The Disordered Cellular Multi-Tasker WIP and Its Protein–Protein Interactions: A Structural View


Abstract
1. Introduction
1.1. Scope
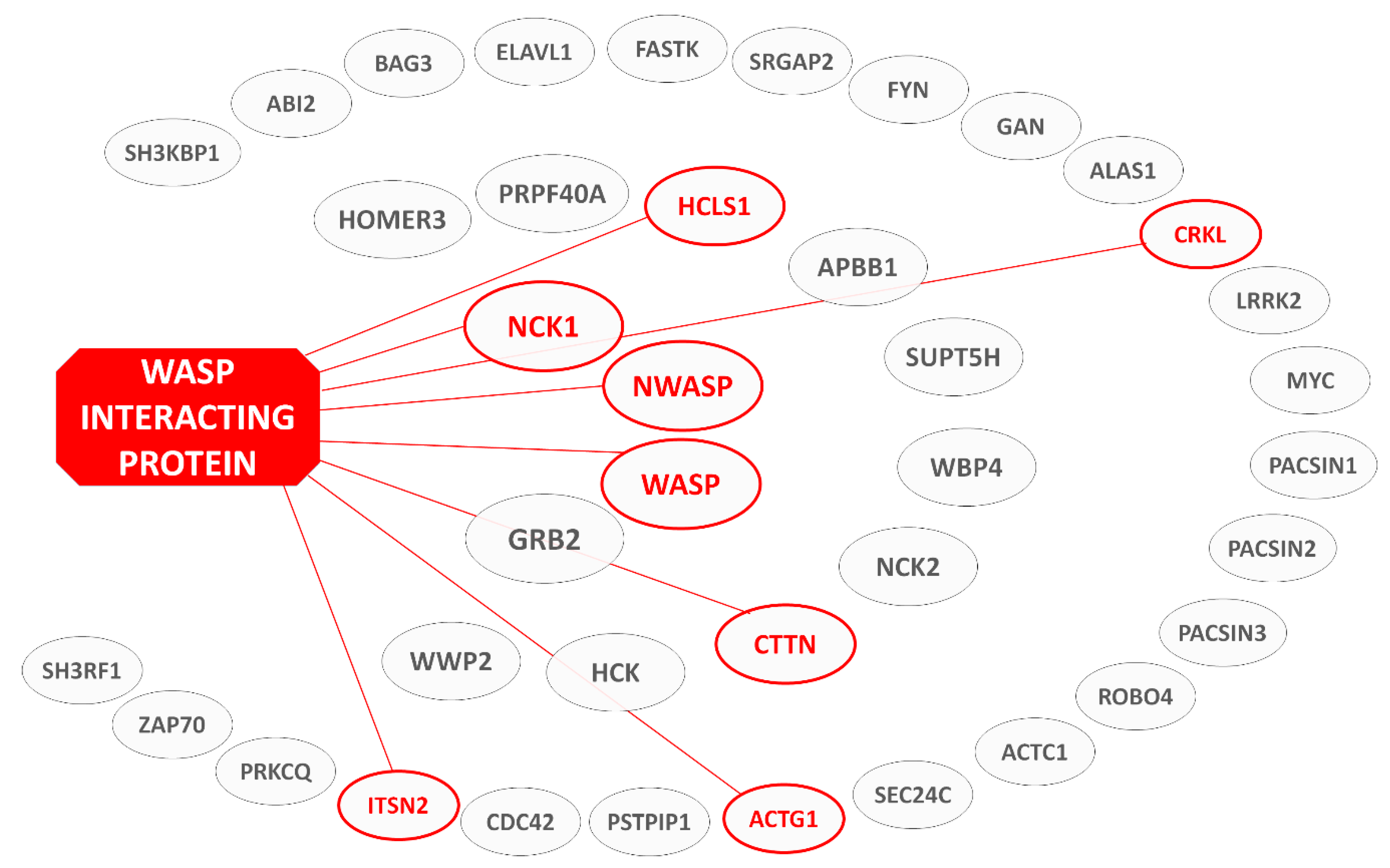
1.2. WIP—Biology and Cellular Roles
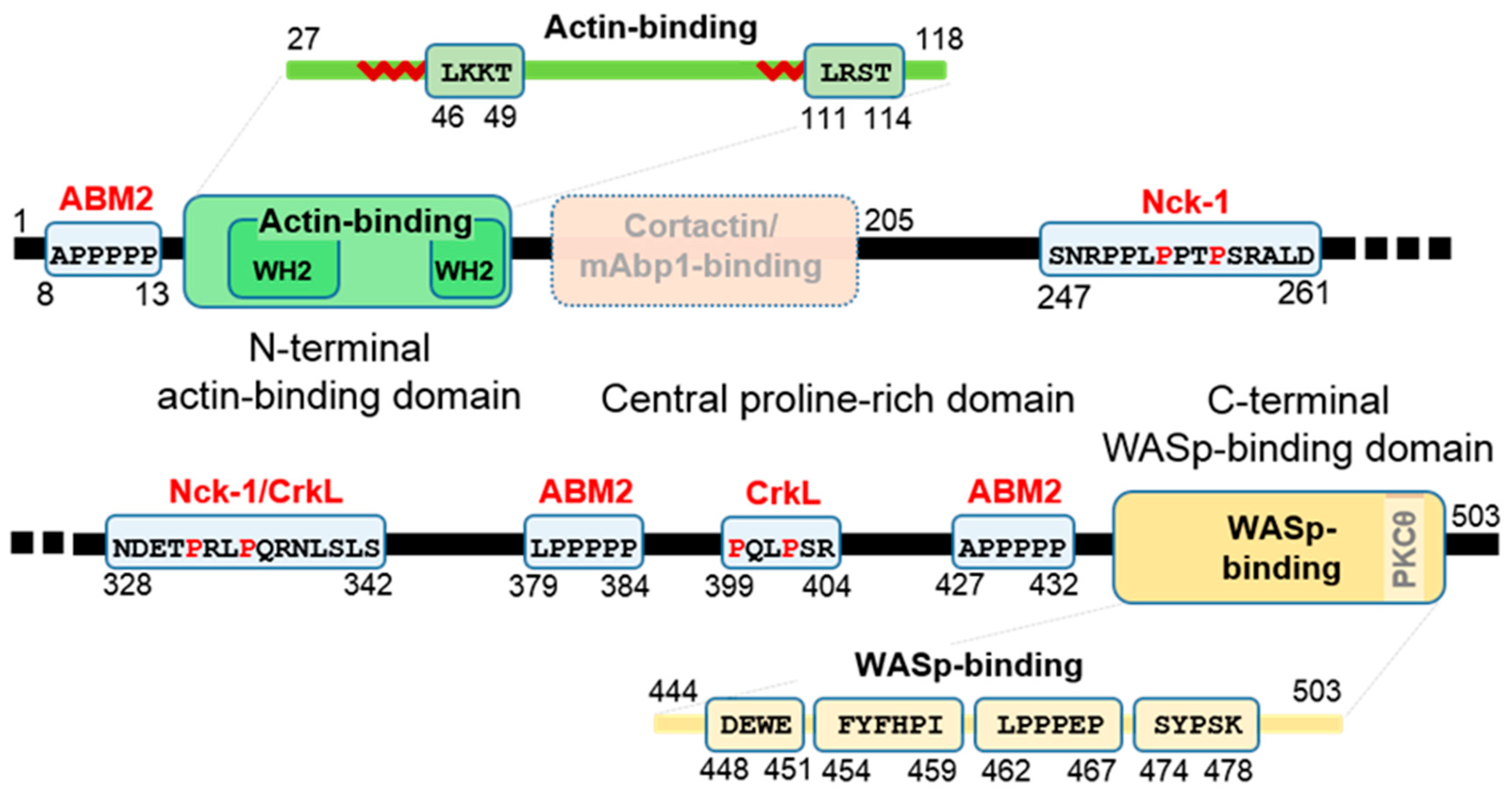
1.3. Functional Domains and Sequences of WIP
1.4. WIP Is a Disordered Polypeptide
1.5. Rationale and Structure of Review—List of WIP Interaction Domains
2. The Actin-Binding Region
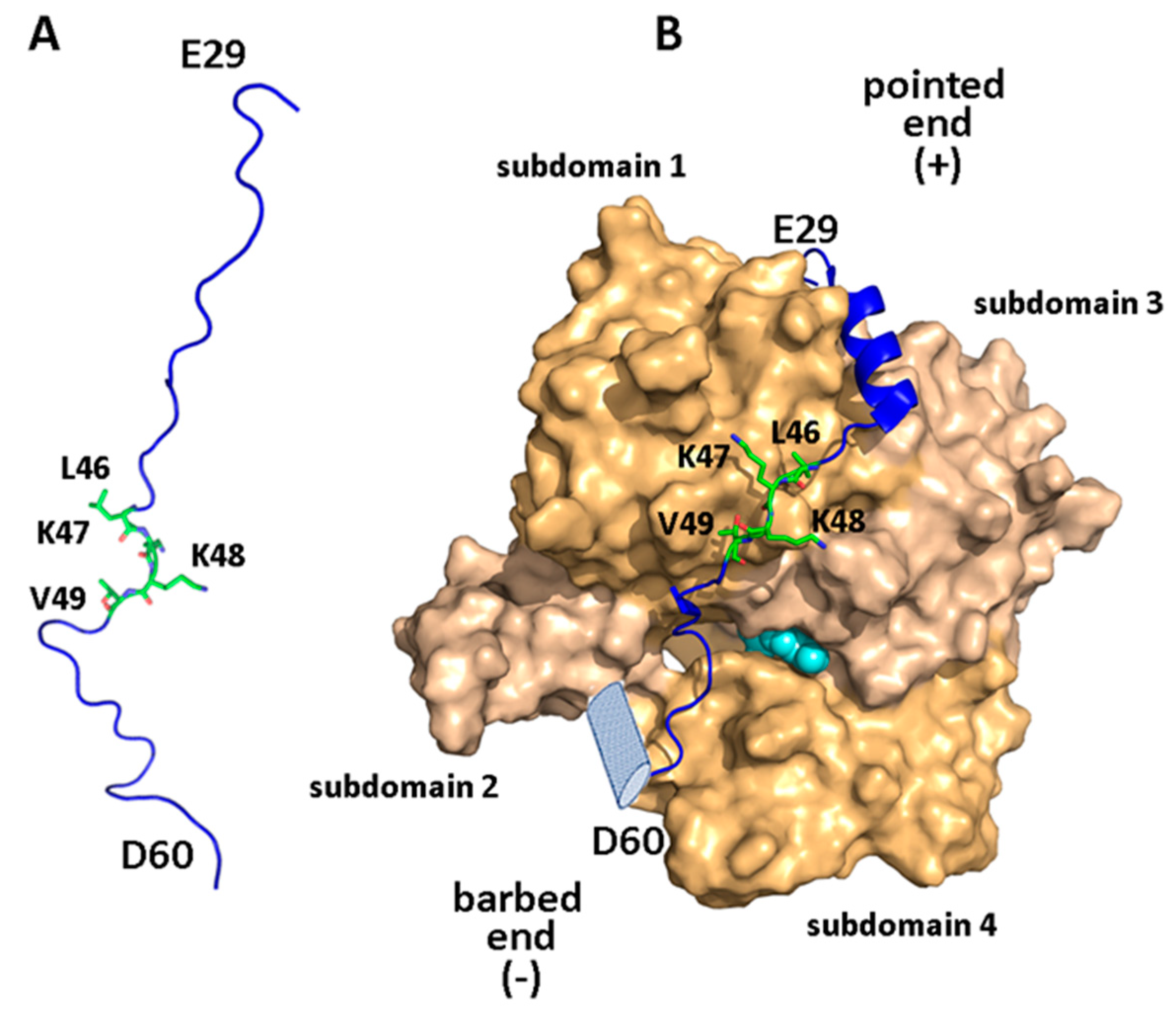
2.1. Actin—A Cytoskeleton Protein—and Actin-Binding Domains
2.2. Structural Aspects of the WIP-Actin Interaction
3. The Proline-Rich Intermediate Region
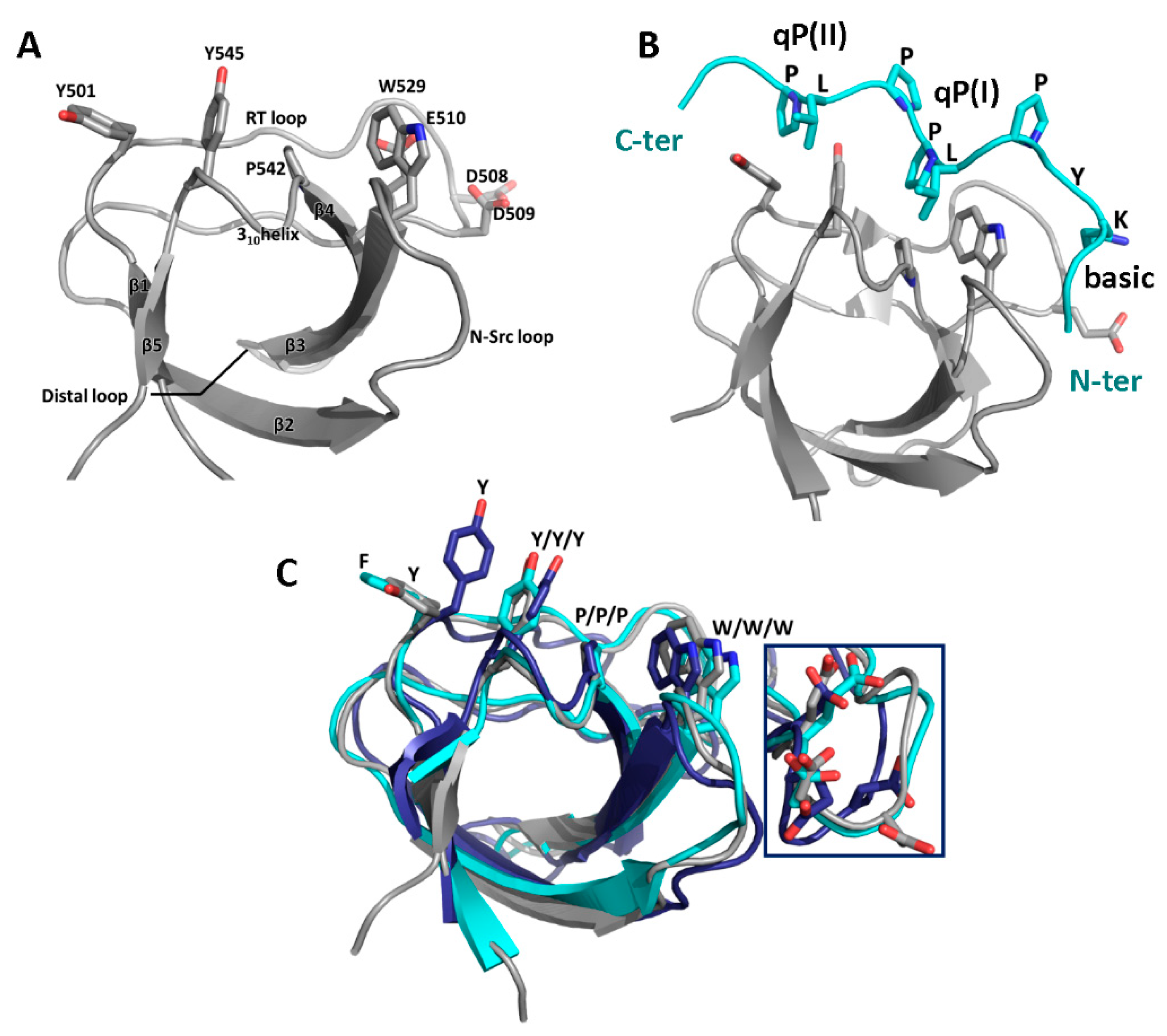
3.1. SH3 Domains and Their Ligands
3.2. Binding Partners and Binding Motifs
4. The WIP-C/WASp Interface
4.1. Structure and Binding Epitopes in the WIP-C/N-WASp Complex
4.2. Functional Implications of the WIP/WASp Interface
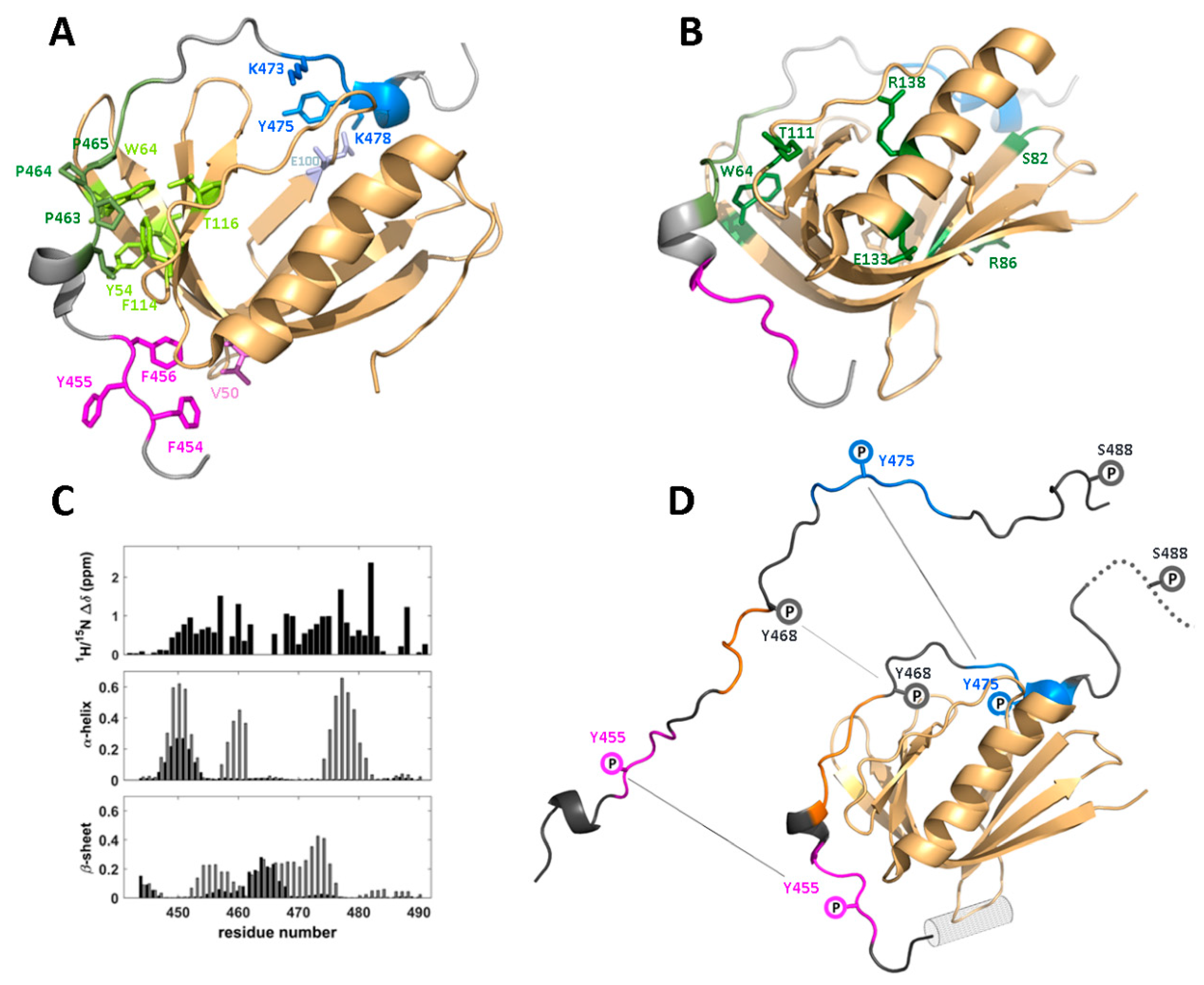
4.3. Phosphorylation-Induced Dissociation of the WIP/WASp Complex
5. Discussion and Summary
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Ramesh, N.; Antón, I.M.; Hartwig, J.H.; Geha, R.S. WIP, a Protein Associated with Wiskott-Aldrich Syndrome Protein, Induces Actin Polymerization and Redistribution in Lymphoid Cells. Proc. Natl. Acad. Sci. USA 1997, 94, 14671–14676. [Google Scholar] [CrossRef]
- Moreau, V.; Frischknecht, F.; Reckmann, I.; Vincentelli, R.; Rabut, G.; Stewart, D.; Way, M. A Complex of N-WASp and WIP Integrates Signalling Cascades That Lead to Actin Polymerization. Nat. Cell Biol. 2000, 2, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Antón, I.M.; Jones, G.E. WIP: A Multifunctional Protein Involved in Actin Cytoskeleton Regulation. Eur. J. Cell Biol. 2006, 85, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Antón, I.M.; Jones, G.E.; Wandosell, F.; Geha, R.; Ramesh, N. WASp-Interacting Protein (WIP): Working in Polymerisation and Much More. Trends Cell Biol. 2007, 17, 555–562. [Google Scholar]
- Ramesh, N.; Geha, R. Recent Advances in the Biology of WASp and WIP. Immunol. Res. 2009, 44, 99–111. [Google Scholar] [CrossRef]
- Noy, E.; Fried, S.; Matalon, O.; Barda-Saad, M. WIP Remodeling Actin behind the Scenes: How WIP Reshapes Immune and Other Functions. Int. J. Mol. Sci. 2012, 13, 7629–7647. [Google Scholar] [CrossRef]
- Sasahara, Y. WASp-WIP Complex in the Molecular Pathogenesis of Wiskott-Aldrich Syndrome. Pediatr. Int. 2016, 58, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Schad, E.; Tantos, A.; Kalmar, L. Intrinsically Disordered Proteins: Emerging Interaction Specialists. Curr. Opin. Struct. Biol. 2015, 35, 49–59. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsic Disorder, Protein–Protein Interactions, and Disease. Adv. Prot. Chem. Struct. Biol. 2010, 110, 85–120. [Google Scholar]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Expanding the Paradigm: Intrinsically Disordered Proteins and Allosteric Regulation. J. Mol. Biol. 2018, 430, 2309–2320. [Google Scholar] [CrossRef]
- Aspenström, P. The Verprolin Family of Proteins: Regulators of Cell Morphogenesis and Endocytosis. FEBS Lett. 2005, 579, 5253–5259. [Google Scholar] [CrossRef] [PubMed]
- Keppler, S.J.; Gasparrini, F.; Burbage, M.; Aggarwal, S.; Frederico, B.; Geha, R.S.; Way, M.; Bruckbauer, A.; Batista, F.D. Wiskott-Aldrich Syndrome Interacting Protein Deficiency Uncovers the Role of the Co-Receptor CD19 as a Generic Hub for PI3 Kinase Signaling in B Cells. Immunity 2015, 43, 660–673. [Google Scholar] [CrossRef] [PubMed]
