; Jentsch, S.; Hochstrasser, M. Multiple ubiquitin-conjugating enzymes participate in the in vivo degradation of the yeast MAT alpha 2 repressor. Cell 1993, 74, 357–369. [Google Scholar] [CrossRef]
- Conrad, M.N.; Lee, C.-Y.; Wilkerson, J.L.; Dresser, M.E. MPS3 mediates meiotic bouquet formation in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2007, 104, 8863–8868. [Google Scholar] [CrossRef]
- Friederichs, J.M.; Ghosh, S.; Smoyer, C.J.; McCroskey, S.; Miller, B.D.; Weaver, K.J.; Delventhal, K.M.; Unruh, J.; Slaughter, B.D.; Jaspersen, S.L. The SUN protein Mps3 is required for spindle pole body insertion into the nuclear membrane and nuclear envelope homeostasis. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef]
- Jaspersen, S.L.; Giddings, T.H.; Winey, M. Mps3p is a novel component of the yeast spindle pole body that interacts with the yeast centrin homologue Cdc31p. J. Cell Biol. 2002, 159, 945–956. [Google Scholar] [CrossRef]
- Koch, B.A.; Jin, H.; Tomko, R.J.; Yu, H.G. The anaphase-promoting complex regulates the degradation of the inner nuclear membrane protein Mps3. J. Cell Biol. 2019, 218, 839–854. [Google Scholar] [CrossRef]
- Pines, J. Cubism and the cell cycle: The many faces of the APC/C. Nat. Rev. Mol. Cell Biol. 2011, 12, 427–438. [Google Scholar] [CrossRef]
- Davey, N.E.; Morgan, D.O. Building a Regulatory Network with Short Linear Sequence Motifs: Lessons from the Degrons of the Anaphase-Promoting Complex. Mol. Cell 2016, 64, 12–23. [Google Scholar] [CrossRef]
- Li, P.; Jin, H.; Koch, B.A.; Abblett, R.L.; Han, X.; Yates, J.R.; Yu, H.G. Cleavage of the SUN-domain protein Mps3 at its N-terminus regulates centrosome disjunction in budding yeast meiosis. PLoS Genet. 2017, 13, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.G.; Nelson, Z.W.; Gottschling, D.E. Degradation-mediated protein quality control in the nucleus. Cell 2005, 120, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, R.; Sandoval, D.; Fredrickson, E.K.; Gardner, R.G.; Kleiger, G. The San1 ubiquitin ligase functions preferentially with ubiquitin-conjugating enzyme Ubc1 during protein quality control. J. Biol. Chem. 2016, 291, 18778–18790. [Google Scholar] [CrossRef] [PubMed]
- Enam, C.; Geffen, Y.; Ravid, T.; Gardner, R.G. Protein Quality Control Degradation in the Nucleus. Annu. Rev. Biochem. 2018, 87, 725–749. [Google Scholar] [CrossRef]
- Jones, R.D.; Gardner, R.G. Protein quality control in the nucleus. Curr. Opin. Cell Biol. 2016, 40, 81–89. [Google Scholar] [CrossRef]
- Fredrickson, E.K.; Rosenbaum, J.C.; Locke, M.N.; Milac, T.I.; Gardner, R.G. Exposed hydrophobicity is a key determinant of nuclear quality control degradation. Mol. Biol. Cell 2011, 22, 2384–2395. [Google Scholar] [CrossRef]
- Fredrickson, E.K.; Gallagher, P.S.; Candadai, S.V.C.; Gardner, R.G. Substrate recognition in nuclear protein quality control degradation is governed by exposed hydrophobicity that correlates with aggregation and insolubility. J. Biol. Chem. 2013, 288, 6130–6139. [Google Scholar] [CrossRef]
- Evans, D.R.H.; Brewster, N.K.; Xu, Q.; Rowley, A.; Altheim, B.A.; Johnston, G.C.; Singer, R.A. The Yeast Protein Complex Containing Cdc68 and Pob3 Mediates Core-Promoter Repression Through the Cdc68 N-Terminal Domain. Genetics 1998, 150, 1393–1405. [Google Scholar]
- Dasgupta, A.; Ramsey, H.L.; Smith, J.S.; Auble, D.T. Sir Antagonist 1 (San1) is a ubiquitin ligase. J. Biol. Chem. 2004, 279, 26830–26838. [Google Scholar] [CrossRef]
- Rosenbaum, J.C.; Fredrickson, E.K.; Oeser, M.L.; Garrett-Engele, C.M.; Locke, M.N.; Richardson, L.A.; Nelson, Z.W.; Hetrick, E.D.; Milac, T.I.; Gottschling, D.E.; et al. Disorder targets misorder in nuclear quality control degradation: A disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol. Cell 2011, 41, 93–106. [Google Scholar] [CrossRef]
