Nickel and GTP Modulate Helicobacter pylori UreG Structural Flexibility

 , , ,
, , ,  ,
, 
 and
and
Abstract
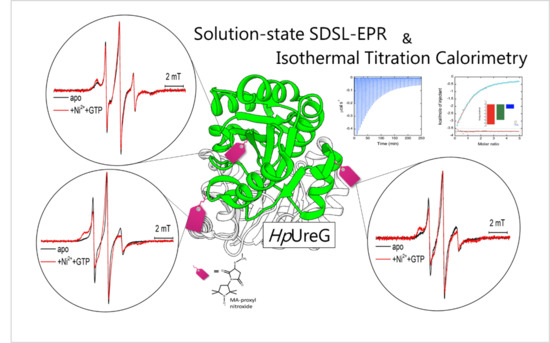
1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
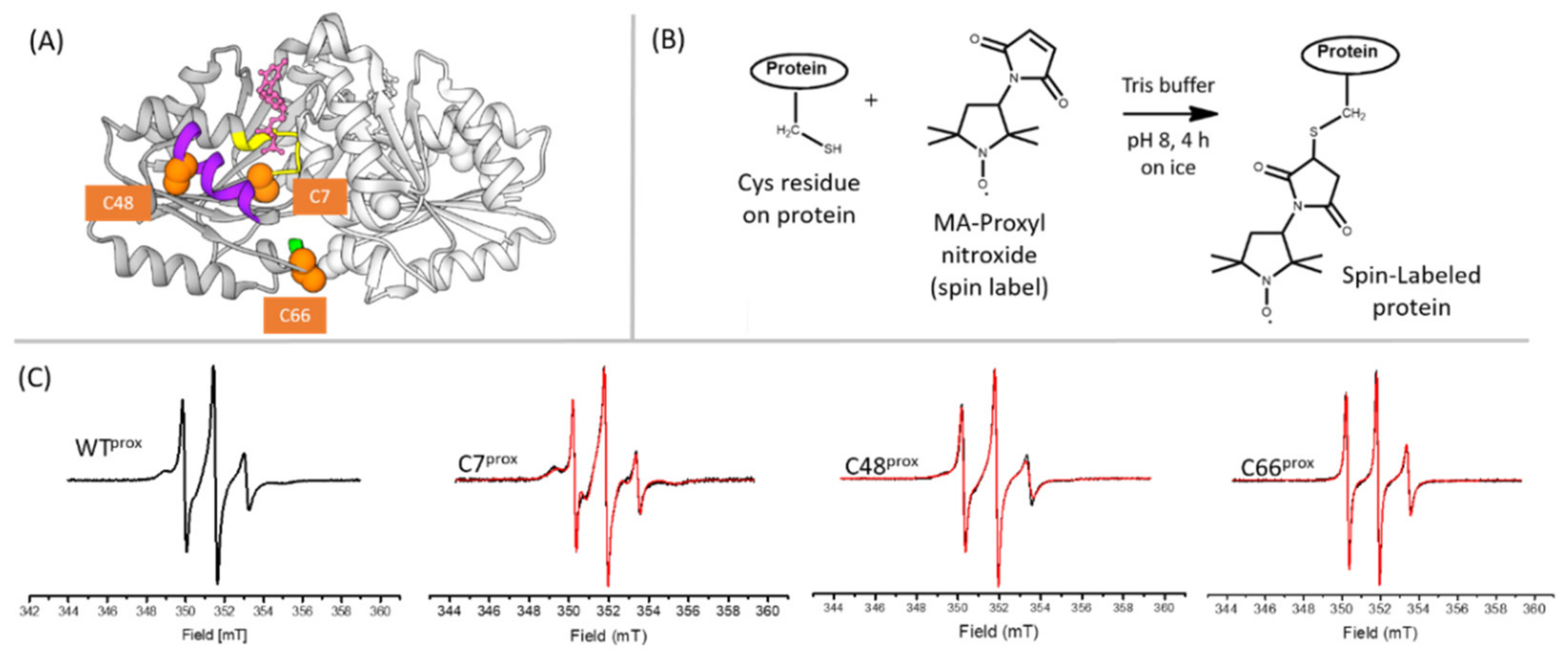
2.2. Isothermal Titration Calorimetry
2.3. HpUreG Mutants Design
2.4. GTP Hydrolase Activity Assays
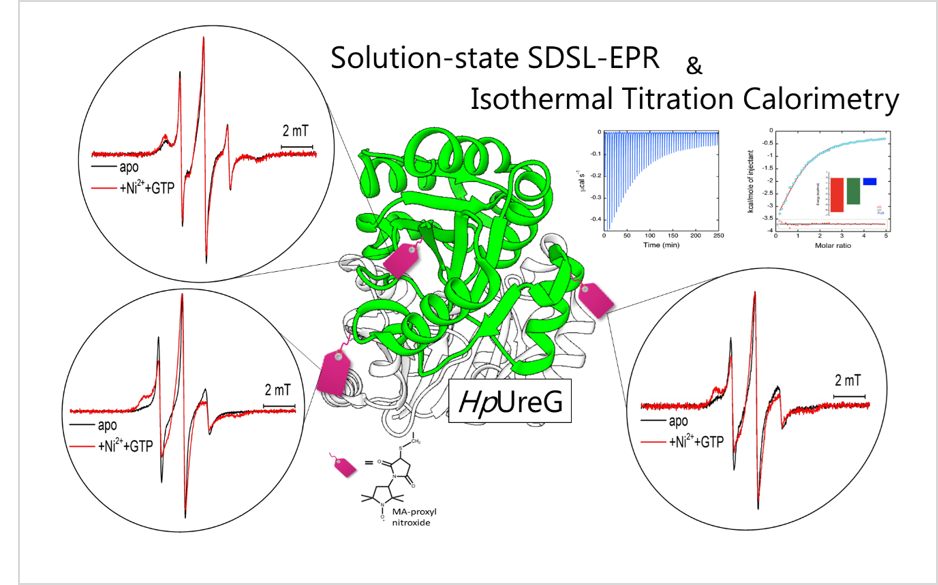
2.5. Protein Labeling with Nitroxide Spin Label
2.6. EPR Spectroscopy
2.7. DEER Measurements
3. Results and Discussion
