Identification of Small Molecules Blocking the Pseudomonas aeruginosa Type III Secretion System Protein PcrV


Abstract
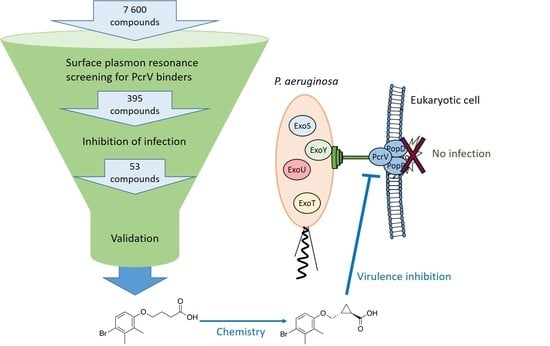
1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Surface Plasmon Resonance
2.3. 1H NMR Binding Experiment
2.4. Cell Viability Assay
2.5. Bacterial Toxicity Assay
3. Results and Discussion
3.1. Screening and Hit Validation
3.2. Design, Synthesis and Structure–Activity Relationships
3.3. Anti-Virulence Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Neill, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations 2014. Available online: https://amr-review.org/sites/default/files/AMR%20Review%20Paper%20-%20Tackling%20a%20crisis%20for%20the%20health%20and%20wealth%20of%20nations_1.pdf (accessed on 11 November 2020).
- Lister, P.D.; Wolter, D.J.; Hanson, N.D. Antibacterial-resistant Pseudomonas aeruginosa: Clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610. [Google Scholar] [CrossRef]
- Breidenstein, E.B.; de la Fuente-Nunez, C.; Hancock, R.E. Pseudomonas aeruginosa: All roads lead to resistance. Trends Microbiol. 2011, 19, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Potron, A.; Poirel, L.; Nordmann, P. Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: Mechanisms and epidemiology. Int. J. Antimicrob. Agents 2015, 45, 568–585. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Huband, M.D.; Castanheira, M.; Flamm, R.K. Pseudomonas aeruginosa antimicrobial susceptibility results from four years (2012 to 2015) of the international network for optimal resistance monitoring program in the United States. Antimicrob. Agents Chemother. 2017, 61, e02252-16. [Google Scholar] [CrossRef] [PubMed]
- Bleves, S.; Viarre, V.; Salacha, R.; Michel, G.P.; Filloux, A.; Voulhoux, R. Protein secretion systems in Pseudomonas aeruginosa: A wealth of pathogenic weapons. Int. J. Med. Microbiol. 2010, 300, 534–543. [Google Scholar] [CrossRef]
- Haiko, J.; Westerlund-Wikström, B. The role of the bacterial flagellum in adhesion and virulence. Biology 2013, 2, 1242–1267. [Google Scholar] [CrossRef]
- Leighton, T.L.; Buensuceso, R.N.; Howell, P.L.; Burrows, L.L. Biogenesis of Pseudomonas aeruginosa type IV pili and regulation of their function. Environ. Microbiol. 2015, 17, 4148–4163. [Google Scholar] [CrossRef]
- Rasmussen, T.B.; Givskov, M. Quorum-sensing inhibitors as anti-pathogenic drugs. Int. J. Med. Microbiol. 2006, 296, 149–161. [Google Scholar] [CrossRef]
- Sundin, C.; Wolfgang, M.C.; Lory, S.; Forsberg, A.; Frithz-Lindsten, E. Type IV pili are not specifically required for contact dependent translocation of exoenzymes by Pseudomonas aeruginosa. Microb. Pathog. 2002, 33, 265–277. [Google Scholar] [CrossRef]
- Shao, X.; Xie, Y.; Zhang, Y.; Liu, J.; Ding, Y.; Wu, M.; Wang, X.; Deng, X. Novel therapeutic strategies for treating Pseudomonas aeruginosa infection. Expert Opin. Drug Discov. 2020, 15, 1403–1423. [Google Scholar] [CrossRef]