- Dunker, A.K.; Oldfield, C.J.; Meng, J.; Romero, P.; Yang, J.Y.; Chen, J.W.; Vacic, V.; Obradovic, Z.; Uversky, V.N. The unfoldomics decade: An update on intrinsically disordered proteins. BMC Genom. 2008, 9, 1–26. [Google Scholar] [CrossRef]
- Jones, R.D.; Enam, C.; Ibarra, R.; Borror, H.R.; Mostoller, K.E.; Fredrickson, E.K.; Lin, J.B.; Chuang, E.; March, Z.; Shorter, J.; et al. The extent of Ssa1/Ssa2 Hsp70 chaperone involvement in nuclear protein quality control degradation varies with the substrate. Mol. Biol. Cell 2020, 31, 221–233. [Google Scholar] [CrossRef]
- Guerriero, C.J.; Weiberth, K.F.; Brodsky, J.L. Hsp70 targets a cytoplasmic quality control substrate to the san1p Ubiquitin ligase. J. Biol. Chem. 2013, 288, 18506–18520. [Google Scholar] [CrossRef]
- Gallagher, P.S.; Candadai, S.V.C.; Gardner, R.G. The requirement for Cdc48/p97 in nuclear protein quality control degradation depends on the substrate and correlates with substrate insolubility. J. Cell Sci. 2014, 127, 1980–1991. [Google Scholar] [CrossRef]
- Prasad, R.; Kawaguchi, S.; Ng, D.T.W. A Nucleus-based Quality Control Mechanism for Cytosolic Proteins. Mol. Biol. Cell 2010, 21, 2117–2127. [Google Scholar] [CrossRef]
- Heck, J.W.; Cheung, S.K.; Hampton, R.Y. Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc. Natl. Acad. Sci. USA 2010, 107, 1106–1111. [Google Scholar] [CrossRef]
- Samant, R.S.; Livingston, C.M.; Sontag, E.M.; Frydman, J. Distinct proteostasis circuits cooperate in nuclear and cytoplasmic protein quality control. Nature 2018, 563, 407–411. [Google Scholar] [CrossRef]
- Prasad, R.; Xu, C.; Ng, D.T.W. Hsp40/70/110 chaperones adapt nuclear protein quality control to serve cytosolic clients. J. Cell Biol. 2018, 217, 2019–2032. [Google Scholar] [CrossRef]
- Varshavsky, A. The N-end rule pathway and regulation by proteolysis. Protein Sci. 2011, 20, 1298–1345. [Google Scholar] [CrossRef]
- Amm, I.; Wolf, D.H. Molecular mass as a determinant for nuclear San1-dependent targeting of misfolded cytosolic proteins to proteasomal degradation. FEBS Lett. 2016, 590, 1765–1775. [Google Scholar] [CrossRef]
- Kriegenburg, F.; Jakopec, V.; Poulsen, E.G.; Nielsen, S.V.; Roguev, A.; Krogan, N.; Gordon, C.; Fleig, U.; Hartmann-Petersen, R. A Chaperone-Assisted Degradation Pathway Targets Kinetochore Proteins to Ensure Genome Stability. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef]
- Miller, S.B.; Ho, C.; Winkler, J.; Khokhrina, M.; Neuner, A.; Mohamed, M.Y.; Guilbride, D.L.; Richter, K.; Lisby, M.; Schiebel, E.; et al. Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition. EMBO J. 2015, 34, 778–797. [Google Scholar] [CrossRef]
- Miller, S.B.M.; Mogk, A.; Bukau, B. Spatially organized aggregation of misfolded proteins as cellular stress defense strategy. J. Mol. Biol. 2015, 427, 1564–1574. [Google Scholar] [CrossRef]
- Hill, S.M.; Hanzén, S.; Nyström, T. Restricted access: Spatial sequestration of damaged proteins during stress and aging. EMBO Rep. 2017, 18, 377–391. [Google Scholar] [CrossRef]
- Malinovska, L.; Kroschwald, S.; Munder, M.C.; Richter, D.; Alberti, S. Molecular chaperones and stress-inducible protein-sorting factors coordinate the spatiotemporal distribution of protein aggregates. Mol. Biol. Cell 2012, 23, 3041–3056. [Google Scholar] [CrossRef]