3.1. Cys Variants Were Generated to Selectively Label Distinct Regions of HpUreG
3.2. The Thermodynamics of Ni(II) and GTP-Driven Dimerization of HpUreG Was Characterized
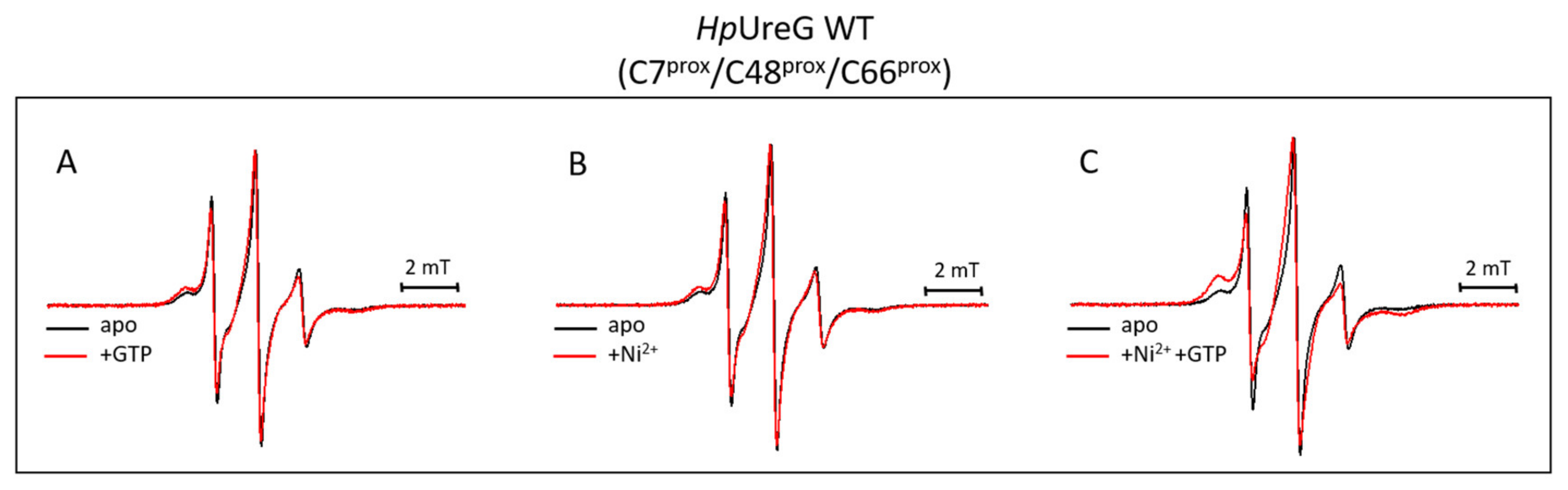
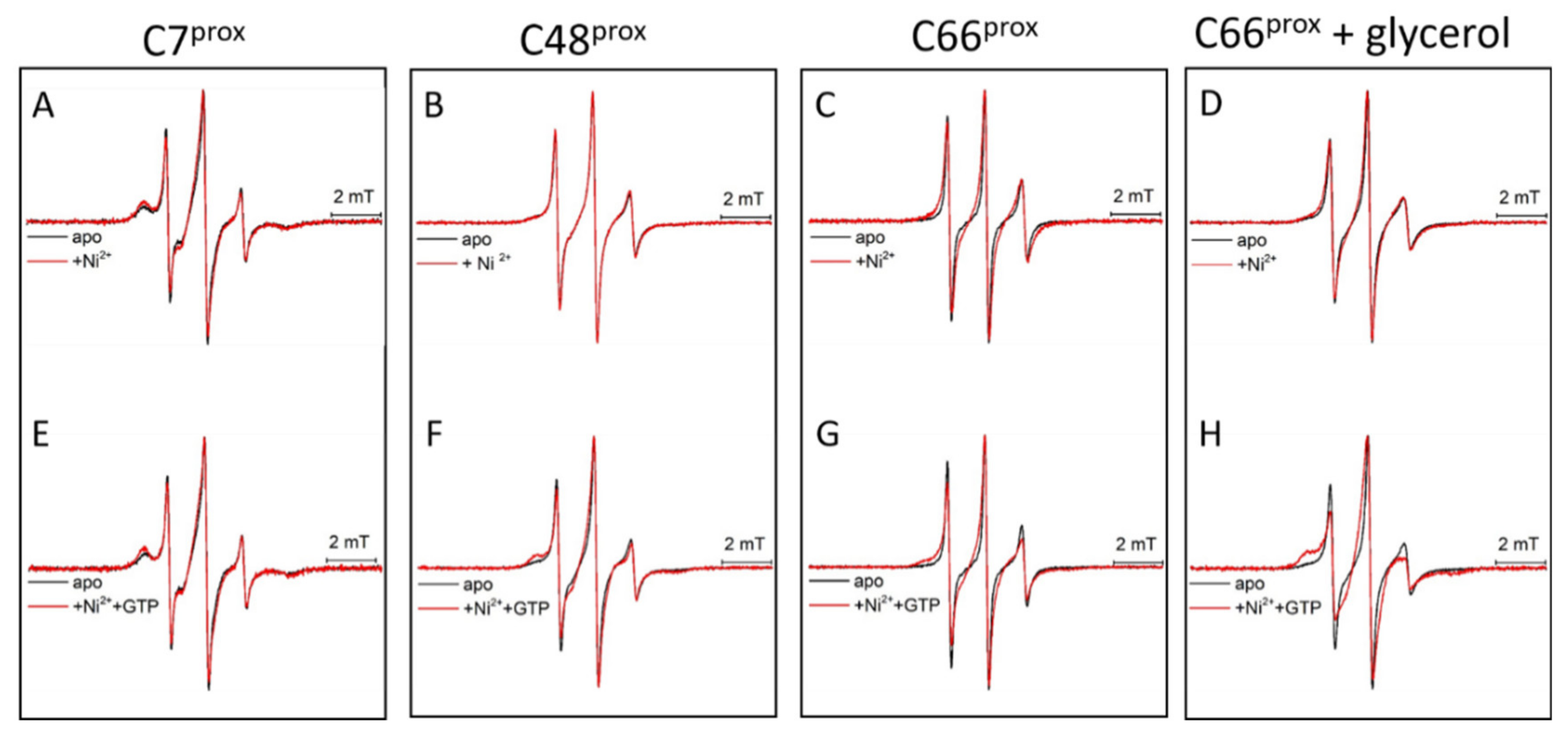
3.3. HpUreG Shows Distinct Flexibility in Different Protein Regions
3.4. Ni(II) Ions and GTP Binding Produce Changes in the Structural Dynamics of Different Protein Regions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.; Wertheim, H.F.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic resistance-the need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef]
- Tornimbene, B.; Eremin, S.; Escher, M.; Griskeviciene, J.; Manglani, S.; Pessoa-Silva, C.L. WHO Global Antimicrobial Resistance Surveillance System early implementation 2016–17. Lancet Infect. Dis. 2018, 18, 241–242. [Google Scholar] [CrossRef]
- Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. Available online: https://www.who.int/medicines/publications/global-priority-list-antibiotic-resistant-bacteria/en/ (accessed on 27 February 2017).
