Rescue of Hepatic Phospholipid Remodeling Defect in iPLA2β-Null Mice Attenuates Obese but Not Non-Obese Fatty Liver


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Obesity and NAFLD
2. Animal Models of Obese and Non-Obese NAFLD/NASH
3. Phospholipids in NAFLD/NASH
4. Phospholipid-Metabolizing Genes and Phospholipases A2 (PLA2) in Obesity and NAFLD
5. iPLA2β in Obesity and NAFLD and Use of iPLA2β-Null Mice
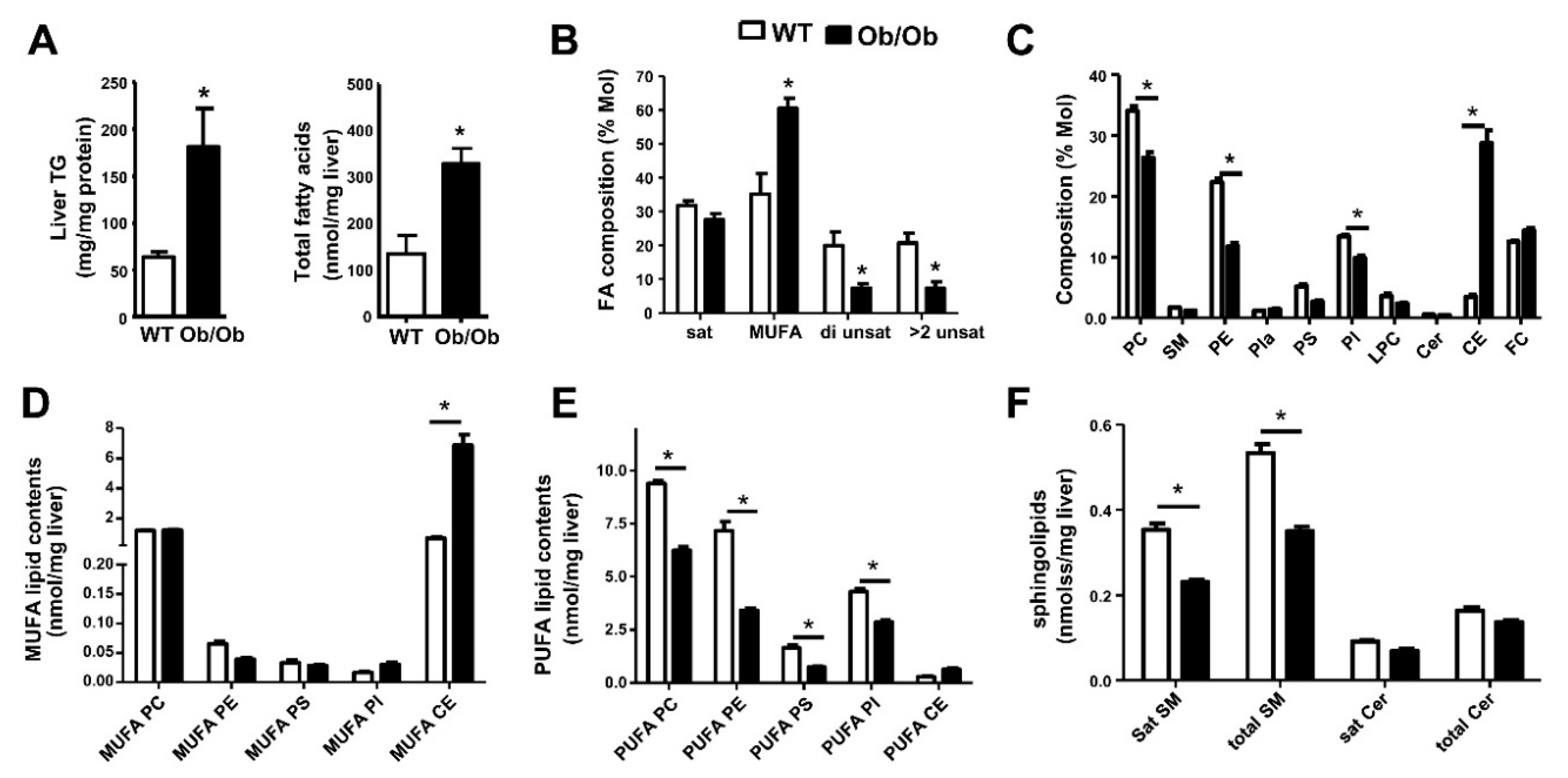
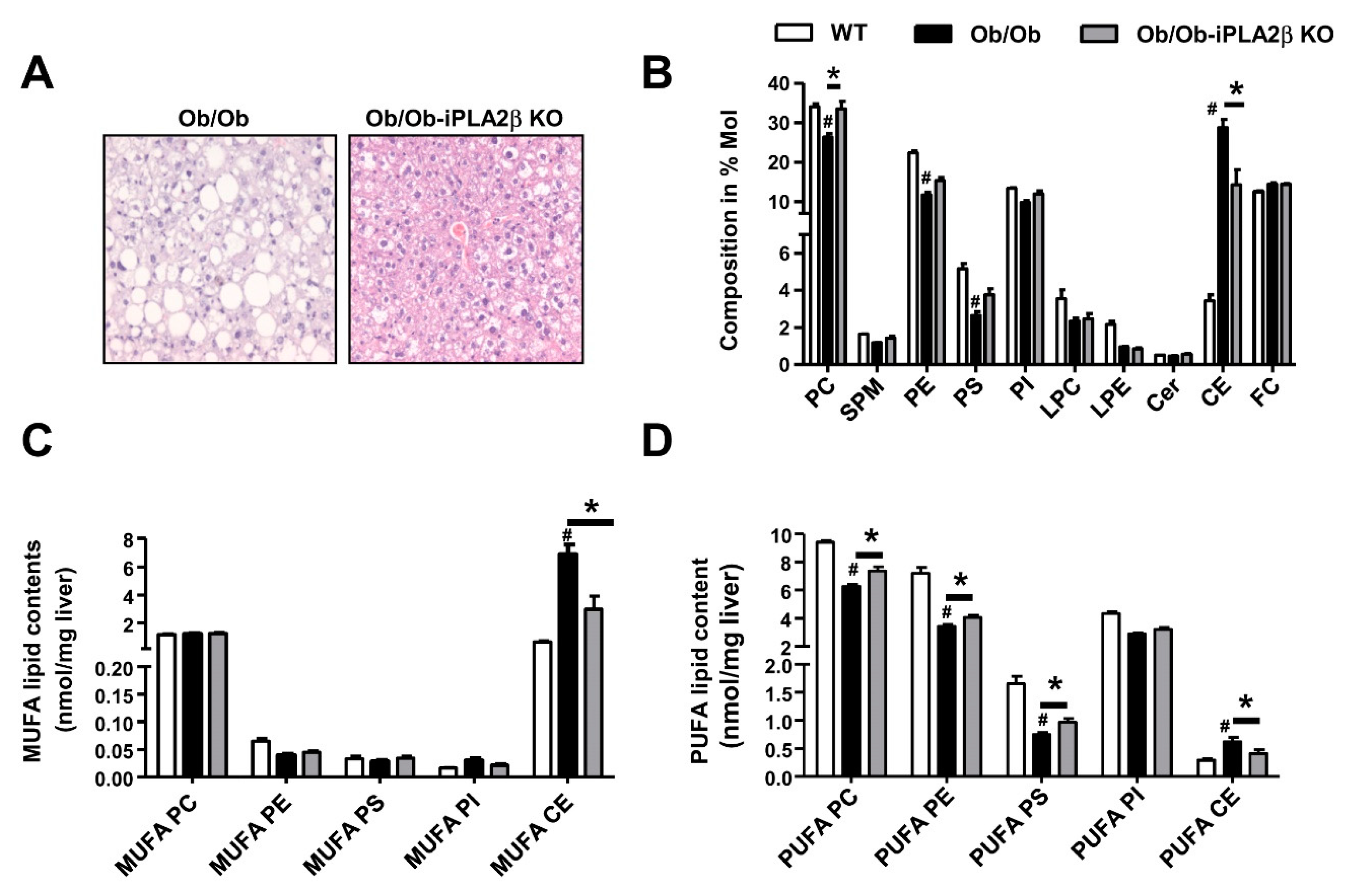
6. Metabolic Lipid Changes in Ob/Ob Mice and Modulation by iPLA2β Deficiency
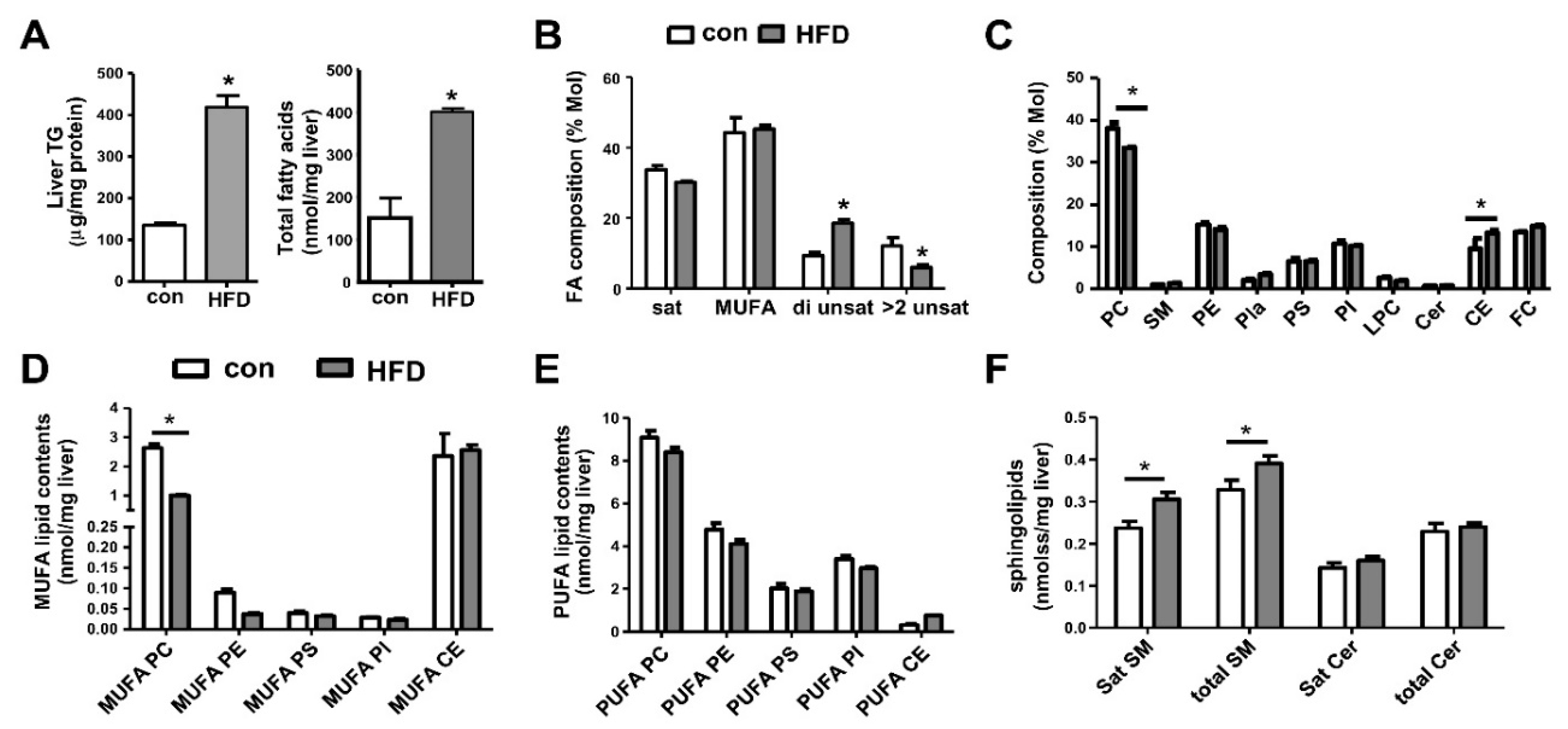
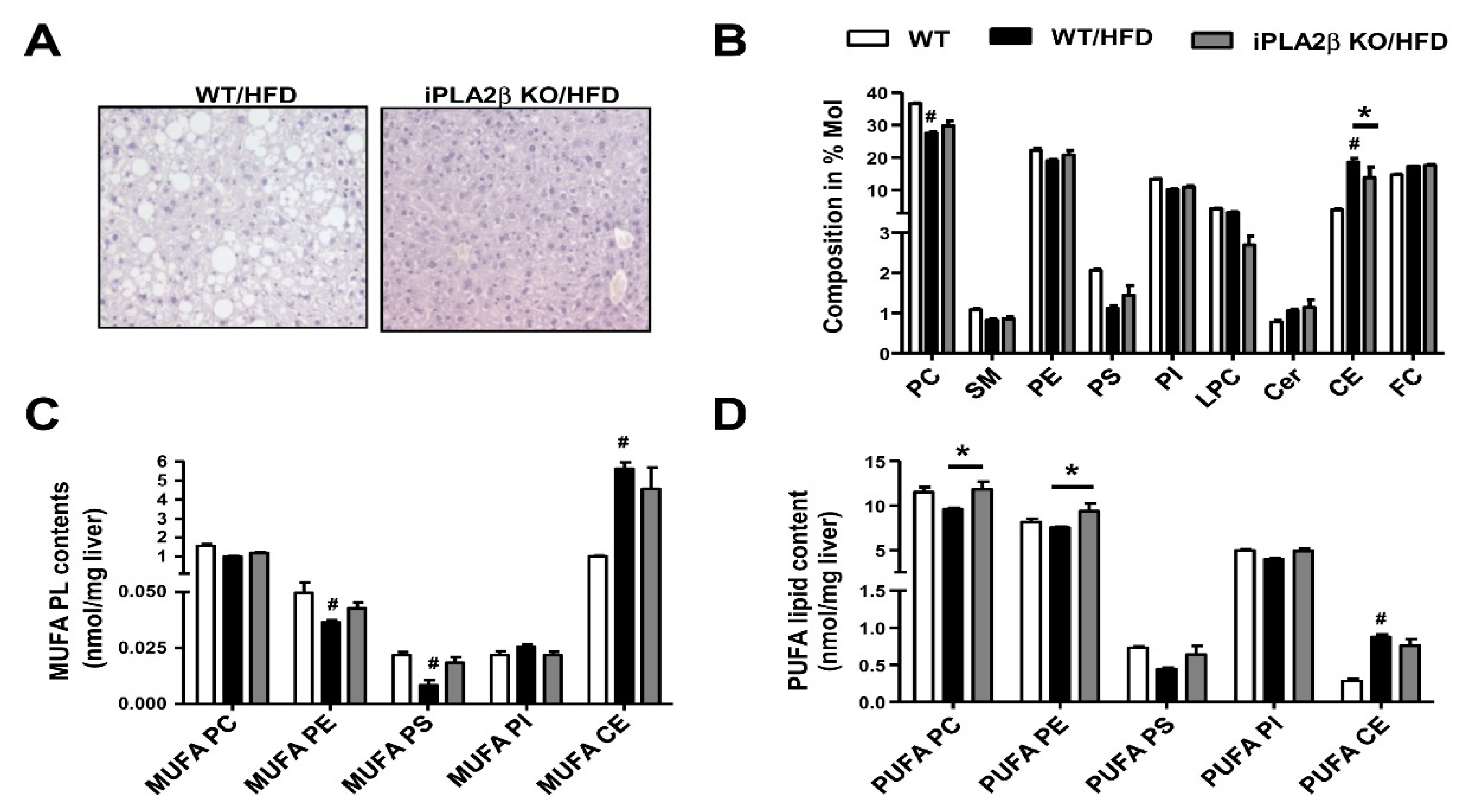
7. Metabolic Lipid Changes in HFD-Fed Mice and Modulation by iPLA2β Deficiency
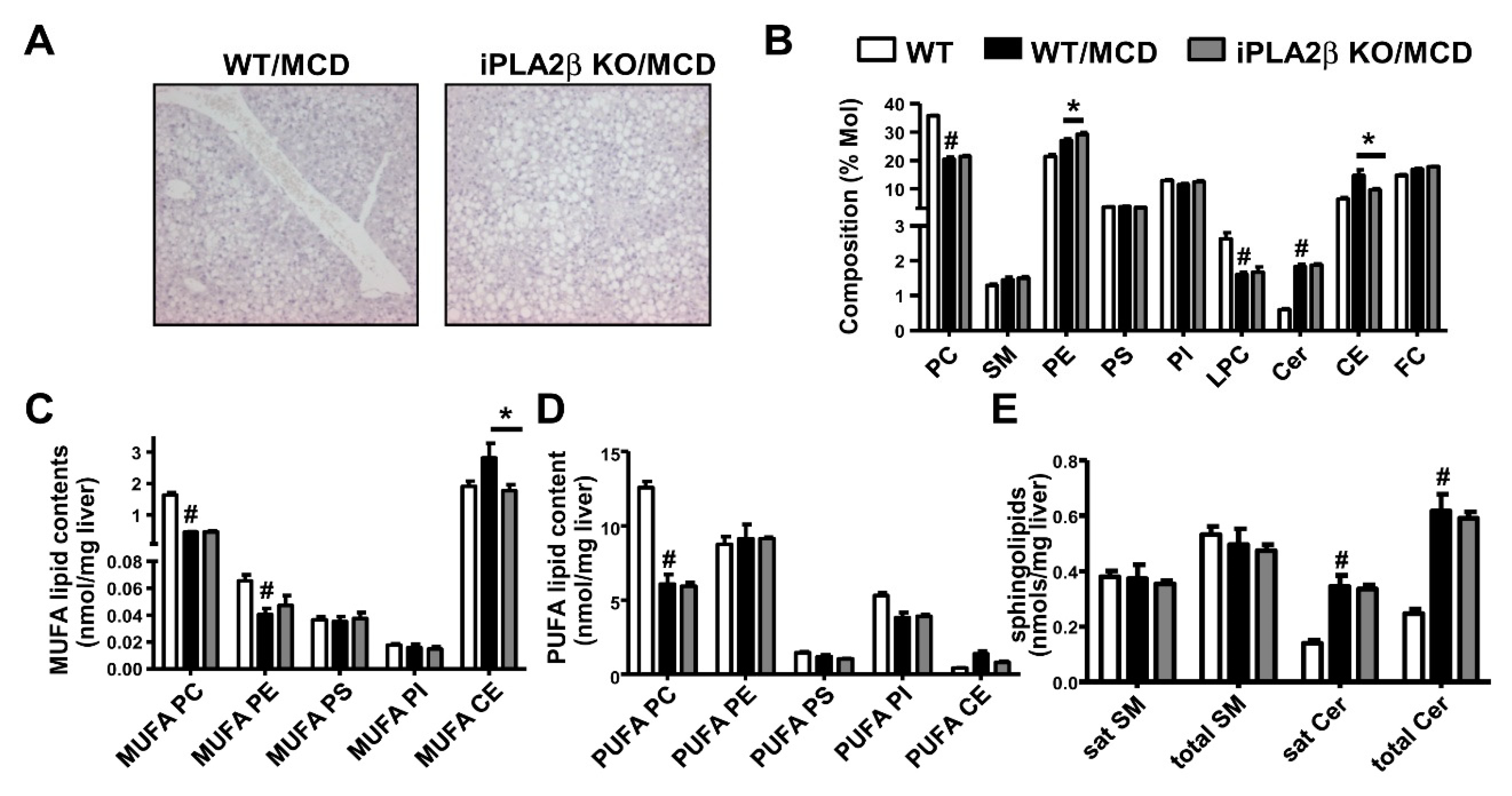
8. Metabolic Lipid Changes in MCD-Fed Mice and Modulation by iPLA2β Deficiency
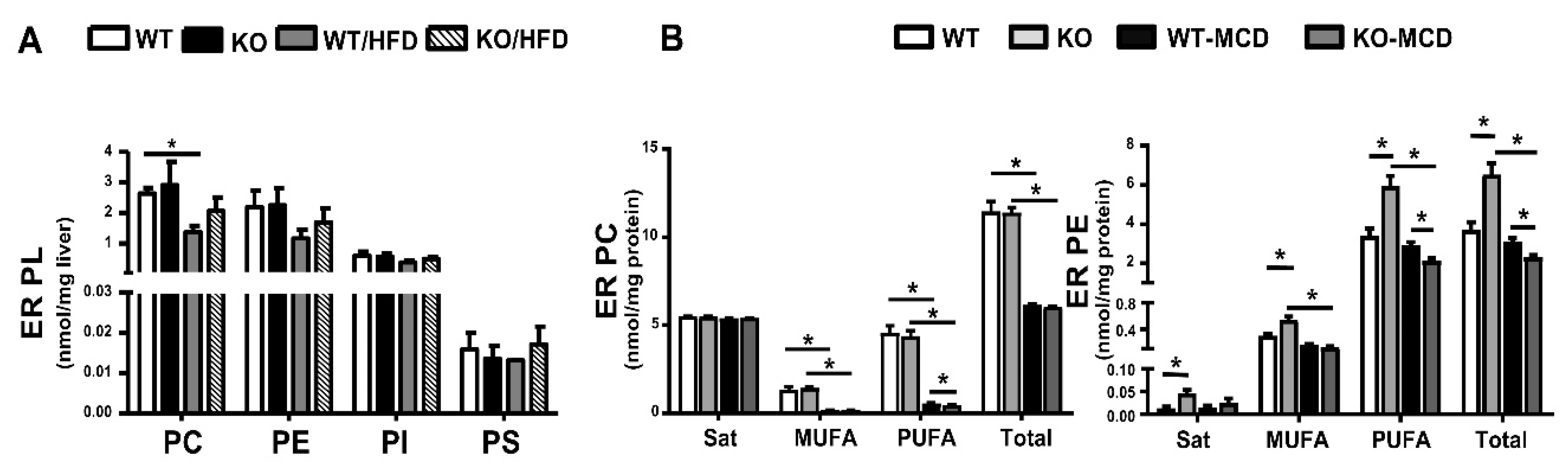
9. PL in Liver Endoplasmic Reticulum of HFD- or MCD-Fed Mice and Modulation by iPLA2β Deficiency
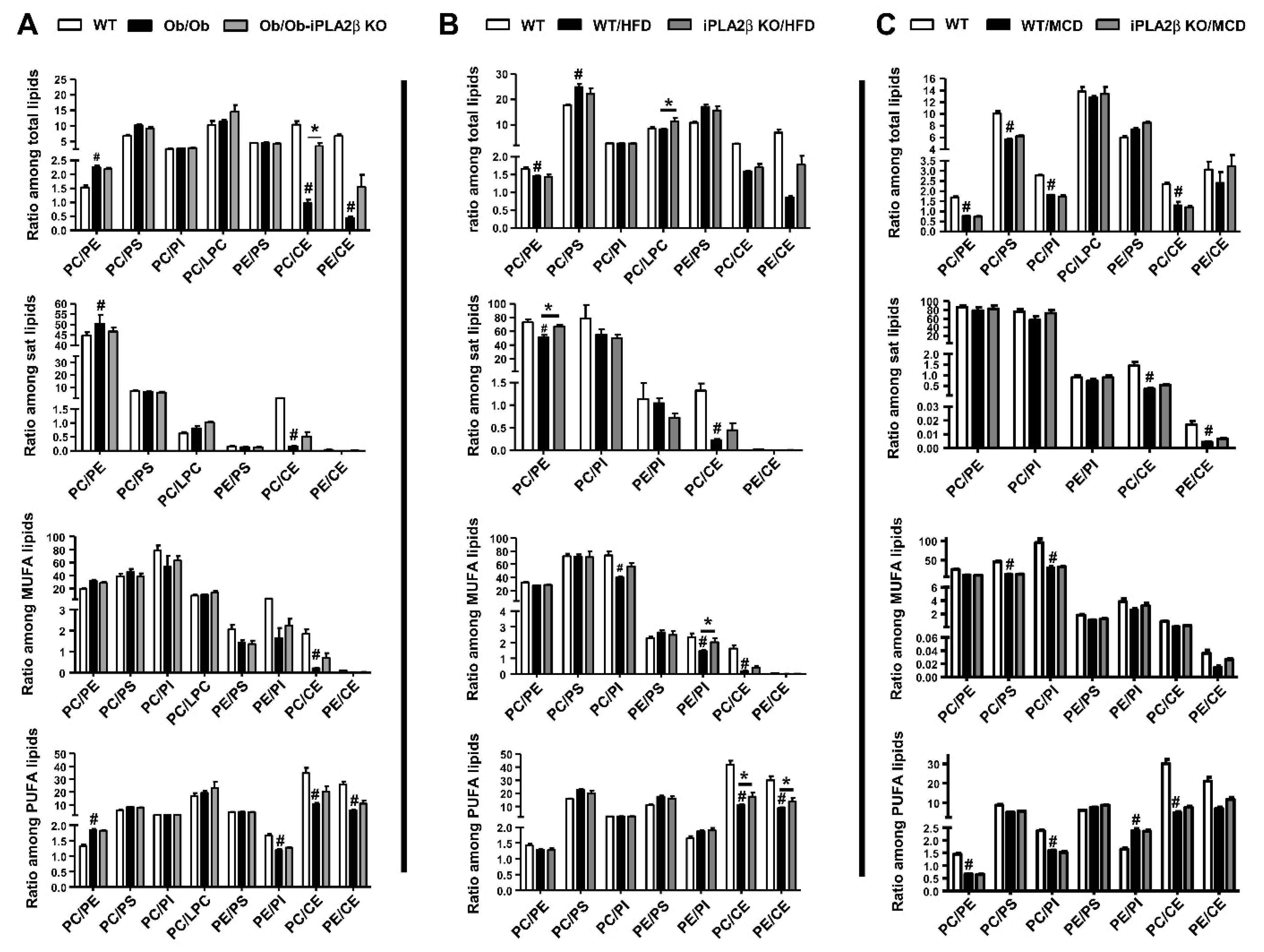
10. Hepatic PL Ratio among Obese and Non-Obese NAFLD and Modulation by iPLA2β Deficiency
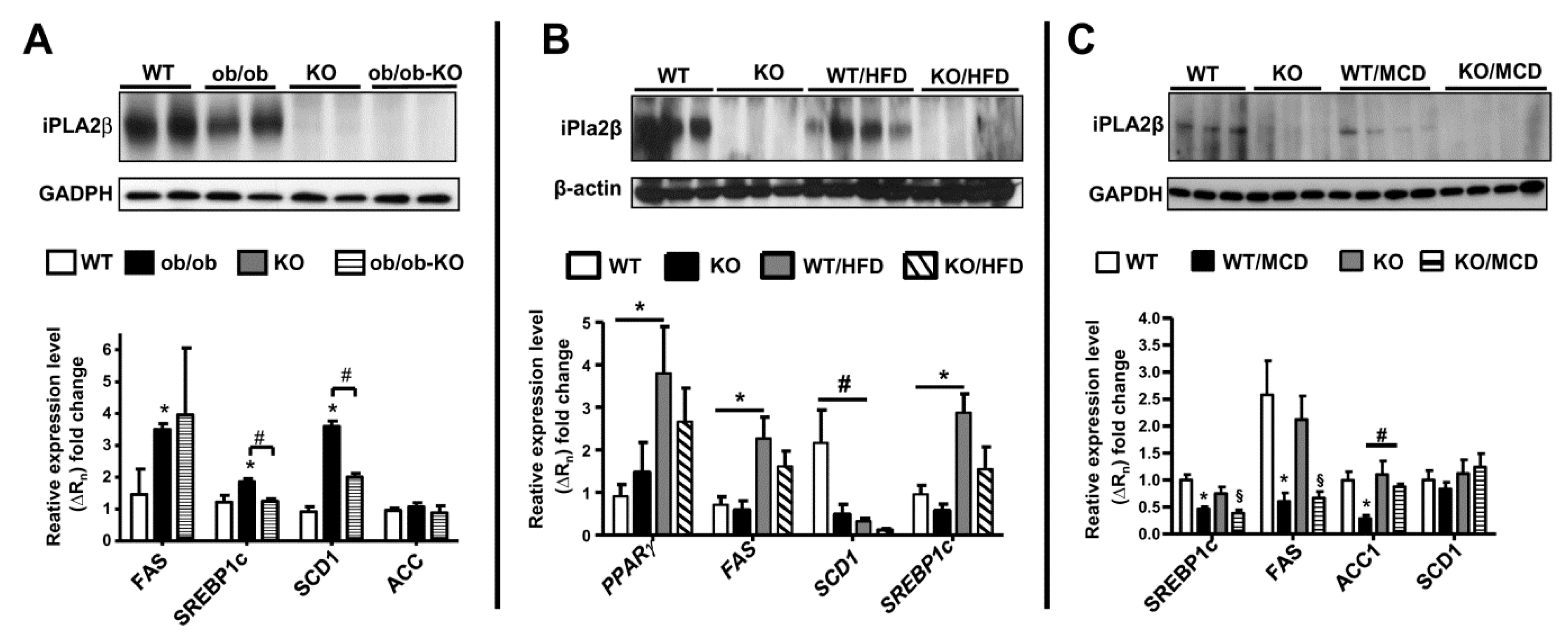
11. iPLA2β and De Novo Lipogenesis Gene Expression in Livers of Mice in 3 NAFLD Models
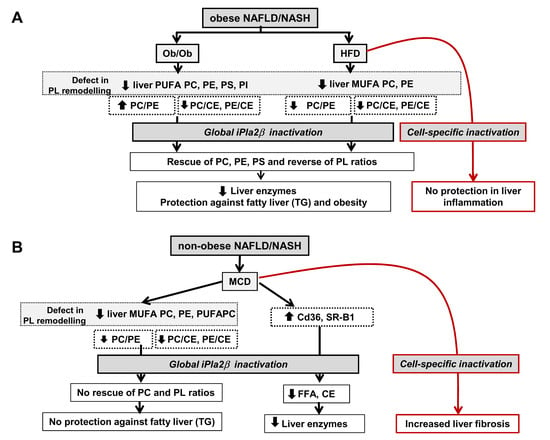
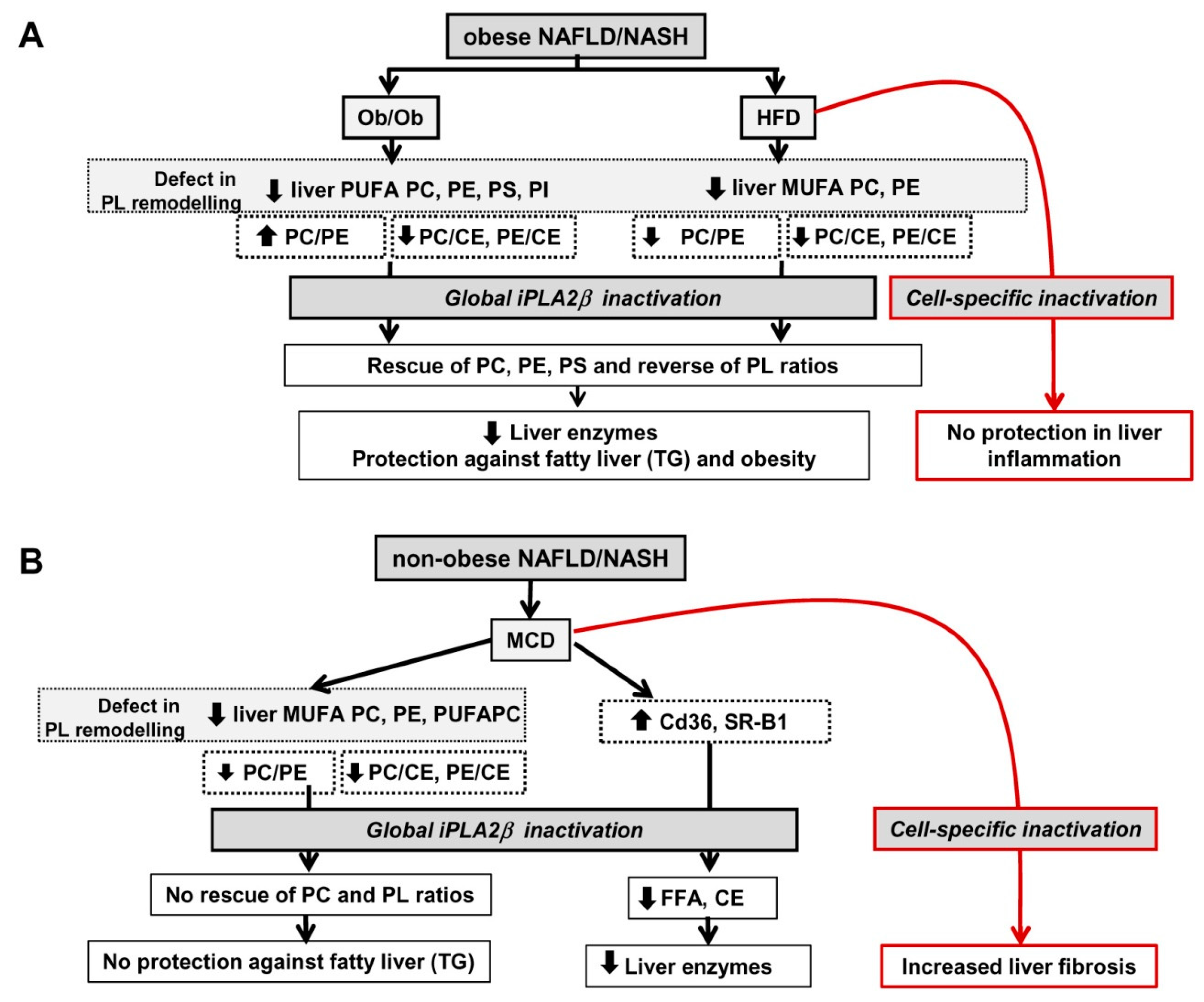
12. Summarized PL Characteristics in Ob/Ob, HFD-, and MCD-Fed Mice and Effects of iPLA2β Deficiency
13. Perspectives
13.1. Consideration of Cell-Type Specificity of iPLA2β
13.2. Use of PLs or iPLA2β Antagonists for Steatosis Protection in Obese Versus Non-Obese NAFLD
14. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BFP | body fat percentage |
| CAD | cardiovascular disease |
| CE | cholesterol esters |
| Cer | ceramides |
| CT | computed tomography |
| ER | endoplasmic reticulum |
| ESI-MS/MS | electrospray ionization tandem mass spectrometry |
| FA | fatty acid |
| FC | free cholesterol |
| FAS | fatty acid synthase |
| GC/MS | gas chromatography mass spectrometry |
| GWAS | genome-wide association studies |
| HFD | high fat diet |
| iPLA2β | group VIA calcium-independent PLA2 |
| LPC | lysophosphatidylcholine |
| LPE | lysophosphatidylethanolamine |
| MCD | methionine- and choline-deficient diet |
| MUFA | monounsaturated fatty acids |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| Ob/Ob mice | leptin-deficient mice |
| PC | phosphatidylcholine |
| PE | phosphatidylethanolamine |
| PEMT | phosphatidylethanolamine N-methyltransferase |
| PI | phosphatidylinositol |
| PL | phospholipid |
| PLA2 | phospholipase A2 |
| Pla | plasmalogens |
| PNPLA | patatin-like phospholipase containing lipase |
| PS | phosphatidylserine |
| PUFA | polyunsaturated fatty acids |
| SR-B1 | scavenger receptor B type 1 |
| SREBP | sterol regulatory element-binding protein |
| SM | sphingomyelin |
| TG | triglyceride |
| WT | wild-type |
| VLDL | very low-density lipoproteins |
References
- Lissner, L.; Visscher, T.L.; Rissanen, A.; Heitmann, B.L.; Prevention and Public Health Task Force of the European Association for the Study of Obesity. Monitoring the obesity epidemic into the 21st century—Weighing the evidence. Obes. Facts 2013, 6, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.J.; LeRoith, D. Epidemiology and molecular mechanisms tying obesity, diabetes, and the metabolic syndrome with cancer. Diabetes Care 2013, 36 (Suppl. 2), S233–S239. [Google Scholar] [CrossRef] [PubMed]
- Astrup, A.; Buemann, B.; Western, P.; Toubro, S.; Raben, A.; Christensen, N.J. Obesity as an adaptation to a high-fat diet: Evidence from a cross-sectional study. Am. J. Clin. Nutr. 1994, 59, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Clement, K.; Garner, C.; Hager, J.; Philippi, A.; LeDuc, C.; Carey, A.; Harris, T.J.; Jury, C.; Cardon, L.R.; Basdevant, A.; et al. Indication for linkage of the human OB gene region with extreme obesity. Diabetes 1996, 45, 687–690. [Google Scholar] [CrossRef] [PubMed][Green Version]
- York, B.; Truett, A.A.; Monteiro, M.P.; Barry, S.J.; Warden, C.H.; Naggert, J.K.; Maddatu, T.P.; West, D.B. Gene-environment interaction: A significant diet-dependent obesity locus demonstrated in a congenic segment on mouse chromosome 7. Mamm. Genome 1999, 10, 457–462. [Google Scholar] [CrossRef]
- Hill, J.O.; Peters, J.C. Environmental contributions to the obesity epidemic. Science 1998, 280, 1371–1374. [Google Scholar] [CrossRef] [PubMed]
- Lissner, L.; Heitmann, B.L.; Bengtsson, C. Population studies of diet and obesity. Br. J. Nutr. 2000, 83 (Suppl. 1), S21–S24. [Google Scholar] [CrossRef]
- Hasselbalch, A.L. Genetics of dietary habits and obesity—A twin study. Dan. Med. Bull. 2010, 57, B4182. [Google Scholar]
- West, D.B.; York, B. Dietary fat, genetic predisposition, and obesity: Lessons from animal models. Am. J. Clin. Nutr. 1998, 67, 505S–512S. [Google Scholar] [CrossRef]
- Collins, S.; Martin, T.L.; Surwit, R.S.; Robidoux, J. Genetic vulnerability to diet-induced obesity in the C57BL/6J mouse: Physiological and molecular characteristics. Physiol. Behav. 2004, 81, 243–248. [Google Scholar] [CrossRef]