- Specht, S.; Miller, S.B.M.; Mogk, A.; Bukau, B. Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae. J. Cell Biol. 2011, 195, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Grousl, T.; Shatz, O.; Jawed, A.; Ruger-Herreros, C.; Semmelink, M.; Zahn, R.; Richter, K.; Bukau, B.; Mogk, A. Cellular sequestrases maintain basal Hsp70 capacity ensuring balanced proteostasis. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Brave, F.D.; Cairo, L.V.; Jagadeesan, C.; Ruger-Herreros, C.; Mogk, A.; Bukau, B.; Jentsch, S. Chaperone-Mediated Protein Disaggregation Triggers Proteolytic Clearance of Intra-nuclear Protein Inclusions. Cell Rep. 2020, 31. [Google Scholar] [CrossRef]
- Johnston, H.E.; Samant, R.S. Alternative systems for misfolded protein clearance: Life beyond the proteasome. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Latonen, L.; Moore, H.M.; Bai, B.; Jäämaa, S.; Laiho, M. Proteasome inhibitors induce nucleolar aggregation of proteasome target proteins and polyadenylated RNA by altering ubiquitin availability. Oncogene 2011, 30, 790–805. [Google Scholar] [CrossRef] [PubMed]
- Frottin, F.; Schueder, F.; Tiwary, S.; Gupta, R.; Körner, R.; Schlichthaerle, T.; Cox, J.; Jungmann, R.; Hartl, F.U.; Hipp, M.S. The nucleolus functions as a phase-separated protein quality control compartment. Science 2019, 365, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Mediani, L.; Guillén-Boixet, J.; Alberti, S.; Carra, S. Nucleoli and Promyelocytic Leukemia Protein (PML) bodies are phase separated nuclear protein quality control compartments for misfolded proteins. Mol. Cell. Oncol. 2019, 6. [Google Scholar] [CrossRef]
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X. SUMO-Mediated Regulation of Nuclear Functions and Signaling Processes. Mol. Cell 2018, 71, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Sriramachandran, A.M.; Dohmen, R.J. SUMO-targeted ubiquitin ligases. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1843, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Ohkuni, K.; Takahashi, Y.; Fulp, A.; Lawrimore, J.; Au, W.C.; Pasupala, N.; Levy-Myers, R.; Warren, J.; Strunnikov, A.; Baker, R.E.; et al. SUMO-targeted ubiquitin ligase (STUbL) Slx5 regulates proteolysis of centromeric histone H3 variant Cse4 and prevents its mislocalization to euchromatin. Mol. Biol. Cell 2016, 27, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Schweiggert, J.; Stevermann, L.; Panigada, D.; Kammerer, D.; Liakopoulos, D. Regulation of a Spindle Positioning Factor at Kinetochores by SUMO-Targeted Ubiquitin Ligases. Dev. Cell 2016, 36, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Thu, Y.M.; van Riper, S.K.; Higgins, L.A.; Zhang, T.; Becker, J.R.; Markowski, T.W.; Nguyen, H.D.; Griffin, T.J.; Bielinsky, A.K. Slx5/Slx8 Promotes Replication Stress Tolerance by Facilitating Mitotic Progression. Cell Rep. 2016, 15, 1254–1265. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Singh, N.; Carlson, C.R.; Albuquerque, C.P.; Corbett, K.D.; Zhou, H. Recruitment of a SUMO isopeptidase to rDNA stabilizes silencing complexes by opposing SUMO targeted ubiquitin ligase activity. Genes Dev. 2017, 31, 802–815. [Google Scholar] [CrossRef]
- Höpfler, M.; Kern, M.J.; Straub, T.; Prytuliak, R.; Habermann, B.H.; Pfander, B.; Jentsch, S. Slx5/Slx8-dependent ubiquitin hotspots on chromatin contribute to stress tolerance. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Drisaldi, B.; Colnaghi, L.; Fioriti, L.; Rao, N.; Myers, C.; Snyder, A.M.; Metzger, D.J.; Tarasoff, J.; Konstantinov, E.; Fraser, P.E.; et al. SUMOylation Is an Inhibitory Constraint that Regulates the Prion-like Aggregation and Activity of CPEB3. Cell Rep. 2015, 11, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Oeser, M.L.; Amen, T.; Nadel, C.M.; Bradley, A.I.; Reed, B.J.; Jones, R.D.; Gopalan, J.; Kaganovich, D.; Gardner, R.G. Dynamic Sumoylation of a Conserved Transcription Corepressor Prevents Persistent Inclusion Formation during Hyperosmotic Stress. PLoS Genet. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Thomas, M.; Dadgar, N.; Lieberman, A.P.; Iñiguez-Lluhi, J.A. Small ubiquitin-like modifier (SUMO) modification of the androgen receptor attenuates polyglutamine-mediated aggregation. J. Biol. Chem. 2009, 284, 21296–21306. [Google Scholar] [CrossRef] [PubMed]