- Mattila, P.K.; Batista, F.D.; Treanor, B. Dynamics of the Actin Cytoskeleton Mediates Receptor Cross Talk: An Emerging Concept in Tuning Receptor Signaling. J. Cell Biol. 2016, 212, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Kettner, A.; Kumar, L.; Antón, I.M.; Sasahara, Y.; De La Fuente, M.; Pivniouk, V.I.; Falet, H.; Hartwig, J.H.; Geha, R.S. WIP Regulates Signaling via the High Affinity Receptor for Immunoglobulin E in Mast Cells. J. Exp. Med. 2004, 199, 357–368. [Google Scholar] [CrossRef]
- Sasahara, Y.; Rachid, R.; Byrne, M.J.; De La Fuente, M.A.; Abraham, R.T.; Ramesh, N.; Geha, R.S. Mechanism of Recruitment of WASp to the Immunological Synapse and of Its Activation Following TCR Ligation. Mol. Cell. 2002, 10, 1269–1281. [Google Scholar] [CrossRef]
- Antón, I.M.; De la Fuente, M.A.; Sims, T.N.; Freeman, S.; Ramesh, N.; Hartwig, J.H.; Dustin, M.L.; Geha, R.S. WIP Deficiency Reveals a Differential Role for WIP and the Actin Cytoskeleton in T and B Cell Activation. Immunity 2002, 16, 193–204. [Google Scholar] [CrossRef]
- Ho, H.Y.H.; Rohatgi, R.; Lebensohn, A.M.; Le, M.; Li, J.; Gygi, S.P.; Kirschner, M.W. Toca-1 Mediates Cdc42-Dependent Actin Nucleation by Activating the N-WASp-WIP Complex. Cell 2004, 118, 203–216. [Google Scholar] [CrossRef]
- Martinez-Quiles, N.; Rohatgi, R.; Antón, I.M.; Medina, M.; Saville, S.P.; Miki, H.; Yamaguchi, H.; Takenawa, T.; Hartwig, J.H.; Geha, R.S.; et al. WIP Regulates N-WASp-Mediated Actin Polymerization and Filopodium Formation. Nat. Cell Biol. 2001, 3, 484–491. [Google Scholar] [CrossRef]
- Calle, Y.; Antón, I.M.; Thrasher, A.J.; Jones, G.E. WASp and WIP Regulate Podosomes in Migrating Leukocytes. J. Microsc. 2008, 231, 494–505. [Google Scholar] [CrossRef]
- De La Fuente, M.A.; Sasahara, Y.; Calamito, M.; Antón, I.M.; Elkhal, A.; Gallego, M.D.; Suresh, K.; Siminovitch, K.; Ochs, H.D.; Anderson, K.C.; et al. WIP Is a Chaperone for Wiskott-Aldrich Syndrome Protein. Proc. Natl. Acad. Sci. USA 2007, 104, 926–931. [Google Scholar] [CrossRef]
- Sun, Y.; Leong, N.; Jiang, T.; Tangara, A.; Darzacq, X.; Drubin, D. Switch-like Arp2/3 Activation upon WASp and WIP Recruitment to an Apparent Threshold Level by Multivalent Linker Proteins in vivo. Elife 2017, 6, e29140. [Google Scholar] [CrossRef] [PubMed]
- Massaad, M.J.; Oyoshi, M.K.; Kane, J.; Koduru, S.; Alcaide, P.; Nakamura, F.; Ramesh, N.; Luscinskas, F.W.; Hartwig, J.; Geha, R.S. Binding of WIP to Actin Is Essential for T Cell Actin Cytoskeleton Integrity and Tissue Homing. Mol. Cell. Biol. 2014, 34, 4343–4354. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Keppler, S.J.; Burbage, M.; Gasparrini, F.; Hartjes, L.; Aggarwal, S.; Massaad, M.J.; Geha, R.S.; Bruckbauer, A.; Batista, F.D. The Lack of WIP Binding to Actin Results in Impaired B Cell Migration and Altered Humoral Immune Responses. Cell Rep. 2018, 24, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.; Knafo, S.; Banon-Rodriguez, I.; Merino-Serrais, P.; Fernaud-Espinosa, I.; Nieto, M.; Garrido, J.J.; Esteban, J.A.; Wandosell, F.; Anton, I.M. WIP is a Negative Regulator of Neuronal Maturation and Synaptic Activity. Cereb. Cortex 2012, 22, 1191–1202. [Google Scholar] [CrossRef]
- García, E.; Machesky, L.M.; Jones, G.E.; Antón, I.M. WIP Is Necessary for Matrix Invasion by Breast Cancer Cells. Eur. J. Cell Biol. 2014, 93, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, S.K.; Weisswange, I.; Zettl, M.; Way, M. WIP Provides an Essential Link between Nck and N-WASp during Arp2/3-Dependent Actin Polymerization. Curr. Biol. 2013, 23, 999–1006. [Google Scholar] [CrossRef]