- Zambelli, B.; Musiani, F.; Benini, S.; Ciurli, S. Chemistry of Ni2+ in urease: Sensing, trafficking, and catalysis. Acc. Chem. Res. 2011, 44, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Beenken, K.E.; Dunman, P.M.; McAleese, F.; Macapagal, D.; Murphy, E.; Projan, S.J.; Blevins, J.S.; Smeltzer, M.S. Global gene expression in Staphylococcus aureus biofilms. J. Bacteriol. 2004, 186, 4665–4684. [Google Scholar] [CrossRef]
- Zhou, C.; Bhinderwala, F.; Lehman, M.K.; Thomas, V.C.; Chaudhari, S.S.; Yamada, K.J.; Foster, K.W.; Powers, R.; Kielian, T.; Fey, P.D. Urease is an essential component of the acid response network of Staphylococcus aureus and is required for a persistent murine kidney infection. PLoS Pathog. 2019, 15, e1007538. [Google Scholar] [CrossRef]
- Jones, B.D.; Lockatell, C.V.; Johnson, D.E.; Warren, J.W.; Mobley, H.L. Construction of a urease-negative mutant of Proteus mirabilis: Analysis of virulence in a mouse model of ascending urinary tract infection. Infect. Immun. 1990, 58, 1120–1123. [Google Scholar] [CrossRef]
- Gatermann, S.; Marre, R. Cloning and expression of Staphylococcus saprophyticus urease gene sequences in Staphylococcus carnosus and contribution of the enzyme to virulence. Infect. Immun. 1989, 57, 2998–3002. [Google Scholar] [CrossRef]
- Ligon, J.V.; Kenny, G.E. Virulence of ureaplasmal urease for mice. Infect. Immun. 1991, 59, 1170–1171. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Testerman, T.L.; Morris, J. Beyond the stomach: An updated view of Helicobacter pylori pathogenesis, diagnosis, and treatment. W. J. Gastroenterol. 2014, 20, 12781–12808. [Google Scholar] [CrossRef]
- Eaton, K.A.; Brooks, C.L.; Morgan, D.R.; Krakowka, S. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect. Immun. 1991, 59, 2470–2475. [Google Scholar] [CrossRef]
- D’Elios, M.M.; Czinn, S.J. Immunity, inflammation, and vaccines for Helicobacter pylori. Helicobacter 2014, 19, 19–26. [Google Scholar] [CrossRef]
- Maroney, M.J.; Ciurli, S. Nonredox nickel enzymes. Chem. Rev. 2014, 114, 4206–4228. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, L.; Musiani, F.; Ciurli, S. CHAPTER 5 Urease. In The Biological Chemistry of Nickel; The Royal Society of Chemistry: London, UK, 2017; pp. 60–97. [Google Scholar]
- Ha, N.-C.; Oh, S.-T.; Sung, J.Y.; Cha, K.A.; Lee, M.H.; Oh, B.-H. Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat. Struct. Biol. 2001, 8, 505–509. [Google Scholar] [CrossRef]
- Tarsia, C.; Danielli, A.; Florini, F.; Cinelli, P.; Ciurli, S.; Zambelli, B. Targeting Helicobacter pylori urease activity and maturation: In-cell high-throughput approach for drug discovery. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2245–2253. [Google Scholar] [CrossRef]