- Janer, A.; Werner, A.; Takahashi-Fujigasaki, J.; Daret, A.L.; Fujigasaki, H.; Takada, K.; Duyckaerts, C.; Brice, A.; Dejean, A.; Sittler, A. SUMOylation attenuates the aggregation propensity and cellular toxicity of the polyglutamine expanded ataxin-7. Hum. Mol. Genet. 2009, 19, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Fei, E.; Jia, N.; Yan, M.; Ying, Z.; Sun, Q.; Wang, H.; Zhang, T.; Ma, X.; Ding, H.; Yao, X.; et al. SUMO-1 modification increases human SOD1 stability and aggregation. Biochem. Biophys. Res. Commun. 2006, 347, 406–412. [Google Scholar] [CrossRef]
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kügler, S.; Melchior, F.; et al. Sumoylation inhibits α-synuclein aggregation and toxicity. J. Cell Biol. 2011, 194, 49–60. [Google Scholar] [CrossRef]
- Ohkuni, K.; Pasupala, N.; Peek, J.; Holloway, G.L.; Sclar, G.D.; Levy-Myers, R.; Baker, R.E.; Basrai, M.A.; Kerscher, O. SUMO-Targeted Ubiquitin Ligases (STUbLs) Reduce the Toxicity and Abnormal Transcriptional Activity Associated with a Mutant, Aggregation-Prone Fragment of Huntingtin. Front. Genet. 2018, 9. [Google Scholar] [CrossRef]
- Gärtner, A.; Muller, S. PML, SUMO, and RNF4: Guardians of nuclear protein quality. Mol. Cell 2014, 55, 1–3. [Google Scholar] [CrossRef]
- Guo, L.; Giasson, B.I.; Glavis-Bloom, A.; Brewer, M.D.; Shorter, J.; Gitler, A.D.; Yang, X. A cellular system that degrades misfolded proteins and protects against neurodegeneration. Mol. Cell 2014, 55, 15–30. [Google Scholar] [CrossRef]
- Hirayama, S.; Sugihara, M.; Morito, D.; Iemura, S.I.; Natsume, T.; Murata, S.; Nagata, K. Nuclear export of ubiquitinated proteins via the UBIN-POST system. Proc. Natl. Acad. Sci. USA 2018, 115, E4199–E4208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kong, H.; Yee, H.; Fung, J.; Fu, S.-C.; Chook, M. Nuclear export receptor CRM1 recognizes diverse conformations in nuclear export signals. eLife 2017, 6, e23961. [Google Scholar] [CrossRef]
- Matsuda, M.; Koide, T.; Yorihuzi, T.; Hosokawa, N.; Nagata, K. Molecular cloning of a novel ubiquitin-like protein, ubin, that binds to er targeting signal sequences. Biochem. Biophys. Res. Commun. 2001, 280, 535–540. [Google Scholar] [CrossRef]
- Boomsma, W.; Nielsen, S.V.; Lindorff-Larsen, K.; Hartmann-Petersen, R.; Ellgaard, L. Bioinformatics analysis identifies several intrinsically disordered human E3 ubiquitin-protein ligases. PeerJ 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- De Magistris, P.; Antonin, W. The Dynamic Nature of the Nuclear Envelope. Curr. Biol. 2018, 28, R487–R497. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franić, D.; Zubčić, K.; Boban, M. Nuclear Ubiquitin-Proteasome Pathways in Proteostasis Maintenance. Biomolecules 2021, 11, 54. https://doi.org/10.3390/biom11010054
Franić D, Zubčić K, Boban M. Nuclear Ubiquitin-Proteasome Pathways in Proteostasis Maintenance. Biomolecules. 2021; 11(1):54. https://doi.org/10.3390/biom11010054
Chicago/Turabian StyleFranić, Dina, Klara Zubčić, and Mirta Boban. 2021. "Nuclear Ubiquitin-Proteasome Pathways in Proteostasis Maintenance" Biomolecules 11, no. 1: 54. https://doi.org/10.3390/biom11010054
APA StyleFranić, D., Zubčić, K., & Boban, M. (2021). Nuclear Ubiquitin-Proteasome Pathways in Proteostasis Maintenance. Biomolecules, 11(1), 54. https://doi.org/10.3390/biom11010054