- Escoll, M.; Gargini, R.; Cuadrado, A.; Antón, I.M.; Wandosell, F. Mutant P53 Oncogenic Functions in Cancer Stem Cells Are Regulated by WIP through YAP/TAZ. Oncogene 2017, 36, 3515–3527. [Google Scholar] [CrossRef]
- Gargini, R.; Escoll, M.; García, E.; García-Escudero, R.; Wandosell, F.; Antón, I.M. WIP Drives Tumor Progression through YAP/TAZ-Dependent Autonomous Cell Growth. Cell Rep. 2016, 17, 1962–1977. [Google Scholar] [CrossRef]
- Rivas, S.; Antón, I.M.; Wandosell, F. WIP-YAP/TAZ as a New pro-Oncogenic Pathway in Glioma. Cancers (Basel) 2018, 10, 191. [Google Scholar] [CrossRef]
- Ramesh, N.; Massaad, M.J.; Kumar, L.; Koduru, S.; Sasahara, Y.; Anton, I.; Bhasin, M.; Libermann, T.; Geha, R. Binding of the WASp/N-WASp-Interacting Protein WIP to Actin Regulates Focal Adhesion Assembly and Adhesion. Mol. Cell. Biol. 2014, 34, 2600–2610. [Google Scholar] [CrossRef]
- Dong, X.; Patino-Lopez, G.; Candotti, F.; Shaw, S. Structure-function analysis of WIP role in TCR-stimulated NFAT activation: Evidence that WIP/WASp dissociation is not required and that the WIP N-terminus is inhibitory. J. Biol. Chem. 2007, 282, 30303–30310. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.P.; Zappacosta, F.; Kim, E.Y.; Annan, R.S.; Miller, W.T. Identification of Novel SH3 Domain Ligands for the Src Family Kinase Hck: WASp, WIP, and ELMO1. J. Biol. Chem. 2002, 277, 28238–28246. [Google Scholar] [CrossRef]
- Linkermann, A.; Gelhaus, C.; Lettau, M.; Qian, J.; Kabelitz, D.; Janssen, O. Identification of Interaction Partners for Individual SH3 Domains of Fas Ligand Associated Members of the PCH Protein Family in T Lymphocytes. Biochim. Biophys. Acta—Proteins Proteom. 2009, 1794, 168–176. [Google Scholar] [CrossRef]
- Volkman, B.F.; Prehoda, K.E.; Scott, J.A.; Peterson, F.C.; Lim, W.A. Structure of the N-WASP EVH1 Domain-WIP Complex: Insight into the Molecular Basis of Wiskott-Aldrich Syndrome. Cell 2002, 111, 565–576. [Google Scholar] [CrossRef]
- Eliezer, D. Biophysical Characterization of Intrinsically Disordered Proteins. Curr. Opin. Struct. Biol. 2009, 19, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.R.; Zweckstetter, M.; Huang, J.R.; Blackledge, M. Exploring Free-Energy Landscapes of Intrinsically Disordered Proteins at Atomic Resolution Using NMR Spectroscopy. Chem. Rev. 2014, 114, 6632–6660. [Google Scholar] [CrossRef]
- Marsh, J.A.; Teichmann, S.A.; Forman-Kay, J.D. Probing the Diverse Landscape of Protein Flexibility and Binding. Curr. Opin. Struct. Biol. 2012, 22, 643–650. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dunker, A.K. Understanding Protein Non-Folding. Biophys. Biochim. Act. 2010, 1804, 1231–1264. [Google Scholar] [CrossRef]
- Tompa, P. The Interplay between Structure and Function in Intrinsically Unstructured Proteins. FEBS Lett. 2005, 579, 3346–3354. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsic Disorder in Proteins Associated with Neurodegenerative Disease. Front. Biosci. 2009, 14, 5188–5238. [Google Scholar] [CrossRef]
- Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Davey, N.E.; Gibson, T.J. Proteome-Wide Analysis of Human Disease Mutations in Short Linear Motifs: Neglected Players in Cancer? Mol. Biosyst. 2014, 10, 2626–2642. [Google Scholar] [CrossRef]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and Structure of Inherently Disordered Proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef]
- Rezaei-Ghaleh, N.; Blackledge, M.; Zweckstetter, M. Intrinsically Disordered Proteins: From Sequence and Conformational Properties toward Drug Discovery. ChemBioChem 2012, 13, 930–950. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Forman-Kay, J.D. Modulation of Intrinsically Disordered Protein Function by Post-Translational Modifications. J. Biol. Chem. 2016, 291, 6696–6705. [Google Scholar] [CrossRef] [PubMed]