- Alexander, J.; Chang, G.Q.; Dourmashkin, J.T.; Leibowitz, S.F. Distinct phenotypes of obesity-prone AKR/J, DBA2J and C57BL/6J mice compared to control strains. Int. J. Obes. 2006, 30, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Suzuki, M. Parental obesity and overweight affect the body-fat accumulation in the offspring: The possible effect of a high-fat diet through epigenetic inheritance. Obes. Rev. 2006, 7, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Parks, B.W.; Nam, E.; Org, E.; Kostem, E.; Norheim, F.; Hui, S.T.; Pan, C.; Civelek, M.; Rau, C.D.; Bennett, B.J.; et al. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab. 2013, 17, 141–152. [Google Scholar] [CrossRef]
- Ghosh, S.; Bouchard, C. Convergence between biological, behavioural and genetic determinants of obesity. Nat. Rev. Genet. 2017, 18, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.B. Non-alcoholic fatty liver disease: The hepatic consequence of obesity and the metabolic syndrome. Proc. Nutr. Soc. 2010, 69, 211–220. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Kim, G.A.; Lee, H.C.; Choe, J.; Kim, M.J.; Lee, M.J.; Chang, H.S.; Bae, I.Y.; Kim, H.K.; An, J.; Shim, J.H.; et al. Association between non-alcoholic fatty liver disease and cancer incidence rate. J. Hepatol. 2017. [Google Scholar] [CrossRef]
- Merriman, R.B.; Aouizerat, B.E.; Bass, N.M. Genetic influences in nonalcoholic fatty liver disease. J. Clin. Gastroenterol. 2006, 40 (Suppl. 1), S30–S33. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Daly, A.K.; Day, C.P. Genetic modifiers of non-alcoholic fatty liver disease progression. Biochim. Biophys. Acta 2011, 1812, 1557–1566. [Google Scholar] [CrossRef]
- Norheim, F.; Hui, S.T.; Kulahcioglu, E.; Mehrabian, M.; Cantor, R.M.; Pan, C.; Parks, B.W.; Lusis, A.J. Genetic and hormonal control of hepatic steatosis in female and male mice. J. Lipid Res. 2017, 58, 178–187. [Google Scholar] [CrossRef]
- Pramfalk, C.; Pavlides, M.; Banerjee, R.; McNeil, C.A.; Neubauer, S.; Karpe, F.; Hodson, L. Sex-Specific Differences in Hepatic Fat Oxidation and Synthesis May Explain the Higher Propensity for NAFLD in Men. J. Clin. Endocrinol. Metab. 2015, 100, 4425–4433. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Guo, J.; Lu, J. Risk factor compositions of nonalcoholic fatty liver disease change with body mass index in males and females. Oncotarget 2016, 7, 35632–35642. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.K.; Bahar, N.; Razlan, H.; Vijayananthan, A.; Sithaneshwar, P.; Goh, K.L. Non-alcoholic fatty liver disease in a young multiracial Asian population: A worrying ethnic predilection in Malay and Indian males. Hepatol. Int. 2014, 8, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Summart, U.; Thinkhamrop, B.; Chamadol, N.; Khuntikeo, N.; Songthamwat, M.; Kim, C.S. Gender differences in the prevalence of nonalcoholic fatty liver disease in the Northeast of Thailand: A population-based cross-sectional study. F1000Research 2017, 6, 1630. [Google Scholar] [CrossRef] [PubMed]
- Katsagoni, C.N.; Georgoulis, M.; Papatheodoridis, G.V.; Fragopoulou, E.; Ioannidou, P.; Papageorgiou, M.; Alexopoulou, A.; Papadopoulos, N.; Deutsch, M.; Kontogianni, M.D. Associations Between Lifestyle Characteristics and the Presence of Nonalcoholic Fatty Liver Disease: A Case-Control Study. Metab. Syndr. Relat. Disord. 2017, 15, 72–79. [Google Scholar] [CrossRef]
- Kumar, R.; Mohan, S. Non-alcoholic Fatty Liver Disease in Lean Subjects: Characteristics and Implications. J. Clin. Transl. Hepatol. 2017, 5, 216–223. [Google Scholar] [CrossRef]
- Yousef, M.H.; Al Juboori, A.; Albarrak, A.A.; Ibdah, J.A.; Tahan, V. Fatty liver without a large “belly”: Magnified review of non-alcoholic fatty liver disease in non-obese patients. World J. Gastrointest. Pathophysiol. 2017, 8, 100–107. [Google Scholar] [CrossRef]
- Liu, C.J. Prevalence and risk factors for non-alcoholic fatty liver disease in Asian people who are not obese. J. Gastroenterol. Hepatol. 2012, 27, 1555–1560. [Google Scholar] [CrossRef]
- Feng, R.N.; Du, S.S.; Wang, C.; Li, Y.C.; Liu, L.Y.; Guo, F.C.; Sun, C.H. Lean-non-alcoholic fatty liver disease increases risk for metabolic disorders in a normal weight Chinese population. World J. Gastroenterol. 2014, 20, 17932–17940. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Systematic review with meta-analysis: The significance of histological disease severity in lean patients with nonalcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2018, 47, 16–25. [Google Scholar] [CrossRef]