- Feng, C.; Huang, Y.; He, W.; Cheng, X.; Liu, H.; Huang, Y.; Ma, B.; Zhang, W.; Liao, C.; Wu, W.; et al. Tanshinones: First-in-class inhibitors of the biogenesis of the type 3 secretion system needle of Pseudomonas aeruginosa for antibiotic therapy. ACS Cent. Sci. 2019, 5, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Roy-Burman, A.; Savel, R.H.; Racine, S.; Swanson, B.L.; Revadigar, N.S.; Fujimoto, J.; Sawa, T.; Frank, D.W.; Wiener-Kronish, J.P. Type III protein secretion is associated with death in lower respiratory and systemic Pseudomonas aeruginosa infections. J. Infect. Dis. 2001, 183, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A. The type III secretion system of Pseudomonas aeruginosa: Infection by injection. Nat. Rev. Microbiol. 2009, 7, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Sundin, C.; Thelaus, J.; Broms, J.E.; Forsberg, A. Polarisation of type III translocation by Pseudomonas aeruginosa requires PcrG, PcrV and PopN. Microb. Pathog. 2004, 37, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, G.R. The type III secretion injectisome, a complex nanomachine for intracellular ‘toxin’delivery. Biol. Chem. 2010, 391, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Portaliou, A.G.; Tsolis, K.C.; Loos, M.S.; Zorzini, V.; Economou, A. Type III Secretion: Building and operating a remarkable nanomachine. Trends Biochem. Sci. 2015, 41, 175–189. [Google Scholar] [CrossRef]
- Sawa, T.; Ito, E.; Nguyen, V.H.; Haight, M. Anti-PcrV antibody strategies against virulent Pseudomonas aeruginosa. Hum. Vaccines Immunother. 2014, 10, 2843–2852. [Google Scholar] [CrossRef]
- Caroline, G.; Eric, F.; Bohn, Y.-S.T.; Sylvie, E.; Attree, I. Oligomerization of PcrV and LcrV, protective antigens of Pseudomonas aeruginosa and Yersinia pestis. J. Biol. Chem. 2008, 283, 23940–23949. [Google Scholar] [CrossRef]
- Frithz-Lindsten, E.; Holmström, A.; Jacobsson, L.; Soltani, M.; Olsson, J.; Rosqvist, R.; Forsberg, Å. Functional conservation of the effector protein translocators PopB/YopB and PopD/YopD of Pseudomonas aeruginosa and Yersinia pseudotuberculosis. Mol. Microbiol. 1998, 29, 1155–1165. [Google Scholar] [CrossRef]
- Dacheux, D.; Goure, J.; Chabert, J.; Usson, Y.; Attree, I. Pore-forming activity of type III system-secreted proteins leads to oncosis of Pseudomonas aeruginosa-infected macrophages. Mol. Microbiol. 2001, 40, 76–85. [Google Scholar] [CrossRef]
- Allmond, L.R.; Karaca, T.J.; Nguyen, V.N.; Nguyen, T.; Wiener-Kronish, J.P.; Sawa, T. Protein binding between PcrG-PcrV and PcrH-PopB/PopD encoded by the pcrGVH-popBD operon of the Pseudomonas aeruginosa type III secretion system. Infect. Immun. 2003, 71, 2230–2233. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mueller, C.; Broz, P.; Cornelis, G. The type III secretion system tip complex and translocon. Mol. Microbiol. 2008, 68, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Schoehn, G.; Di Guilmi, A.M.; Lemaire, D.; Attree, I.; Weissenhorn, W.; Dessen, A. Oligomerization of type III secretion proteins PopB and PopD precedes pore formation in Pseudomonas. EMBO J. 2003, 22, 4957–4967. [Google Scholar] [CrossRef] [PubMed]
- Goure, J.; Broz, P.; Attree, O.; Cornelis, G.R.; Attree, I. Protective anti-V antibodies inhibit Pseudomonas and Yersinia translocon assembly within host membranes. J. Infect. Dis. 2005, 192, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Goure, J.; Pastor, A.; Faudry, E.; Chabert, J.; Dessen, A.; Attree, I. The V antigen of Pseudomonas aeruginosa is required for assembly of the functional PopB/PopD translocation pore in host cell membranes. Infect. Immun. 2004, 72, 4741–4750. [Google Scholar] [CrossRef]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef]
- Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [Google Scholar] [CrossRef]
- Marra, A. Targeting virulence for antibacterial chemotherapy: Identifying and characterising virulence factors for lead discovery. Drugs R D 2006, 7, 1–16. [Google Scholar] [CrossRef]
- Lyons, B.J.E.; Strynadka, N.C.J. On the road to structure-based development of anti-virulence therapeutics targeting the type III secretion system injectisome. Medchemcomm 2019, 10, 1273–1289. [Google Scholar] [CrossRef]
- Anantharajah, A.; Mingeot-Leclercq, M.P.; Van Bambeke, F. Targeting the type three secretion system in Pseudomonas aeruginosa. Trends Pharmacol. Sci. 2016, 37, 734–749. [Google Scholar] [CrossRef]
- Anantharajah, A.; Faure, E.; Buyck, J.M.; Sundin, C.; Lindmark, T.; Mecsas, J.; Yahr, T.L.; Tulkens, P.M.; Mingeot-Leclercq, M.P.; Guery, B.; et al. Inhibition of the injectisome and flagellar type III secretion systems by INP1855 impairs Pseudomonas aeruginosa pathogenicity and inflammasome activation. J. Infect. Dis. 2016, 214, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Uusitalo, P.; Hagglund, U.; Rhoos, E.; Scherman Norberg, H.; Elofsson, M.; Sundin, C. The salicylidene acylhydrazide INP0341 attenuates Pseudomonas aeruginosa virulence in vitro and in vivo. J. Antibiot. (Tokyo) 2017, 70, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Zetterstrom, C.E.; Hasselgren, J.; Salin, O.; Davis, R.A.; Quinn, R.J.; Sundin, C.; Elofsson, M. The resveratrol tetramer (-)-hopeaphenol inhibits type III secretion in the gram-negative pathogens Yersinia pseudotuberculosis and Pseudomonas aeruginosa. PLoS ONE 2013, 8, e81969. [Google Scholar] [CrossRef] [PubMed]
- Sundin, C.; Zetterstrom, C.E.; Vo, D.D.; Brkljaca, R.; Urban, S.; Elofsson, M. Exploring resveratrol dimers as virulence blocking agents—Attenuation of type III secretion in Yersinia pseudotuberculosis and Pseudomonas aeruginosa. Sci. Rep. 2020, 10, 2103. [Google Scholar] [CrossRef] [PubMed]
- Ngo, T.D.; Ple, S.; Thomas, A.; Barette, C.; Fortune, A.; Bouzidi, Y.; Fauvarque, M.O.; Pereira de Freitas, R.; Francisco Hilario, F.; Attree, I.; et al. Chimeric protein-protein interface inhibitors allow efficient inhibition of type III secretion machinery and Pseudomonas aeruginosa virulence. ACS Infect. Dis. 2019, 5, 1843–1854. [Google Scholar] [CrossRef]
- Aiello, D.; Williams, J.D.; Majgier-Baranowska, H.; Patel, I.; Peet, N.P.; Huang, J.; Lory, S.; Bowlin, T.L.; Moir, D.T. Discovery and characterization of inhibitors of Pseudomonas aeruginosa type III secretion. Antimicrob. Agents Chemother. 2010, 54, 1988–1999. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.P.; Lindsey, A.S.; Housley, N.A.; Ochoa, C.R.; Zhou, C.; Toba, M.; Oka, M.; Annamdevula, N.S.; Fitzgerald, M.S.; Frank, D.W. In the absence of effector proteins, the Pseudomonas aeruginosa type three secretion system needle tip complex contributes to lung injury and systemic inflammatory responses. PLoS ONE 2013, 8, e81792. [Google Scholar] [CrossRef]