- Schramm, A.; Bignon, C.; Brocca, S.; Grandori, R.; Santambrogio, C.; Longhi, S. An Arsenal of Methods for the Experimental Characterization of Intrinsically Disordered Proteins – How to Choose and Combine Them? Arch. Biochem. Biophys. 2019, 676, 108055. [Google Scholar] [CrossRef] [PubMed]
- Selenko, P. Quo Vadis Biomolecular NMR Spectroscopy? Int. J. Mol. Sci. 2019, 20, 1278. [Google Scholar] [CrossRef]
- Milles, S.; Salvi, N.; Blackledge, M.; Jensen, M.R. Characterization of Intrinsically Disordered Proteins and Their Dynamic Complexes: From in Vitro to Cell-like Environments. Prog. Nucl. Magn. Reson. Spectrosc. 2018, 109, 79–100. [Google Scholar] [CrossRef] [PubMed]
- Gomes, G.N.; Gradinaru, C.C. Insights into the Conformations and Dynamics of Intrinsically Disordered Proteins Using Single-Molecule Fluorescence. Biochim. Biophys. Acta—Proteins Proteom. 2017, 1865, 1696–1706. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B. Computational and Theoretical Advances in Studies of Intrinsically Disordered Proteins. Curr. Opin. Struct. Biol. 2017, 42, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Kachala, M.; Valentini, E.; Svergun, D.I. Application of SAXS for the Structural Characterization of IDPs. Adv. Exp. Med. Biol. 2015, 870, 261–289. [Google Scholar]
- Schuler, B.; Soranno, A.; Hofmann, H.; Nettels, D. Single-Molecule FRET Spectroscopy and the Polymer Physics of Unfolded and Intrinsically Disordered Proteins. Annu. Rev. Biophys. 2016, 45, 207–231. [Google Scholar] [CrossRef] [PubMed]
- Stuchfield, D.; Barran, P. Unique Insights to Intrinsically Disordered Proteins Provided by Ion Mobility Mass Spectrometry. Curr. Opin. Chem. Biol. 2018, 42, 177–185. [Google Scholar] [CrossRef]
- Ma, B.; Kumar, S.; Tsai, C.; Nussinov, R. Folding Funnels and Binding Mechanisms. Protein Eng. 1999, 12, 713–720. [Google Scholar] [CrossRef]
- Koshland, D.E. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef]
- Fuxreiter, M.; Tompa, P. Fuzzy complexes: A more stochastic view of protein function. Adv. Exp. Med. Biol. 2012, 725, 1–14. [Google Scholar]
- Csermely, P.; Palotai, R.; Nussinov, R. Induced Fit, Conformational Selection and Independent Dynamic Segments: An Extended View of Binding Events. Trends Biochem. Sci. 2010, 35, 539–546. [Google Scholar] [CrossRef]
- Olsen, J.G.; Teilum, K.; Kragelund, B.B. Behaviour of Intrinsically Disordered Proteins in Protein–Protein Complexes with an Emphasis on Fuzziness. Cell. Mol. Life Sci. 2017, 74, 3175–3183. [Google Scholar] [CrossRef]
- Alanis-Lobato, G.; Andrade-Navarro, M.A.; Schaefer, M.H. HIPPIE v2.0: Enhancing Meaningfulness and Reliability of Protein-Protein Interaction Networks. Nucleic Acids Res. 2017, 45, D408–D414. [Google Scholar] [CrossRef] [PubMed]
- Roberto Dominguez and Kenneth, C. Holmes. Actin Structure & Function. Annu Rev. Biophys. 2011, 45, 169–186. [Google Scholar]
- Lassing, I.; Schmitzberger, F.; Björnstedt, M.; Holmgren, A.; Nordlund, P.; Schutt, C.E.; Lindberg, U. Molecular and Structural Basis for Redox Regulation of β-Actin. J. Mol. Biol. 2007, 370, 331–348. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Iwasa, M.; Aihara, T.; Maéda, Y.; Narita, A. The Nature of the Globular- to Fibrous-Actin Transition. Nature 2009, 457, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, R. Actin-Binding Proteins—A Unifying Hypothesis. Trends Biochem. Sci. 2004, 29, 572–578. [Google Scholar] [CrossRef]