- Bellucci, M.; Zambelli, B.; Musiani, F.; Turano, P.; Ciurli, S. Helicobacter pylori UreE, a urease accessory protein: Specific Ni(2+)- and Zn(2+)-binding properties and interaction with its cognate UreG. Biochem. J. 2009, 422, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Musiani, F.; Gioia, D.; Masetti, M.; Falchi, F.; Cavalli, A.; Recanatini, M.; Ciurli, S. Protein Tunnels: The Case of Urease Accessory Proteins. J. Chem. Theory Computat. 2017, 13, 2322–2331. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, B.; Turano, P.; Musiani, F.; Neyroz, P.; Ciurli, S. Zn2+-linked dimerization of UreG from Helicobacter pylori, a chaperone involved in nickel trafficking and urease activation. Proteins Struct. Funct. Bioinform. 2009, 74, 222–239. [Google Scholar] [CrossRef]
- Fong, Y.H.; Wong, H.C.; Yuen, M.H.; Lau, P.H.; Chen, Y.W.; Wong, K.-B. Structure of UreG/UreF/UreH Complex Reveals How Urease Accessory Proteins Facilitate Maturation of Helicobacter pylori Urease. PLoS Biol. 2013, 11, e1001678. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.H.; Fong, Y.H.; Nim, Y.S.; Lau, P.H.; Wong, K.-B. Structural insights into how GTP-dependent conformational changes in a metallochaperone UreG facilitate urease maturation. Proc. Natl. Acad. Sci. USA 2017, 114, E10890–E10898. [Google Scholar] [CrossRef] [PubMed]
- Miraula, M.; Ciurli, S.; Zambelli, B. Intrinsic disorder and metal binding in UreG proteins from Archae hyperthermophiles: GTPase enzymes involved in the activation of Ni(II) dependent urease. J. Biol. Inorg. Chem. 2015, 20, 739–755. [Google Scholar] [CrossRef] [PubMed]
- Real-Guerra, R.; Staniscuaski, F.; Zambelli, B.; Musiani, F.; Ciurli, S.; Carlini, C.R. Biochemical and structural studies on native and recombinant Glycine max UreG: A detailed characterization of a plant urease accessory protein. Plant Mol. Biol. 2012, 78, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, B.; Musiani, F.; Savini, M.; Tucker, P.; Ciurli, S. Biochemical studies on Mycobacterium tuberculosis UreG and comparative modeling reveal structural and functional conservation among the bacterial UreG family. Biochemistry 2007, 46, 3171–3182. [Google Scholar] [CrossRef]
- Zambelli, B.; Stola, M.; Musiani, F.; De Vriendt, K.; Samyn, B.; Devreese, B.; Van Beeumen, J.; Turano, P.; Dikiy, A.; Bryant, D.A.; et al. UreG, a chaperone in the urease assembly process, is an intrinsically unstructured GTPase that specifically binds Zn2+. J. Biol. Chem. 2005, 280, 4684–4695. [Google Scholar] [CrossRef] [PubMed]
- Palombo, M.; Bonucci, A.; Etienne, E.; Ciurli, S.; Uversky, V.N.; Guigliarelli, B.; Belle, V.; Mileo, E.; Zambelli, B. The relationship between folding and activity in UreG, an intrinsically disordered enzyme. Sci. Rep. 2017, 7, 5977. [Google Scholar] [CrossRef]
- D’Urzo, A.; Santambrogio, C.; Grandori, R.; Ciurli, S.; Zambelli, B. The conformational response to Zn(II) and Ni(II) binding of Sporosarcina pasteurii UreG, an intrinsically disordered GTPase. J. Biol. Inorg. Chem. 2014, 19, 1341–1354. [Google Scholar] [CrossRef]