- Feldman, A.; Eder, S.K.; Felder, T.K.; Kedenko, L.; Paulweber, B.; Stadlmayr, A.; Huber-Schonauer, U.; Niederseer, D.; Stickel, F.; Auer, S.; et al. Clinical and Metabolic Characterization of Lean Caucasian Subjects With Non-alcoholic Fatty Liver. Am. J. Gastroenterol. 2017, 112, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Bril, F.; Sninsky, J.J.; Baca, A.M.; Superko, H.R.; Portillo Sanchez, P.; Biernacki, D.; Maximos, M.; Lomonaco, R.; Orsak, B.; Suman, A.; et al. Hepatic Steatosis and Insulin Resistance, But Not Steatohepatitis, Promote Atherogenic Dyslipidemia in NAFLD. J. Clin. Endocrinol. Metab. 2016, 101, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Zivkovic, A.M.; German, J.B.; Sanyal, A.J. Comparative review of diets for the metabolic syndrome: Implications for nonalcoholic fatty liver disease. Am. J. Clin. Nutr. 2007, 86, 285–300. [Google Scholar] [CrossRef]
- Veena, J.; Muragundla, A.; Sidgiddi, S.; Subramaniam, S. Non-alcoholic fatty liver disease: Need for a balanced nutritional source. Br. J. Nutr. 2014, 112, 1858–1872. [Google Scholar] [CrossRef]
- Larter, C.Z.; Yeh, M.M. Animal models of NASH: Getting both pathology and metabolic context right. J. Gastroenterol. Hepatol. 2008, 23, 1635–1648. [Google Scholar] [CrossRef]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; Almeida Pereira, T.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef]
- Nakatsuka, A.; Matsuyama, M.; Yamaguchi, S.; Katayama, A.; Eguchi, J.; Murakami, K.; Teshigawara, S.; Ogawa, D.; Wada, N.; Yasunaka, T.; et al. Insufficiency of phosphatidylethanolamine N-methyltransferase is risk for lean non-alcoholic steatohepatitis. Sci. Rep. 2016, 6, 21721. [Google Scholar] [CrossRef]
- Deng, X.; Wang, J.; Jiao, L.; Utaipan, T.; Tuma-Kellner, S.; Schmitz, G.; Liebisch, G.; Stremmel, W.; Chamulitrat, W. iPLA2beta deficiency attenuates obesity and hepatic steatosis in ob/ob mice through hepatic fatty-acyl phospholipid remodeling. Biochim. Biophys. Acta 2016, 1861, 449–461. [Google Scholar] [CrossRef]
- Otto, A.C.; Gan-Schreier, H.; Zhu, X.; Tuma-Kellner, S.; Staffer, S.; Ganzha, A.; Liebisch, G.; Chamulitrat, W. Group VIA phospholipase A2 deficiency in mice chronically fed with high-fat-diet attenuates hepatic steatosis by correcting a defect of phospholipid remodeling. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 662–676. [Google Scholar] [CrossRef]
- Zhu, X.; Gan-Schreier, H.; Otto, A.C.; Cheng, Y.; Staffer, S.; Tuma-Kellner, S.; Ganzha, A.; Liebisch, G.; Chamulitrat, W. iPla2beta deficiency in mice fed with MCD diet does not correct the defect of phospholipid remodeling but attenuates hepatocellular injury via an inhibition of lipid uptake genes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Tauchi-Sato, K.; Ozeki, S.; Houjou, T.; Taguchi, R.; Fujimoto, T. The surface of lipid droplets is a phospholipid monolayer with a unique Fatty Acid composition. J. Biol. Chem. 2002, 277, 44507–44512. [Google Scholar] [CrossRef] [PubMed]
- Krahmer, N.; Guo, Y.; Wilfling, F.; Hilger, M.; Lingrell, S.; Heger, K.; Newman, H.W.; Schmidt-Supprian, M.; Vance, D.E.; Mann, M.; et al. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholine cytidylyltransferase. Cell Metab. 2011, 14, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K.; Jacobs, R.L.; Watts, J.L.; Rottiers, V.; Jiang, K.; Finnegan, D.M.; Shioda, T.; Hansen, M.; Yang, F.; Niebergall, L.J.; et al. A conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell 2011, 147, 840–852. [Google Scholar] [CrossRef]
- Lim, H.Y.; Wang, W.; Wessells, R.J.; Ocorr, K.; Bodmer, R. Phospholipid homeostasis regulates lipid metabolism and cardiac function through SREBP signaling in Drosophila. Genes Dev. 2011, 25, 189–200. [Google Scholar] [CrossRef]
- Fullerton, M.D.; Hakimuddin, F.; Bonen, A.; Bakovic, M. The development of a metabolic disease phenotype in CTP:phosphoethanolamine cytidylyltransferase-deficient mice. J. Biol. Chem. 2009, 284, 25704–25713. [Google Scholar] [CrossRef]
- Leonardi, R.; Frank, M.W.; Jackson, P.D.; Rock, C.O.; Jackowski, S. Elimination of the CDP-ethanolamine pathway disrupts hepatic lipid homeostasis. J. Biol. Chem. 2009, 284, 27077–27089. [Google Scholar] [CrossRef]
- Higgins, J.A. Evidence that during very low density lipoprotein assembly in rat hepatocytes most of the triacylglycerol and phospholipid are packaged with apolipoprotein B in the Golgi complex. FEBS Lett. 1988, 232, 405–408. [Google Scholar] [CrossRef]
- Testerink, N.; van der Sanden, M.H.; Houweling, M.; Helms, J.B.; Vaandrager, A.B. Depletion of phosphatidylcholine affects endoplasmic reticulum morphology and protein traffic at the Golgi complex. J. Lipid Res. 2009, 50, 2182–2192. [Google Scholar] [CrossRef]
- Gaspar, M.L.; Jesch, S.A.; Viswanatha, R.; Antosh, A.L.; Brown, W.J.; Kohlwein, S.D.; Henry, S.A. A block in endoplasmic reticulum-to-Golgi trafficking inhibits phospholipid synthesis and induces neutral lipid accumulation. J. Biol. Chem. 2008, 283, 25735–25751. [Google Scholar] [CrossRef]
- Tran, K.; Sun, F.; Cui, Z.; Thorne-Tjomsland, G.; St Germain, C.; Lapierre, L.R.; McLeod, R.S.; Jamieson, J.C.; Yao, Z. Attenuated secretion of very low density lipoproteins from McA-RH7777 cells treated with eicosapentaenoic acid is associated with impaired utilization of triacylglycerol synthesized via phospholipid remodeling. Biochim. Biophys. Acta 2006, 1761, 463–473. [Google Scholar] [CrossRef]