- Otterbein, L.R.; Graceffa, P.; Dominguez, R. The Crystal Structure of Uncomplexed Actin in the ADP State. Science 2001, 293, 708–711. [Google Scholar] [CrossRef]
- Durer, Z.A.O.; Kudryashov, D.S.; Sawaya, M.R.; Altenbach, C.; Hubbell, W.; Reisler, E. Structural States and Dynamics of the D-Loop in Actin. Biophys. J. 2012, 103, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Fixe, P. Actin-Binding Proteins (ABPs) review. 2014. Available online: https://www.tebu-bio.com/blog/2014/06/12/actin-binding-proteins-abps-review/ (accessed on 17 July 2020).
- Paavilainen, V.O.; Bertling, E.; Falck, S.; Lappalainen, P. Regulation of Cytoskeletal Dynamics by Actin-Monomer-Binding Proteins. Trends Cell Biol. 2004, 14, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Lappalainen, P.; Mattila, P. Are β-Thymosins WH2 Domains? FEBS Lett. 2004, 573, 231–232. [Google Scholar] [CrossRef]
- Chereau, D.; Kerff, F.; Graceffa, P.; Grabarek, Z.; Langsetmo, K.; Dominguez, R. Actin-Bound Structures of Wiskott-Aldrich Syndrome Protein (WASp)-Homology Domain 2 and the Implications for Filament Assembly. Proc. Natl. Acad. Sci. USA 2005, 102, 16644–16649. [Google Scholar] [CrossRef]
- Aguda, A.H.; Xue, B.; Irobi, E.; Préat, T.; Robinson, R.C. The Structural Basis of Actin Interaction with Multiple WH2/β-Thymosin Motif-Containing Proteins. Structure 2006, 14, 469–476. [Google Scholar] [CrossRef]
- Paunola, E.; Mattila, P.K.; Lappalainen, P. WH2 Domain: A Small, Versatile Adapter for Actin Monomers. FEBS Lett. 2002, 513, 92–97. [Google Scholar] [CrossRef]
- Husson, C.; Cantrelle, F.X.; Roblin, P.; Didry, D.; Le, K.H.D.; Perez, J.; Guittet, E.; Van Heijenoort, C.; Renault, L.; Carlier, M.F. Multifunctionality of the β-Thymosin/WH2 Module: G-Actin Sequestration, Actin Filament Growth, Nucleation, and Severing. Ann. N. Y. Acad. Sci. 2010, 1194, 44–52. [Google Scholar] [CrossRef]
- Elazari-Shalom, H.; Shaked, H.; Esteban-Martin, S.; Salvatella, X.; Barda-Saad, M.; Chill, J.H. New Insights into the Role of the Disordered WIP N-Terminal Domain Revealed by NMR Structural Characterization. FEBS J. 2015, 282, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Rozentur-Shkop, E.; Goobes, G.; Chill, J.H. A J-Modulated Protonless NMR Experiment Characterizes the Conformational Ensemble of the Intrinsically Disordered Protein WIP. J. Biomol. NMR 2016, 66, 243–257. [Google Scholar] [CrossRef] [PubMed]
- García, E.; Ragazzini, C.; Yu, X.; Cuesta-García, E.; Bernardino De La Serna, J.; Zech, T.; Sarrió, D.; Machesky, L.M.; Antón, I.M. WIP and WICH/WIRE Co-Ordinately Control Invadopodium Formation and Maturation in Human Breast Cancer Cell Invasion. Sci. Rep. 2016, 6, 23590. [Google Scholar] [CrossRef] [PubMed]
- Van Audenhove, I.; Boucherie, C.; Pieters, L.; Zwaenepoel, O.; Vanloo, B.; Martens, E.; Verbrugge, C.; Hassanzadeh-Ghassabeh, G.; Vandekerckhove, J.; Cornelissen, M.; et al. Stratifying Fascin and Cortactin Function in Invadopodium Formation Using Inhibitory Nanobodies and Targeted Subcellular Delocalization. FASEB J. 2014, 28, 1805–1818. [Google Scholar] [CrossRef]
- Bañón-Rodríguez, I.; Monypenny, J.; Ragazzini, C.; Franco, A.; Calle, Y.; Jones, G.E.; Antón, I.M. The Cortactin-Binding Domain of WIP Is Essential for Podosome Formation and Extracellular Matrix Degradation by Murine Dendritic Cells. Eur. J. Cell Biol. 2011, 90, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, J.K.; Feng, S.; Dalgarno, D.C.; Brauer, A.W.; Schreiber, S.L. Structural Basis for the Binding of Proline-Rich Peptides to SH3 Domains. Cell 1994, 76, 933–945. [Google Scholar] [CrossRef]