- Yang, X.; Li, H.; Lai, T.-P.; Sun, H. UreE-UreG Complex Facilitates Nickel Transfer and Preactivates GTPase of UreG in Helicobacter pylori. J. Biol. Chem. 2015, 290, 12474–12485. [Google Scholar] [CrossRef] [PubMed]
- Merloni, A.; Dobrovolska, O.; Zambelli, B.; Agostini, F.; Bazzani, M.; Musiani, F.; Ciurli, S. Molecular landscape of the interaction between the urease accessory proteins UreE and UreG. Biochim. Biophys. Acta 2014, 1844, 1662–1674. [Google Scholar] [CrossRef]
- Etienne, E.; Le Breton, N.; Martinho, M.; Mileo, E.; Belle, V. SimLabel: A graphical user interface to simulate continuous wave EPR spectra from site-directed spin labeling experiments. Magn. Reson. Chem. MRC 2017, 55, 714–719. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reason. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Pannier, M.; Veit, S.; Godt, A.; Jeschke, G.; Spiess, H.W. Dead-Time Free Measurement of Dipole–Dipole Interactions between Electron Spins. J. Magn. Reason. 2000, 142, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, G. MMM: A toolbox for integrative structure modeling. Protein Sci. Publ. Protein Soc. 2018, 27, 76–85. [Google Scholar] [CrossRef]
- Jeschke, G. The contribution of modern EPR to structural biology. Emerg. Topics Life Sci. 2018. [Google Scholar] [CrossRef]
- Bordignon, E. EPR Spectroscopy of Nitroxide Spin Probes. In eMagRes; Harris, R.K., Wasylishen, R.L., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2017. [Google Scholar]
- Roser, P.; Schmidt, M.J.; Drescher, M.; Summerer, D. Site-directed spin labeling of proteins for distance measurements in vitro and in cells. Organ. Biomol. Chem. 2016, 14, 5468–5476. [Google Scholar] [CrossRef] [PubMed]
- Potapov, A.; Yagi, H.; Huber, T.; Jergic, S.; Dixon, N.E.; Otting, G.; Goldfarb, D. Nanometer-Scale Distance Measurements in Proteins Using Gd3+ Spin Labeling. J. Am. Chem. Soc. 2010, 132, 9040–9048. [Google Scholar] [CrossRef]
- Bonucci, A.; Ouari, O.; Guigliarelli, B.; Belle, V.; Mileo, E. In-Cell EPR: Progress towards Structural Studies Inside Cells. Chem. Bio. Chem. 2020, 21, 451–460. [Google Scholar] [CrossRef]
- Hubbell, W.L.; McHaourab, H.S.; Altenbach, C.; Lietzow, M.A. Watching proteins move using site-directed spin labeling. Structure 1996, 4, 779–783. [Google Scholar] [CrossRef]
- Hubbell, W.L.; Cafiso, D.S.; Altenbach, C. Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 2000, 7, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Hubbell, W.L.; Lopez, C.J.; Altenbach, C.; Yang, Z. Technological advances in site-directed spin labeling of proteins. Curr. Opin. Struct. Biol. 2013, 23, 725–733. [Google Scholar] [CrossRef]
- Le Breton, N.; Martinho, M.; Mileo, E.; Etienne, E.; Gerbaud, G.; Guigliarelli, B.; Belle, V. Exploring intrinsically disordered proteins using site-directed spin labeling electron paramagnetic resonance spectroscopy. Front. Mol. Biosci. 2015, 2, 21. [Google Scholar] [CrossRef]