- Sawa, T.; Yahr, T.L.; Ohara, M.; Kurahashi, K.; Gropper, M.A.; Wiener-Kronish, J.P.; Frank, D.W. Active and passive immunization with the Pseudomonas V antigen protects against type III intoxication and lung injury. Nat. Med. 1999, 5, 392. [Google Scholar] [CrossRef]
- Imamura, Y.; Yanagihara, K.; Fukuda, Y.; Kaneko, Y.; Seki, M.; Izumikawa, K.; Miyazaki, Y.; Hirakata, Y.; Sawa, T.; Wiener-Kronish, J. Effect of anti-PcrV antibody in a murine chronic airway Pseudomonas aeruginosa infection model. Eur. Respir. J. 2007, 29, 965–968. [Google Scholar] [CrossRef]
- Faure, K.; Fujimoto, J.; Shimabukuro, D.W.; Ajayi, T.; Shime, N.; Moriyama, K.; Spack, E.G.; Wiener-Kronish, J.P.; Sawa, T. Effects of monoclonal anti-PcrV antibody on Pseudomonas aeruginosa-induced acute lung injury in a rat model. J. Immune Based Ther. Vaccines 2003, 1, 2. [Google Scholar] [CrossRef]
- Song, Y.; Baer, M.; Srinivasan, R.; Lima, J.; Yarranton, G.; Bebbington, C.; Lynch, S. PcrV antibody–antibiotic combination improves survival in Pseudomonas aeruginosa-infected mice. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Shime, N.; Sawa, T.; Fujimoto, J.; Faure, K.; Allmond, L.R.; Karaca, T.; Swanson, B.L.; Spack, E.G.; Wiener-Kronish, J.P. Therapeutic administration of anti-PcrV F (ab′) 2 in sepsis associated with Pseudomonas aeruginosa. J. Immun. 2001, 167, 5880–5886. [Google Scholar] [CrossRef] [PubMed]
- Thanabalasuriar, A.; Surewaard, B.G.; Willson, M.E.; Neupane, A.S.; Stover, C.K.; Warrener, P.; Wilson, G.; Keller, A.E.; Sellman, B.R.; DiGiandomenico, A. Bispecific antibody targets multiple Pseudomonas aeruginosa evasion mechanisms in the lung vasculature. J. Clin. Investig. 2017, 127, 2249–2261. [Google Scholar] [CrossRef] [PubMed]
- Milla, C.E.; Chmiel, J.F.; Accurso, F.J.; VanDevanter, D.R.; Konstan, M.W.; Yarranton, G.; Geller, D.E.; Group, K.S. Anti-PcrV antibody in cystic fibrosis: A novel approach targeting Pseudomonas aeruginosa airway infection. Pediatr. Pulmonol. 2014, 49, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Hunt, M.L.; Weiner, J.J.; Hansen, A.T.; Frank, D.W. Modified needle-tip PcrV proteins reveal distinct phenotypes relevant to the control of type III secretion and intoxication by Pseudomonas aeruginosa. PLoS ONE 2011, 6, e18356. [Google Scholar] [CrossRef] [PubMed]
- McShan, A.C.; Anbanandam, A.; Patnaik, S.; De Guzman, R.N. Characterization of the binding of hydroxyindole, indoleacetic acid, and morpholinoaniline to the Salmonella type III secretion system proteins SipD and SipB. ChemMedChem 2016, 11, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Anbanandam, A.; Mumford, B.E.; De Guzman, R.N. Characterization of small molecule scaffolds that bind to the Shigella type III secretion system protein IpaD. ChemMedChem 2017, 12, 1534–1541. [Google Scholar] [CrossRef]
- Neumann, T.; Junker, H.; Schmidt, K.; Sekul, R. SPR-based fragment screening: Advantages and applications. Curr. Top. Med. Chem. 2007, 7, 1630–1642. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Olejniczak, E.T.; Fesik, S.W. One-dimensional relaxation- and diffusion-edited NMR methods for screening compounds that bind to macromolecules. J. Am. Chem. Soc. 1997, 119, 12257–12261. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2011, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Antoulinakis, E.G. Intermolecular metal-catalyzed carbenoid cyclopropanations. In Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004; pp. 1–326. [Google Scholar]