- Kurochkina, N.; Guha, U. SH3 Domains: Modules of Protein-Protein Interactions. Biophys. Rev. 2013, 5, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Saksela, K.; Permi, P. SH3 Domain Ligand Binding: What’s the Consensus and Where’s the Specificity? FEBS Lett. 2012, 586, 2609–2614. [Google Scholar] [CrossRef]
- Kay, B.K.; Williamson, M.P.; Sudol, M. The Importance of Being Proline: The Interaction of Proline-rich Motifs in Signaling Proteins with Their Cognate Domains. FASEB J. 2000, 14, 231–241. [Google Scholar] [CrossRef]
- Lee, C.H.; Leung, B.; Lemmon, M.A.; Zheng, J.; Cowburn, D.; Kuriyan, J.; Saksela, K. A Single Amino Acid in the SH3 Domain of Hck Determines Its High Affinity and Specificity in Binding to HIV-1 Nef Protein. EMBO J. 1995, 14, 5006–5015. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.C. Specificity and Versatility of SH3 and Other Proline-Recognition Domains: Structural Basis and Implications for Cellular Signal Transduction. Biochem. J. 2005, 390, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Dalgarno, D.C.; Botfield, M.C.; Rickles, R.J. SH3 Domains and Drug Design: Ligands, Structure, and Biological Function. Biopolymers 1997, 43, 383–400. [Google Scholar] [CrossRef]
- Mayer, B.J. SH3 Domains: Complexity in Moderation. J. Cell. Sci. 2001, 114, 1253–1263. [Google Scholar] [PubMed]
- Zarrinpar, A.; Bhattacharyya, R.P.; Lim, W.A. The Structure and Function of Proline Recognition Domains. Sci. STKE 2003, 2003, 1–10. [Google Scholar] [CrossRef]
- Teyra, J.; Huang, H.; Jain, S.; Guan, X.; Dong, A.; Liu, Y.; Tempel, W.; Min, J.; Tong, Y.; Kim, P.M.; et al. Comprehensive Analysis of the Human SH3 Domain Family Reveals a Wide Variety of Non-Canonical Specificities. Structure 2017, 25, 1598–1610. [Google Scholar] [CrossRef]
- Twafra, S.; Gil-Henn, H.; Dessau, M. PDB ID: 5NVJ. Available online: https://pdbj.org/emnavi/quick.php?id=pdb-5nvj (accessed on 21 July 2020).
- Schmidt, H.; Hoffmann, S.; Tran, T.; Stoldt, M.; Stangler, T.; Wiesehan, K.; Willbold, D. Solution Structure of a Hck SH3 Domain Ligand Complex Reveals Novel Interaction Modes. J. Mol. Biol. 2007, 365, 1517–1532. [Google Scholar] [CrossRef]
- Hake, M.J.; Choowongkomon, K.; Kostenko, O.; Carlin, C.R.; Sönnichsen, F.D. Specificity Determinants of a Novel Nck Interaction with the Juxtamembrane Domain of the Epidermal Growth Factor Receptor. Biochemistry 2008, 47, 3096–3108. [Google Scholar] [CrossRef]
- Jankowski, W.; Saleh, T.; Pai, M.T.; Sriram, G.; Birge, R.B.; Kalodimos, C.G. Domain Organization Differences Explain Bcr-Abl’s Preference for CrkL over CrkII. Nat. Chem. Biol. 2012, 8, 590–596. [Google Scholar] [CrossRef]
- Kessels, M.M.; Engqvist-Goldstein, Å.E.Y.; Drubin, D.G. Association of Mouse Actin-Binding Protein 1 (MAbp1/SH3P7), an Src Kinase Target, with Dynamic Regions of the Cortical Actin Cytoskeleton in Response to Rac1 Activation. Mol. Biol. Cell 2000, 11, 393–412. [Google Scholar] [CrossRef]
- Kinley, A.W.; Weed, S.A.; Weaver, A.M.; Karginov, A.V.; Bissonette, E.; Cooper, J.A.; Parsons, J.T. Cortactin Interacts with WIP in Regulating Arp2/3 Activation and Membrane Protrusion. Curr. Biol. 2003, 13, 384–393. [Google Scholar] [CrossRef]
- Cortesio, C.L.; Perrin, B.J.; Bennin, D.A.; Huttenlocher, A. Actin-Binding Protein-1 Interacts with WASp-Interacting Protein to Regulate Growth Factor-Induced Dorsal Ruffle Formation. Mol. Biol. Cell 2010, 21, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Klos Dehring, D.A.; Clarke, F.; Ricart, B.G.; Huang, Y.; Gomez, T.S.; Williamson, E.K.; Hammer, D.A.; Billadeau, D.D.; Argon, Y.; Burkhardt, J.K. Hematopoietic Lineage Cell-Specific Protein 1 Functions in Concert with the Wiskott–Aldrich Syndrome Protein To Promote Podosome Array Organization and Chemotaxis in Dendritic Cells. J. Immunol. 2011, 186, 4805–4818. [Google Scholar] [CrossRef] [PubMed]