- Belle, V.; Rouger, S.; Costanzo, S.; Liquiere, E.; Strancar, J.; Guigliarelli, B.; Fournel, A.; Longhi, S. Mapping alpha-helical induced folding within the intrinsically disordered C-terminal domain of the measles virus nucleoprotein by site-directed spin-labeling EPR spectroscopy. Proteins 2008, 73, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Martinho, M.; Fournier, E.; Le Breton, N.; Mileo, E.; Belle, V. Nitroxide spin labels: Fabulous spy spins for biostructural EPR applications. In Electron Paramagnetic Resonance: Volume 26; The Royal Society of Chemistry: London, UK, 2019; Volume 26, pp. 66–88. [Google Scholar]
- Jeschke, G. DEER distance measurements on proteins. Ann. Rev. Phys. Chem. 2012, 63, 419–446. [Google Scholar] [CrossRef] [PubMed]
- Schiemann, O.; Prisner, T.F. Long-range distance determinations in biomacromolecules by EPR spectroscopy. Quart. Rev. Biophys. 2007, 40, 1–53. [Google Scholar] [CrossRef]
- Schmidt, T.; Walti, M.A.; Baber, J.L.; Hustedt, E.J.; Clore, G.M. Long Distance Measurements up to 160 A in the GroEL Tetradecamer Using Q-Band DEER EPR Spectroscopy. Angew. Chem. Int. Ed. Engl. 2016, 55, 15905–15909. [Google Scholar] [CrossRef]
- Pineiro, A.; Munoz, E.; Sabin, J.; Costas, M.; Bastos, M.; Velazquez-Campoy, A.; Garrido, P.F.; Dumas, P.; Ennifar, E.; Garcia-Rio, L.; et al. AFFINImeter: A software to analyze molecular recognition processes from experimental data. Anal. Biochem. 2019, 577, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, B.; Bellucci, M.; Danielli, A.; Scarlato, V.; Ciurli, S. The Ni2+ binding properties of Helicobacter pylori NikR. Chem. Commun. 2007, 21, 3649–3651. [Google Scholar] [CrossRef]
- López, C.J.; Fleissner, M.R.; Guo, Z.; Kusnetzow, A.K.; Hubbell, W.L. Osmolyte perturbation reveals conformational equilibria in spin-labeled proteins. Protein Sci. Publ. Protein Soc. 2009, 18, 1637–1652. [Google Scholar] [CrossRef]
- Flores Jiménez, R.H.; Do Cao, M.A.; Kim, M.; Cafiso, D.S. Osmolytes modulate conformational exchange in solvent-exposed regions of membrane proteins. Protein Sci. Publ. Protein Soc. 2010, 19, 269–278. [Google Scholar] [CrossRef]
- Bolen, D.W. Effects of naturally occurring osmolytes on protein stability and solubility: Issues important in protein crystallization. Methods 2004, 34, 312–322. [Google Scholar] [CrossRef]
- Mileo, E.; Lorenzi, M.; Erales, J.; Lignon, S.; Puppo, C.; Le Breton, N.; Etienne, E.; Marque, S.R.; Guigliarelli, B.; Gontero, B.; et al. Dynamics of the intrinsically disordered protein CP12 in its association with GAPDH in the green alga Chlamydomonas reinhardtii: A fuzzy complex. Mol. Biosyst. 2013, 9, 2869–2876. [Google Scholar] [CrossRef]
- Musiani, F.; Ippoliti, E.; Micheletti, C.; Carloni, P.; Ciurli, S. Conformational fluctuations of UreG, an intrinsically disordered enzyme. Biochemistry 2013, 52, 2949–2954. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, G.; Chechik, V.; Ionita, P.; Godt, A.; Zimmermann, H.; Banham, J.E.; Timmel, C.R.; Hilger, D.; Jung, H. DeerAnalysis2006—A Comprehensive Software Package for Analyzing Pulsed ELDOR Data. Appl. Magn. Reson. 2006, 30, 473–498. [Google Scholar] [CrossRef]