- Li, Z.; Agellon, L.B.; Allen, T.M.; Umeda, M.; Jewell, L.; Mason, A.; Vance, D.E. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006, 3, 321–331. [Google Scholar] [CrossRef]
- Niebergall, L.J.; Jacobs, R.L.; Chaba, T.; Vance, D.E. Phosphatidylcholine protects against steatosis in mice but not non-alcoholic steatohepatitis. Biochim. Biophys. Acta 2011, 1811, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Mallampalli, R.K.; Ryan, A.J.; Salome, R.G.; Jackowski, S. Tumor necrosis factor-alpha inhibits expression of CTP:phosphocholine cytidylyltransferase. J. Biol. Chem. 2000, 275, 9699–9708. [Google Scholar] [CrossRef] [PubMed]
- Kienesberger, P.C.; Oberer, M.; Lass, A.; Zechner, R. Mammalian patatin domain containing proteins: A family with diverse lipolytic activities involved in multiple biological functions. J. Lipid Res. 2009, 50, S63–S68. [Google Scholar] [CrossRef]
- Ii, H.; Yokoyama, N.; Yoshida, S.; Tsutsumi, K.; Hatakeyama, S.; Sato, T.; Ishihara, K.; Akiba, S. Alleviation of high-fat diet-induced fatty liver damage in group IVA phospholipase A2-knockout mice. PLoS ONE 2009, 4, e8089. [Google Scholar] [CrossRef]
- Song, H.; Wohltmann, M.; Bao, S.; Ladenson, J.H.; Semenkovich, C.F.; Turk, J. Mice deficient in group VIB phospholipase A2 (iPLA2gamma) exhibit relative resistance to obesity and metabolic abnormalities induced by a Western diet. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1097–E1114. [Google Scholar] [CrossRef]
- Mancuso, D.J.; Sims, H.F.; Yang, K.; Kiebish, M.A.; Su, X.; Jenkins, C.M.; Guan, S.; Moon, S.H.; Pietka, T.; Nassir, F.; et al. Genetic ablation of calcium-independent phospholipase A2gamma prevents obesity and insulin resistance during high fat feeding by mitochondrial uncoupling and increased adipocyte fatty acid oxidation. J. Biol. Chem. 2010, 285, 36495–36510. [Google Scholar] [CrossRef]
- Pena, L.; Meana, C.; Astudillo, A.M.; Lorden, G.; Valdearcos, M.; Sato, H.; Murakami, M.; Balsinde, J.; Balboa, M.A. Critical role for cytosolic group IVA phospholipase A2 in early adipocyte differentiation and obesity. Biochim. Biophys. Acta 2016, 1861, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Hadad, N.; Burgazliev, O.; Elgazar-Carmon, V.; Solomonov, Y.; Wueest, S.; Item, F.; Konrad, D.; Rudich, A.; Levy, R. Induction of cytosolic phospholipase a2alpha is required for adipose neutrophil infiltration and hepatic insulin resistance early in the course of high-fat feeding. Diabetes 2013, 62, 3053–3063. [Google Scholar] [CrossRef] [PubMed]
- Kuefner, M.S.; Pham, K.; Redd, J.R.; Stephenson, E.J.; Harvey, I.; Deng, X.; Bridges, D.; Boilard, E.; Elam, M.B.; Park, E.A. Secretory phospholipase A2 group IIA modulates insulin sensitivity and metabolism. J. Lipid Res. 2017, 58, 1822–1833. [Google Scholar] [CrossRef]
- Xu, J.; Bourgeois, H.; Vandermeulen, E.; Vlaeminck, B.; Meyer, E.; Demeyere, K.; Hesta, M. Secreted phospholipase A2 inhibitor modulates fatty acid composition and reduces obesity-induced inflammation in Beagle dogs. Vet. J. 2015, 204, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Lim, J.; Poudyal, H.; Reid, R.C.; Suen, J.Y.; Webster, J.; Prins, J.B.; Whitehead, J.P.; Fairlie, D.P.; Brown, L. An inhibitor of phospholipase A2 group IIA modulates adipocyte signaling and protects against diet-induced metabolic syndrome in rats. Diabetes 2012, 61, 2320–2329. [Google Scholar] [CrossRef]
- Hui, D.Y.; Cope, M.J.; Labonte, E.D.; Chang, H.T.; Shao, J.; Goka, E.; Abousalham, A.; Charmot, D.; Buysse, J. The phospholipase A(2) inhibitor methyl indoxam suppresses diet-induced obesity and glucose intolerance in mice. Br. J. Pharmacol. 2009, 157, 1263–1269. [Google Scholar] [CrossRef]
- Hollie, N.I.; Hui, D.Y. Group 1B phospholipase A(2) deficiency protects against diet-induced hyperlipidemia in mice. J. Lipid Res. 2011, 52, 2005–2011. [Google Scholar] [CrossRef]
- Hollie, N.I.; Konaniah, E.S.; Goodin, C.; Hui, D.Y. Group 1B phospholipase A(2) inactivation suppresses atherosclerosis and metabolic diseases in LDL receptor-deficient mice. Atherosclerosis 2014, 234, 377–380. [Google Scholar] [CrossRef]
- Cash, J.G.; Kuhel, D.G.; Goodin, C.; Hui, D.Y. Pancreatic acinar cell-specific overexpression of group 1B phospholipase A2 exacerbates diet-induced obesity and insulin resistance in mice. Int. J. Obes. 2011, 35, 877–881. [Google Scholar] [CrossRef][Green Version]
- Sato, H.; Taketomi, Y.; Ushida, A.; Isogai, Y.; Kojima, T.; Hirabayashi, T.; Miki, Y.; Yamamoto, K.; Nishito, Y.; Kobayashi, T.; et al. The adipocyte-inducible secreted phospholipases PLA2G5 and PLA2G2E play distinct roles in obesity. Cell Metab. 2014, 20, 119–132. [Google Scholar] [CrossRef]