- Gryaznova, T.; Kropyvko, S.; Burdyniuk, M.; Gubar, O.; Kryklyva, V.; Tsyba, L.; Rynditch, A. Intersectin Adaptor Proteins Are Associated with Actin-Regulating Protein WIP in Invadopodia. Cell. Signal. 2015, 27, 1499–1508. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.A.; Wilson, J.; Russo, A.; Wang, L.; Okur, M.N.; Wang, X.; Martin, N.P.; Scappini, E.; Carnegie, G.K.; O’Bryan, J.P. Intersectin (ITSN) Family of Scaffolds Function as Molecular Hubs in Protein Interaction Networks. PLoS ONE 2012, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M.; Salgia, R. Hematopoietic and BCR/ABL-Transformed Cells. Leukemia 1998, 12, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Antón, I.M.; Lu, W.; Mayer, B.J.; Ramesh, N.; Geha, R.S. The Wiskott-Aldrich Syndrome Protein-Interacting Protein (WIP) Binds to the Adaptor Protein Nck. J. Biol. Chem. 1998, 273, 20992–20995. [Google Scholar] [CrossRef]
- Zhao, Z.-S.; Manser, E.; Lim, L. Interaction between PAK and Nck: A Template for Nck Targets and Role of PAK Autophosphorylation. Mol. Cell. Biol. 2000, 20, 3906–3917. [Google Scholar] [CrossRef]
- Gryaznova, T.; Gubar, O.; Burdyniuk, M.; Kropyvko, S.; Rynditch, A. WIP/ITSN1 Complex Is Involved in Cellular Vesicle Trafficking and Formation of Filopodia-like Protrusions. Gene 2018, 674, 49–56. [Google Scholar] [CrossRef]
- Derry, J.M.; Ochs, H.D.; Francke, U. Isolation of a Novel Gene Mutated in Wiskott-Aldrich Syndrome. Cell 1994, 79, 635–644. [Google Scholar] [CrossRef]
- Schindelhauer, D.; Weiss, M.; Hellebrand, H.; Golla, A.; Hergersberg, M.; Seger, R.; Belohradsky, B.H.; Meindl, A. Wiskott-Aldrich Syndrome: No Strict Genotype-Phenotype Correlations but Clustering of Missense Mutations in the Amino-Terminal Part of the WASP Gene Product. Hum. Genet. 1996, 98, 68–76. [Google Scholar] [CrossRef]
- Stewart, D.M.; Tian, L.; Nelson, D.L. Mutations That Cause the Wiskott-Aldrich Syndrome Impair the Interaction of Wiskott-Aldrich Syndrome Protein (WASp) with WASp Interacting Protein. J. Immunol. 1999, 162, 5019–5024. [Google Scholar] [PubMed]
- Peterson, F.C.; Deng, Q.; Zettl, M.; Prehoda, K.E.; Lim, W.A.; Way, M.; Volkman, B.F. Multiple WASp-Interacting Protein Recognition Motifs Are Required for a Functional Interaction with N-WASp. J. Biol. Chem. 2007, 282, 8446–8453. [Google Scholar] [CrossRef] [PubMed]
- Zettl, M.; Way, M. The WH1 and EVH1 Domains of WASp and Ena/VASP Family Members Bind Distinct Sequence Motifs. Curr. Biol. 2002, 12, 1617–1622. [Google Scholar] [CrossRef][Green Version]
- Haba, N.Y.; Gross, R.; Novacek, J.; Shaked, H.; Zidek, L.; Barda-Saad, M.; Chill, J.H. NMR Determines Transient Structure and Dynamics in the Disordered C-Terminal Domain of WASp Interacting Protein. Biophys. J. 2013, 105, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Halle-Bikovski, A.; Fried, S.; Rozentur-Shkop, E.; Biber, G.; Shaked, H.; Joseph, N.; Barda-Saad, M.; Chill, J.H. New Structural Insights into Formation of the Key Actin Regulating WIP-WASp Complex Determined by NMR and Molecular Imaging. ACS Chem. Biol. 2018, 13, 100–109. [Google Scholar] [CrossRef]
- Ferguson, K.M.; Lemmon, M.A.; Schlessinger, J.; Sigler, P.B. Structure of the High Affinity Complex of Inositol Trisphosphate with a Phospholipase C Pleckstrin Homology Domain. Cell 1995, 83, 1037–1046. [Google Scholar] [CrossRef]