- Sarver, J.L.; Zhang, M.; Liu, L.; Nyenhuis, D.; Cafiso, D.S. A Dynamic Protein-Protein Coupling between the TonB-Dependent Transporter FhuA and TonB. Biochemistry 2018, 57, 1045–1053. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample in the ITC Cell | N | Ka (M−1) | Kd (µM) | ΔH (kcal mol−1) | ΔS (cal mol−1K-1) |
|---|---|---|---|---|---|
| HpUreG | 2 | 1.38 ± 0.02 × 104 | 72 ± 1 | −5.90 ± 0.06 | −0.86 |
| C66A-HpUreG | 1 | 4.23 ± 0.02 × 103 | 236 ± 1 | −5.52 ± 0.01 | −1.93 |
| WTprox-HpUreG | 1 | 4.33 ± 0.04 × 104 | 23.0 ± 0.3 | −4.99 ± 0.02 | 4.47 |
| HpUreG-GTPγS | HpUreG-Ni | 3.45 ± 0.02 × 104 | 30.0 ± 0.2 | −7.14 ± 0.06 | −4.99 |
| HpUreG2-Ni | 1.40 ± 0.04 × 104 | 71 ± 2 | +11.2 ± 0.3 | +56.77 | |
| C66AHpUreG-GTPγS | C66AHpUreG-Ni | 6.1 ± 0.8 × 103 | 160 ± 20 | −61 ± 7 | −187 |
| C66AHpUreG2-Ni | 4.5 ± 0.7 × 104 | 22 ± 3 | +99 ± 9 | +353 | |
| WTprox-HpUreG-GTPγS | WTProx-HpUreG-Ni | 4.20 ± 0.04 × 104 | 23.8 ± 0.2 | −2.63 ± 0.03 | +12.3 |
| WTProx-HpUreG2-Ni | 3.8 ± 0.3 × 103 | 260 ± 20 | +9.1 ± 0.5 | +46.8 |
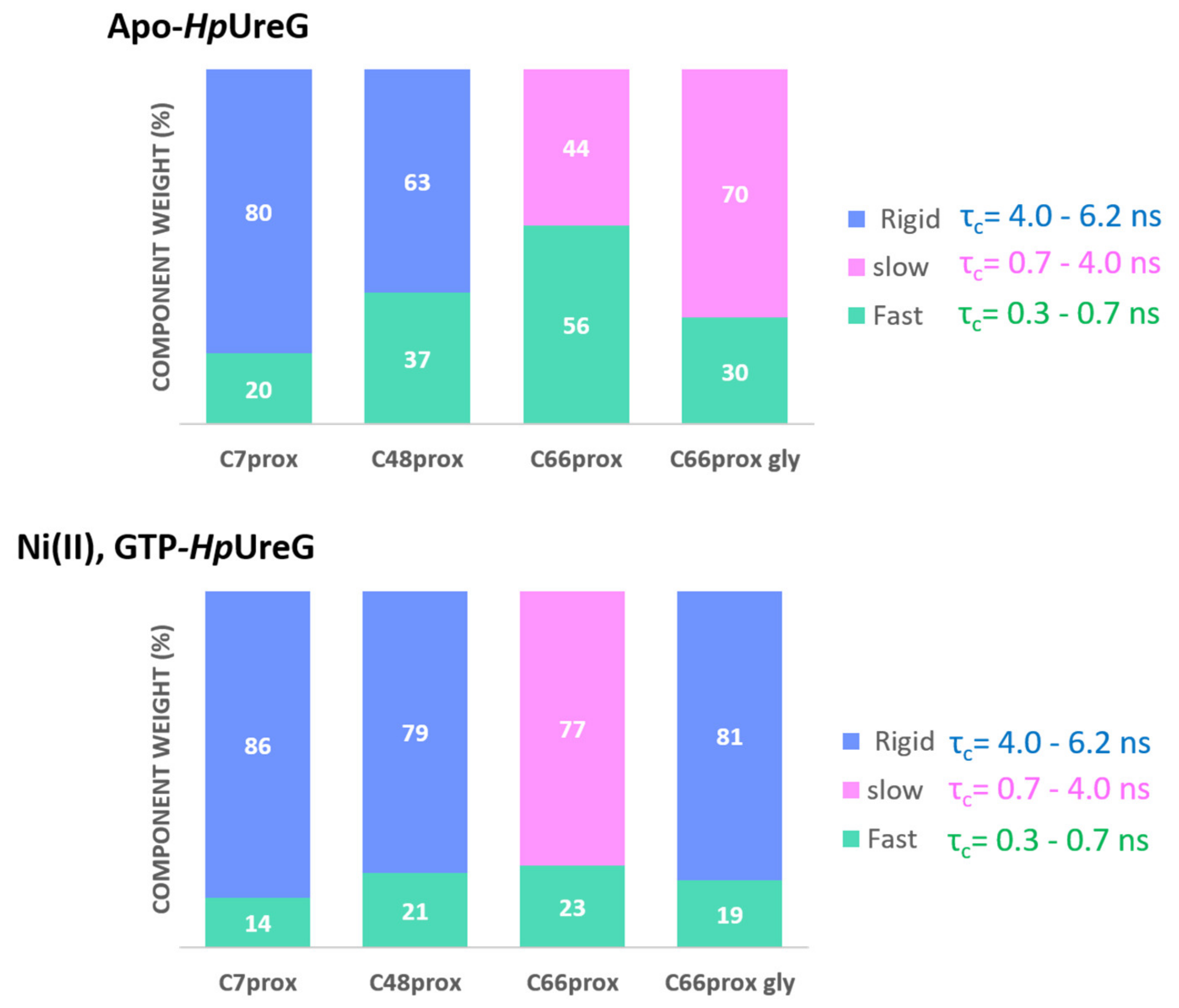
| Apo-Form | +Ni(II) | +Ni(II) and GTP | |||||
|---|---|---|---|---|---|---|---|
| Labeled Site | Component | Weight % | τc (ns) | Weight % | τc (ns) | Weight % | τc (ns) |
| C66prox | Fast | 56 | 0.6 | 34 | 0.6 | 28 | 0.7 |
| Slow | 44 | 2.4 | 66 | 2.4 | 72 | 3.4 | |
| C66prox | Fast | 30 | 0.6 | 22 | 0.6 | 19 | 0.8 |
| +gly 30% | Slow/Rigid * | 70 | 2.4 | 78 | 2.4 | 81 * | 4.4 * |
| C48prox | Fast | 37 | 0.6 | 39 | 0.6 | 21 | 0.6 |
| Rigid | 63 | 4.9 | 61 | 4.9 | 79 | 4.9 | |
| C7prox | Fast | 20 | 0.3 | 15 | 0.3 | 14 | 0.3 |
| Rigid | 80 | 6.1 | 85 | 6.1 | 86 | 6.1 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pierro, A.; Etienne, E.; Gerbaud, G.; Guigliarelli, B.; Ciurli, S.; Belle, V.; Zambelli, B.; Mileo, E. Nickel and GTP Modulate Helicobacter pylori UreG Structural Flexibility. Biomolecules 2020, 10, 1062. https://doi.org/10.3390/biom10071062
Pierro A, Etienne E, Gerbaud G, Guigliarelli B, Ciurli S, Belle V, Zambelli B, Mileo E. Nickel and GTP Modulate Helicobacter pylori UreG Structural Flexibility. Biomolecules. 2020; 10(7):1062. https://doi.org/10.3390/biom10071062
Chicago/Turabian StylePierro, Annalisa, Emilien Etienne, Guillaume Gerbaud, Bruno Guigliarelli, Stefano Ciurli, Valérie Belle, Barbara Zambelli, and Elisabetta Mileo. 2020. "Nickel and GTP Modulate Helicobacter pylori UreG Structural Flexibility" Biomolecules 10, no. 7: 1062. https://doi.org/10.3390/biom10071062
APA StylePierro, A., Etienne, E., Gerbaud, G., Guigliarelli, B., Ciurli, S., Belle, V., Zambelli, B., & Mileo, E. (2020). Nickel and GTP Modulate Helicobacter pylori UreG Structural Flexibility. Biomolecules, 10(7), 1062. https://doi.org/10.3390/biom10071062