- Jha, P.; Claudel, T.; Baghdasaryan, A.; Mueller, M.; Halilbasic, E.; Das, S.K.; Lass, A.; Zimmermann, R.; Zechner, R.; Hoefler, G.; et al. Role of adipose triglyceride lipase (PNPLA2) in protection from hepatic inflammation in mouse models of steatohepatitis and endotoxemia. Hepatology 2014, 59, 858–869. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chang, B.; Li, L.; Chan, L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 2010, 52, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Ochi, T.; Munekage, K.; Ono, M.; Higuchi, T.; Tsuda, M.; Hayashi, Y.; Okamoto, N.; Toda, K.; Sakamoto, S.; Oben, J.A.; et al. Patatin-like phospholipase domain-containing protein 3 is involved in hepatic fatty acid and triglyceride metabolism through X-box binding protein 1 and modulation of endoplasmic reticulum stress in mice. Hepatol. Res. 2016, 46, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Garces, F.; Lopez, F.; Nino, C.; Fernandez, A.; Chacin, L.; Hurt-Camejo, E.; Camejo, G.; Apitz-Castro, R. High plasma phospholipase A2 activity, inflammation markers, and LDL alterations in obesity with or without type 2 diabetes. Obesity 2010, 18, 2023–2029. [Google Scholar] [CrossRef] [PubMed]
- Konukoglu, D.; Uzun, H.; Firtina, S.; Cigdem Arica, P.; Kocael, A.; Taskin, M. Plasma adhesion and inflammation markers: Asymmetrical dimethyl-L-arginine and secretory phospholipase A2 concentrations before and after laparoscopic gastric banding in morbidly obese patients. Obes. Surg. 2007, 17, 672–678. [Google Scholar] [CrossRef]
- Misso, N.L.; Petrovic, N.; Grove, C.; Celenza, A.; Brooks-Wildhaber, J.; Thompson, P.J. Plasma phospholipase A2 activity in patients with asthma: Association with body mass index and cholesterol concentration. Thorax 2008, 63, 21–26. [Google Scholar] [CrossRef]
- Speliotes, E.K.; Yerges-Armstrong, L.M.; Wu, J.; Hernaez, R.; Kim, L.J.; Palmer, C.D.; Gudnason, V.; Eiriksdottir, G.; Garcia, M.E.; Launer, L.J.; et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011, 7, e1001324. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Linden, D.; Ahnmark, A.; Pingitore, P.; Ciociola, E.; Ahlstedt, I.; Andreasson, A.C.; Sasidharan, K.; Madeyski-Bengtson, K.; Zurek, M.; Mancina, R.M.; et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol. Metab. 2019, 22, 49–61. [Google Scholar] [CrossRef]
- Sevastianova, K.; Kotronen, A.; Gastaldelli, A.; Perttila, J.; Hakkarainen, A.; Lundbom, J.; Suojanen, L.; Orho-Melander, M.; Lundbom, N.; Ferrannini, E.; et al. Genetic variation in PNPLA3 (adiponutrin) confers sensitivity to weight loss-induced decrease in liver fat in humans. Am. J. Clin. Nutr. 2011, 94, 104–111. [Google Scholar] [CrossRef]
- Lu, Y.; Day, F.R.; Gustafsson, S.; Buchkovich, M.L.; Na, J.; Bataille, V.; Cousminer, D.L.; Dastani, Z.; Drong, A.W.; Esko, T.; et al. New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nat. Commun. 2016, 7, 10495. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.O.; Loos, R.J.F.; Kilpelainen, T.O. Evidence of genetic predisposition for metabolically healthy obesity and metabolically obese normal weight. Physiol. Genom. 2018, 50, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Bauer, R.C.; Khetarpal, S.A.; Hand, N.J.; Rader, D.J. Therapeutic Targets of Triglyceride Metabolism as Informed by Human Genetics. Trends Mol. Med. 2016, 22, 328–340. [Google Scholar] [CrossRef]
- Johansen, C.T.; Hegele, R.A. Allelic and phenotypic spectrum of plasma triglycerides. Biochim. Biophys. Acta 2012, 1821, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.Y.; Wang, Y.; Hu, H.; Zhang, X.J.; Li, Q. Identification of the molecular subgroups in coronary artery disease by gene expression profiles. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef]
- Klimentidis, Y.C.; Chougule, A.; Arora, A.; Frazier-Wood, A.C.; Hsu, C.H. Triglyceride-Increasing Alleles Associated with Protection against Type-2 Diabetes. PLoS Genet. 2015, 11, e1005204. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Mora, S.; Ridker, P.M.; Hu, F.B.; Chasman, D.I. Gene-Based Elevated Triglycerides and Type 2 Diabetes Mellitus Risk in the Women’s Genome Health Study. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 97–106. [Google Scholar] [CrossRef]
- Yan, J.; Hu, C.; Jiang, F.; Zhang, R.; Wang, J.; Tang, S.; Peng, D.; Chen, M.; Bao, Y.; Jia, W. Genetic variants of PLA2G6 are associated with Type 2 diabetes mellitus and triglyceride levels in a Chinese population. Diabet. Med. J. Br. Diabet. Assoc. 2015, 32, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Song, H.; Wohltmann, M.; Ramanadham, S.; Jin, W.; Bohrer, A.; Turk, J. Insulin secretory responses and phospholipid composition of pancreatic islets from mice that do not express Group VIA phospholipase A2 and effects of metabolic stress on glucose homeostasis. J. Biol. Chem. 2006, 281, 20958–20973. [Google Scholar] [CrossRef]
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Ali, T.; Ashley, J.W.; Bone, R.N.; Hancock, W.D.; Lei, X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 2015, 56, 1643–1668. [Google Scholar] [CrossRef]