- Li, S.C.; Zwahlen, C.; Vincent, S.J.F.; Jane McGlade, C.; Kay, L.E.; Pawson, T.; Forman-Kay, J.D. Structure of a Numb PTB Domain-Peptide Complex Suggests a Basis for Diverse Binding Specificity. Nat. Struct. Biol. 1998, 5, 1075–1083. [Google Scholar] [CrossRef]
- Luthi, J.N.; Gandhi, M.J.; Drachman, J.G. X-Linked Thrombocytopenia Caused by a Mutation in the Wiskott-Aldrich Syndrome (WAS) Gene That Disrupts Interaction with the WAS Protein (WASp)-Interacting Protein (WIP). Exp. Hematol. 2003, 31, 150–158. [Google Scholar] [CrossRef]
- Reicher, B.; Joseph, N.; David, A.; Pauker, M.H.; Perl, O.; Barda-Saad, M. Ubiquitylation-Dependent Negative Regulation of WASp Is Essential for Actin Cytoskeleton Dynamics. Mol. Cell. Biol. 2012, 32, 3153–3163. [Google Scholar] [CrossRef]
- Fried, S.; Reicher, B.; Pauker, M.H.; Eliyahu, S.; Matalon, O.; Noy, E.; Chill, J.; Barda-Saad, M. Triple-Color FRET Analysis Reveals Conformational Changes in the WIP-WASp Actin-Regulating Complex. Sci. Signal. 2014, 7, ra60. [Google Scholar] [CrossRef]
- Peterson, F.C.; Volkman, B.F. Diversity of Polyproline Recognition by EVH1 Domains. Front. Biosci. 2009, 14, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, V.; Monypenny, J.; Chen, X.J.; Machesky, L.M.; Lilla, S.; Thrasher, A.J.; Antón, I.M.; Calle, Y.; Jones, G.E. Tyrosine Phosphorylation of WIP Releases Bound WASp and Impairs Podosome Assembly in Macrophages. J. Cell Sci. 2015, 128, 251–265. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Partner | WIP Segment | Binding Motif | Effect | Ref. |
|---|---|---|---|---|
| Cortactin SH3 (NPF) | 136–205 | Not determined (ND) | Increases cortactin’s activation of the Arp2/3 complex, cortactin recruits WIP in invadopodium formation | [75,92] |
| mAbp1 SH3 (adaptor) | 110–170 | ND | Regulates dorsal ruffle formation | [93] |
| ITSN1-S/ ITSN1-L 1st/3rd/5th of 5 SH3 domains (adaptor) | 318–450 | ND | enhances association of ITSN1 with N-WASp and β-actin, facilitates formation of filopodia-like protrusions, regulates intra-cellular vesicle trafficking | [95,100] |
| ITSN2 1st/3rd/5th of 5 (adaptor) | 13–450 | ND | [95,96] | |
| CrkL 1st SH3 of 2 (adaptor) | 321–415 | 332PRLPQR337 (class 2) 399PQLPSR404 (class 2) (comply with Crk SH3.1 consensus binding motif PxLPxK/R) | Presumably preformed CrkL-WIP-WASp complex associates with phos-ZAP70 after T cell receptor (TCR) ligation | [15] |
| Nck-1 2nd SH3 of 3 (adaptor) | 247–261 328–342 | 247SNRPPLPPTPSRALD261 328NDETPRLPQRNLSLS342 (both class 2, 328–342 complies with the consensus motif for Nck SH3.2 PxxPxRxxS) | Couples extracellular signals to cytoskeleton assembly system | [26,98,99] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sokolik, C.G.; Qassem, N.; Chill, J.H. The Disordered Cellular Multi-Tasker WIP and Its Protein–Protein Interactions: A Structural View. Biomolecules 2020, 10, 1084. https://doi.org/10.3390/biom10071084
Sokolik CG, Qassem N, Chill JH. The Disordered Cellular Multi-Tasker WIP and Its Protein–Protein Interactions: A Structural View. Biomolecules. 2020; 10(7):1084. https://doi.org/10.3390/biom10071084
Chicago/Turabian StyleSokolik, Chana G., Nasrin Qassem, and Jordan H. Chill. 2020. "The Disordered Cellular Multi-Tasker WIP and Its Protein–Protein Interactions: A Structural View" Biomolecules 10, no. 7: 1084. https://doi.org/10.3390/biom10071084
APA StyleSokolik, C. G., Qassem, N., & Chill, J. H. (2020). The Disordered Cellular Multi-Tasker WIP and Its Protein–Protein Interactions: A Structural View. Biomolecules, 10(7), 1084. https://doi.org/10.3390/biom10071084