- Sirimulla, S.; Bailey, J.B.; Vegesna, R.; Narayan, M. Halogen Interactions in Protein-Ligand Complexes: Implications of halogen bonding for rational drug design. J. Chem. Inf. Model 2013, 53, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Bradberry, S.M.; Watt, B.E.; Proudfoot, A.T.; Vale, J.A. Mechanisms of toxicity, clinical features, and management of acute chlorophenoxy herbicide poisoning: A review. J. Toxicol. Clin. Toxicol. 2000, 38, 111–122. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Structure | SPR KD (µM) a | Viability Assay (% of Uninfected Control) | |||
|---|---|---|---|---|---|---|
| 200 µM | 100 µM | 50 µM | 25 µM | |||
| H1 |  | 106 ± 36 | 28% | 27% | 20% | 12% |
| 1 |  | 95 ± 41 | 16% b | 21% | 12% | 14% |
| 2 |  | 102, 127 | nd | |||
| 3 |  | >600 | nd | |||
| 4 |  | >600 | nd | |||
| 5 |  | >600 | nd | |||
| 6 |  | >600 | nd | |||
| 7 |  | >600 | nd | |||
| 8 |  | >600 | nd | |||
| 9 |  | >600 | nd | |||
| 10 |  | >600 | nd | |||
| 11 |  | >600 | nd | |||
| 12 |  | >600 | nd | |||
| 13 |  | 431 ± 110 | 20% | 21% | 18% | 19% |
| 14 |  | 148 ± 62 | 32% | 26% | 23% | 13% |
| 15 |  | 153 | 17% c | 24% c | 17% c | 12% c |
| 16 |  | >600 | nd | |||
| 17 |  | 536 | −2% b | 7% | 3% | 10% |
| 18 |  | 42 ± 12 | Toxic e | |||
| 19 |  | 121 ± 45 | Toxic e | |||
| 20 |  | 71, 131 | 9% | 15% | 15% | 15% |
| 21 |  | 67, 88 | 30% c | 36% c | 26% c | 12% c |
| 22 |  | 62, 77 | 28% c | 29% c | 27% c | 18% c |
| 23 |  | 94, 103 | 9% b | 20% | 18% | 17% |
| 24 |  | >600 | nd | |||
| 25 |  | 102, 164 | 34% | 22% | 11% | 9% |
| 26 |  | 93 ± 31 | 13% | 17% | 11% | 10% |
| 27 |  | 118 ± 62 | −6% b | 25% | 15% | 13% |
| 28 |  | 127, 351 | Toxic e | |||
| 29 |  | 110 ± 83 | 14% c | 23% c | 12% c | 8% c |
| 30 |  | 113 ± 31 | −16% b | 21% | 25% | 16% |
| 31 |  | 120, 138 | 17% | 18% | 17% | 11% |
| 32d |  | 61 ± 10 | 38% | 33% | 21% | 22% |
| 33d |  | 63 ± 4 | 36% | 38% | 24% | 24% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sundin, C.; Saleeb, M.; Spjut, S.; Qin, L.; Elofsson, M. Identification of Small Molecules Blocking the Pseudomonas aeruginosa Type III Secretion System Protein PcrV. Biomolecules 2021, 11, 55. https://doi.org/10.3390/biom11010055
Sundin C, Saleeb M, Spjut S, Qin L, Elofsson M. Identification of Small Molecules Blocking the Pseudomonas aeruginosa Type III Secretion System Protein PcrV. Biomolecules. 2021; 11(1):55. https://doi.org/10.3390/biom11010055
Chicago/Turabian StyleSundin, Charlotta, Michael Saleeb, Sara Spjut, Liena Qin, and Mikael Elofsson. 2021. "Identification of Small Molecules Blocking the Pseudomonas aeruginosa Type III Secretion System Protein PcrV" Biomolecules 11, no. 1: 55. https://doi.org/10.3390/biom11010055
APA StyleSundin, C., Saleeb, M., Spjut, S., Qin, L., & Elofsson, M. (2021). Identification of Small Molecules Blocking the Pseudomonas aeruginosa Type III Secretion System Protein PcrV. Biomolecules, 11(1), 55. https://doi.org/10.3390/biom11010055