- Turk, J.; White, T.D.; Nelson, A.J.; Lei, X.; Ramanadham, S. iPLA2beta and its role in male fertility, neurological disorders, metabolic disorders, and inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J.; Bianco, I.D.; Ackermann, E.J.; Conde-Frieboes, K.; Dennis, E.A. Inhibition of calcium-independent phospholipase A2 prevents arachidonic acid incorporation and phospholipid remodeling in P388D1 macrophages. Proc. Natl. Acad. Sci. USA 1995, 92, 8527–8531. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J. Roles of various phospholipases A2 in providing lysophospholipid acceptors for fatty acid phospholipid incorporation and remodelling. Biochem. J. 2002, 364, 695–702. [Google Scholar] [CrossRef]
- Zhang, X.H.; Zhao, C.; Ma, Z.A. The increase of cell-membranous phosphatidylcholines containing polyunsaturated fatty acid residues induces phosphorylation of p53 through activation of ATR. J. Cell Sci. 2007, 120, 4134–4143. [Google Scholar] [CrossRef]
- Bao, S.; Miller, D.J.; Ma, Z.; Wohltmann, M.; Eng, G.; Ramanadham, S.; Moley, K.; Turk, J. Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J. Biol. Chem. 2004, 279, 38194–38200. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhong, S.; Li, Y.; Ji, G.; Sundaram, M.; Yao, Z. Global Inactivation of the Pla2g6 Gene in Mice Does Not Cause Dyslipidemia under Chow or High-fat Diet Conditions. J. Cancer Prev. 2013, 18, 235–248. [Google Scholar] [CrossRef][Green Version]
- Su, X.; Mancuso, D.J.; Bickel, P.E.; Jenkins, C.M.; Gross, R.W. Small interfering RNA knockdown of calcium-independent phospholipases A2 beta or gamma inhibits the hormone-induced differentiation of 3T3-L1 preadipocytes. J. Biol. Chem. 2004, 279, 21740–21748. [Google Scholar] [CrossRef]
- Honda, Y.; Yoneda, M.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Mawatari, H.; Fujita, K.; Hyogo, H.; Ueno, T.; et al. Characteristics of non-obese non-alcoholic fatty liver disease: Effect of genetic and environmental factors. Hepatol. Res. 2016, 46, 1011–1018. [Google Scholar] [CrossRef]
- Ecker, J.; Scherer, M.; Schmitz, G.; Liebisch, G. A rapid GC-MS method for quantification of positional and geometric isomers of fatty acid methyl esters. J. Chromatogr. Banal. Technol. Biomed. Life Sci. 2012, 897, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef] [PubMed]
- Liebisch, G.; Lieser, B.; Rathenberg, J.; Drobnik, W.; Schmitz, G. High-throughput quantification of phosphatidylcholine and sphingomyelin by electrospray ionization tandem mass spectrometry coupled with isotope correction algorithm. Biochim. Biophys. Acta 2004, 1686, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Liebisch, G.; Binder, M.; Schifferer, R.; Langmann, T.; Schulz, B.; Schmitz, G. High throughput quantification of cholesterol and cholesteryl ester by electrospray ionization tandem mass spectrometry (ESI-MS/MS). Biochim. Biophys. Acta 2006, 1761, 121–128. [Google Scholar] [CrossRef]
- Liebisch, G.; Drobnik, W.; Lieser, B.; Schmitz, G. High-throughput quantification of lysophosphatidylcholine by electrospray ionization tandem mass spectrometry. Clin. Chem. 2002, 48, 2217–2224. [Google Scholar] [CrossRef]
- Liebisch, G.; Drobnik, W.; Reil, M.; Trumbach, B.; Arnecke, R.; Olgemoller, B.; Roscher, A.; Schmitz, G. Quantitative measurement of different ceramide species from crude cellular extracts by electrospray ionization tandem mass spectrometry (ESI-MS/MS). J. Lipid Res. 1999, 40, 1539–1546. [Google Scholar]
- Song, H.; Bao, S.; Lei, X.; Jin, C.; Zhang, S.; Turk, J.; Ramanadham, S. Evidence for proteolytic processing and stimulated organelle redistribution of iPLA(2)beta. Biochim. Biophys. Acta 2010, 1801, 547–558. [Google Scholar] [CrossRef]
- Gao, X.; van der Veen, J.N.; Vance, J.E.; Thiesen, A.; Vance, D.E.; Jacobs, R.L. Lack of phosphatidylethanolamine N-methyltransferase alters hepatic phospholipid composition and induces endoplasmic reticulum stress. Biochim. Biophys. Acta 2015, 1852, 2689–2699. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef]
- Ming, Y.; Zhu, X.; Tuma-Kellner, S.; Ganzha, A.; Liebisch, G.; Gan-Schreier, H.; Chamulitrat, W. iPla2beta Deficiency Suppresses Hepatic ER UPR, Fxr, and Phospholipids in Mice Fed with MCD Diet, Resulting in Exacerbated Hepatic Bile Acids and Biliary Cell Proliferation. Cells 2019, 8, 879. [Google Scholar] [CrossRef]
- Tu, L.N.; Showalter, M.R.; Cajka, T.; Fan, S.; Pillai, V.V.; Fiehn, O.; Selvaraj, V. Metabolomic characteristics of cholesterol-induced non-obese nonalcoholic fatty liver disease in mice. Sci. Rep. 2017, 7, 6120. [Google Scholar] [CrossRef] [PubMed]
- Gurtovenko, A.A.; Vattulainen, I. Membrane potential and electrostatics of phospholipid bilayers with asymmetric transmembrane distribution of anionic lipids. J. Phys. Chem. B 2008, 112, 4629–4634. [Google Scholar] [CrossRef] [PubMed]
- Sena, A.; Rebel, G.; Bieth, R.; Hubert, P.; Waksman, A. Lipid composition in liver and brain of genetically obese (ob/ob), heterozygote (ob/+)and normal (+/+) mice. Biochim. Biophys. Acta 1982, 710, 290–296. [Google Scholar] [CrossRef]
- Van Rooyen, D.M.; Larter, C.Z.; Haigh, W.G.; Yeh, M.M.; Ioannou, G.; Kuver, R.; Lee, S.P.; Teoh, N.C.; Farrell, G.C. Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 2011, 141, 1393–1403.e5. [Google Scholar] [CrossRef] [PubMed]
- Patelski, J.; Piorunska-Stolzmann, M. Effects of substrate fatty acids on products of lecithin hydrolysis and acyl-CoA-independent transacylation with cholesterol by aortic enzyme preparations. Enzyme 1985, 34, 217–219. [Google Scholar] [CrossRef]
- Zhao, G.; Wakabayashi, R.; Shimoda, S.; Fukunaga, Y.; Kumagai, M.; Tanaka, M.; Nakano, K. Impaired activities of cyclic adenosine monophosphate-responsive element binding protein, protein kinase A and calcium-independent phospholipase A2 are involved in deteriorated regeneration of cirrhotic liver after partial hepatectomy in rats. Hepatol. Res. 2011, 41, 1110–1119. [Google Scholar] [CrossRef]
- Lei, X.; Zhang, S.; Barbour, S.E.; Bohrer, A.; Ford, E.L.; Koizumi, A.; Papa, F.R.; Ramanadham, S. Spontaneous development of endoplasmic reticulum stress that can lead to diabetes mellitus is associated with higher calcium-independent phospholipase A2 expression: A role for regulation by SREBP-1. J. Biol. Chem. 2010, 285, 6693–6705. [Google Scholar] [CrossRef]
- Gorden, D.L.; Myers, D.S.; Ivanova, P.T.; Fahy, E.; Maurya, M.R.; Gupta, S.; Min, J.; Spann, N.J.; McDonald, J.G.; Kelly, S.L.; et al. Biomarkers of NAFLD progression: A lipidomics approach to an epidemic. J. Lipid Res. 2015, 56, 722–736. [Google Scholar] [CrossRef]
- Chiappini, F.; Coilly, A.; Kadar, H.; Gual, P.; Tran, A.; Desterke, C.; Samuel, D.; Duclos-Vallee, J.C.; Touboul, D.; Bertrand-Michel, J.; et al. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci. Rep. 2017, 7, 46658. [Google Scholar] [CrossRef]
- Arendt, B.M.; Ma, D.W.; Simons, B.; Noureldin, S.A.; Therapondos, G.; Guindi, M.; Sherman, M.; Allard, J.P. Nonalcoholic fatty liver disease is associated with lower hepatic and erythrocyte ratios of phosphatidylcholine to phosphatidylethanolamine. Appl. Physiol. Nutr. Metab. 2013, 38, 334–340. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Pacana, T. A Lipidomic Readout of Disease Progression in A Diet-Induced Mouse Model of Nonalcoholic Fatty Liver Disease. Trans. Am. Clin. Climatol. Assoc. 2015, 126, 271–288. [Google Scholar] [PubMed]
- Chiappini, F.; Desterke, C.; Bertrand-Michel, J.; Guettier, C.; Le Naour, F. Hepatic and serum lipid signatures specific to nonalcoholic steatohepatitis in murine models. Sci. Rep. 2016, 6, 31587. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; McAdoo, K.R.; Horrobin, D.F. n-3 Essential fatty acids decrease weight gain in genetically obese mice. Br. J. Nutr. 1986, 56, 87–95. [Google Scholar] [CrossRef]
- Sekiya, M.; Yahagi, N.; Matsuzaka, T.; Najima, Y.; Nakakuki, M.; Nagai, R.; Ishibashi, S.; Osuga, J.; Yamada, N.; Shimano, H. Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology 2003, 38, 1529–1539. [Google Scholar] [CrossRef]
- Stubbs, C.D.; Smith, A.D. The modification of mammalian membrane polyunsaturated fatty acid composition in relation to membrane fluidity and function. Biochim. Biophys. Acta 1984, 779, 89–137. [Google Scholar] [CrossRef]
- Ii, H.; Oka, M.; Yamashita, A.; Waku, K.; Uozumi, N.; Shimizu, T.; Sato, T.; Akiba, S. Inhibition of cytosolic phospholipase A(2) suppresses production of cholesteryl ester through the reesterification of free cholesterol but not formation of foam cells in oxidized LDL-stimulated macrophages. Biol. Pharm. Bull. 2008, 31, 6–12. [Google Scholar] [CrossRef]
- Gubern, A.; Barcelo-Torns, M.; Casas, J.; Barneda, D.; Masgrau, R.; Picatoste, F.; Balsinde, J.; Balboa, M.A.; Claro, E. Lipid droplet biogenesis induced by stress involves triacylglycerol synthesis that depends on group VIA phospholipase A2. J. Biol. Chem. 2009, 284, 5697–5708. [Google Scholar] [CrossRef] [PubMed]
- Ashley, J.W.; Hancock, W.D.; Nelson, A.J.; Bone, R.N.; Tse, H.M.; Wohltmann, M.; Turk, J.; Ramanadham, S. Polarization of Macrophages toward M2 Phenotype Is Favored by Reduction in iPLA2beta (Group VIA Phospholipase A2). J. Biol. Chem. 2016, 291, 23268–23281. [Google Scholar] [CrossRef]
- Monge, P.; Garrido, A.; Rubio, J.M.; Magrioti, V.; Kokotos, G.; Balboa, M.A.; Balsinde, J. The Contribution of Cytosolic Group IVA and Calcium-Independent Group VIA Phospholipase A2s to Adrenic Acid Mobilization in Murine Macrophages. Biomolecules 2020, 10, 542. [Google Scholar] [CrossRef]
- Inhoffen, J.; Tuma-Kellner, S.; Straub, B.; Stremmel, W.; Chamulitrat, W. Deficiency of iPLA(2)beta Primes Immune Cells for Proinflammation: Potential Involvement in Age-Related Mesenteric Lymph Node Lymphoma. Cancers 2015, 7, 2427–2442. [Google Scholar] [CrossRef]
- Jiao, L.; Gan-Schreier, H.; Tuma-Kellner, S.; Stremmel, W.; Chamulitrat, W. Sensitization to autoimmune hepatitis in group VIA calcium-independent phospholipase A2-null mice led to duodenal villous atrophy with apoptosis, goblet cell hyperplasia and leaked bile acids. Biochim. Biophys. Acta 2015, 1852, 1646–1657. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Gan-Schreier, H.; Zhu, X.; Wei, W.; Tuma-Kellner, S.; Liebisch, G.; Stremmel, W.; Chamulitrat, W. Ageing sensitized by iPLA2beta deficiency induces liver fibrosis and intestinal atrophy involving suppression of homeostatic genes and alteration of intestinal lipids and bile acids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1520–1533. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Blase, J.R.; Hoft, D.F.; Marentette, J.O.; Turk, J.; McHowat, J. Mice with Genetic Deletion of Group VIA Phospholipase A2beta Exhibit Impaired Macrophage Function and Increased Parasite Load in Trypanosoma cruzi-Induced Myocarditis. Infect. Immun. 2016, 84, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, K.; Ahmadian, M.; Duncan, R.E.; Sarkadi-Nagy, E.; Varady, K.A.; Hellerstein, M.K.; Lee, H.Y.; Samuel, V.T.; Shulman, G.I.; Kim, K.H.; et al. AdPLA ablation increases lipolysis and prevents obesity induced by high-fat feeding or leptin deficiency. Nat. Med. 2009, 15, 159–168. [Google Scholar] [CrossRef]
- Shimabukuro, M.; Koyama, K.; Chen, G.; Wang, M.Y.; Trieu, F.; Lee, Y.; Newgard, C.B.; Unger, R.H. Direct antidiabetic effect of leptin through triglyceride depletion of tissues. Proc. Natl. Acad. Sci. USA 1997, 94, 4637–4641. [Google Scholar] [CrossRef] [PubMed]
- Metlakunta, A.; Huang, W.; Stefanovic-Racic, M.; Dedousis, N.; Sipula, I.; O’Doherty, R.M. Kupffer cells facilitate the acute effects of leptin on hepatic lipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E11–E18. [Google Scholar] [CrossRef]
- Bone, R.N.; Gai, Y.; Magrioti, V.; Kokotou, M.G.; Ali, T.; Lei, X.; Tse, H.M.; Kokotos, G.; Ramanadham, S. Inhibition of Ca2+-independent phospholipase A2beta (iPLA2beta) ameliorates islet infiltration and incidence of diabetes in NOD mice. Diabetes 2015, 64, 541–554. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Limnios, D.; Kokotou, M.G.; Barbayianni, E.; Kokotos, G.; McCammon, J.A.; Dennis, E.A. Development of Potent and Selective Inhibitors for Group VIA Calcium-Independent Phospholipase A2 Guided by Molecular Dynamics and Structure-Activity Relationships. J. Med. Chem. 2016, 59, 4403–4414. [Google Scholar] [CrossRef]
- Guo, H.H.; Feng, C.L.; Zhang, W.X.; Luo, Z.G.; Zhang, H.J.; Zhang, T.T.; Ma, C.; Zhan, Y.; Li, R.; Wu, S.; et al. Liver-target nanotechnology facilitates berberine to ameliorate cardio-metabolic diseases. Nat. Commun. 2019, 10, 1981. [Google Scholar] [CrossRef]
- Sharma, Y.; Ahmad, A.; Yavvari, P.S.; Kumar Muwal, S.; Bajaj, A.; Khan, F. Targeted SHP-1 Silencing Modulates the Macrophage Phenotype, Leading to Metabolic Improvement in Dietary Obese Mice. Mol. Ther. Nucleic Acids 2019, 16, 626–636. [Google Scholar] [CrossRef]
- Cao, J.; Peterson, S.J.; Sodhi, K.; Vanella, L.; Barbagallo, I.; Rodella, L.F.; Schwartzman, M.L.; Abraham, N.G.; Kappas, A. Heme oxygenase gene targeting to adipocytes attenuates adiposity and vascular dysfunction in mice fed a high-fat diet. Hypertension 2012, 60, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Buchman, A.L.; Dubin, M.D.; Moukarzel, A.A.; Jenden, D.J.; Roch, M.; Rice, K.M.; Gornbein, J.; Ament, M.E. Choline deficiency: A cause of hepatic steatosis during parenteral nutrition that can be reversed with intravenous choline supplementation. Hepatology 1995, 22, 1399–1403. [Google Scholar]
- Corbin, K.D.; Zeisel, S.H. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr. Opin. Gastroenterol. 2012, 28, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.D.; Hamp, T.J.; Reid, R.W.; Fischer, L.M.; Zeisel, S.H.; Fodor, A.A. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 2011, 140, 976–986. [Google Scholar] [CrossRef]
- Sha, W.; da Costa, K.A.; Fischer, L.M.; Milburn, M.V.; Lawton, K.A.; Berger, A.; Jia, W.; Zeisel, S.H. Metabolomic profiling can predict which humans will develop liver dysfunction when deprived of dietary choline. FASEB J. 2010, 24, 2962–2975. [Google Scholar] [CrossRef] [PubMed]
- Cordero, P.; Gomez-Uriz, A.M.; Campion, J.; Milagro, F.I.; Martinez, J.A. Dietary supplementation with methyl donors reduces fatty liver and modifies the fatty acid synthase DNA methylation profile in rats fed an obesogenic diet. Genes Nutr. 2013, 8, 105–113. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamulitrat, W.; Jansakun, C.; Li, H.; Liebisch, G. Rescue of Hepatic Phospholipid Remodeling Defect in iPLA2β-Null Mice Attenuates Obese but Not Non-Obese Fatty Liver. Biomolecules 2020, 10, 1332. https://doi.org/10.3390/biom10091332
Chamulitrat W, Jansakun C, Li H, Liebisch G. Rescue of Hepatic Phospholipid Remodeling Defect in iPLA2β-Null Mice Attenuates Obese but Not Non-Obese Fatty Liver. Biomolecules. 2020; 10(9):1332. https://doi.org/10.3390/biom10091332
Chicago/Turabian StyleChamulitrat, Walee, Chutima Jansakun, Huili Li, and Gerhard Liebisch. 2020. "Rescue of Hepatic Phospholipid Remodeling Defect in iPLA2β-Null Mice Attenuates Obese but Not Non-Obese Fatty Liver" Biomolecules 10, no. 9: 1332. https://doi.org/10.3390/biom10091332
APA StyleChamulitrat, W., Jansakun, C., Li, H., & Liebisch, G. (2020). Rescue of Hepatic Phospholipid Remodeling Defect in iPLA2β-Null Mice Attenuates Obese but Not Non-Obese Fatty Liver. Biomolecules, 10(9), 1332. https://doi.org/10.3390/biom10091332