Structures of FOX-4 Cephamycinase in Complex with Transition-State Analog Inhibitors

 ,
, 

Abstract
1. Introduction
2. Materials and Methods
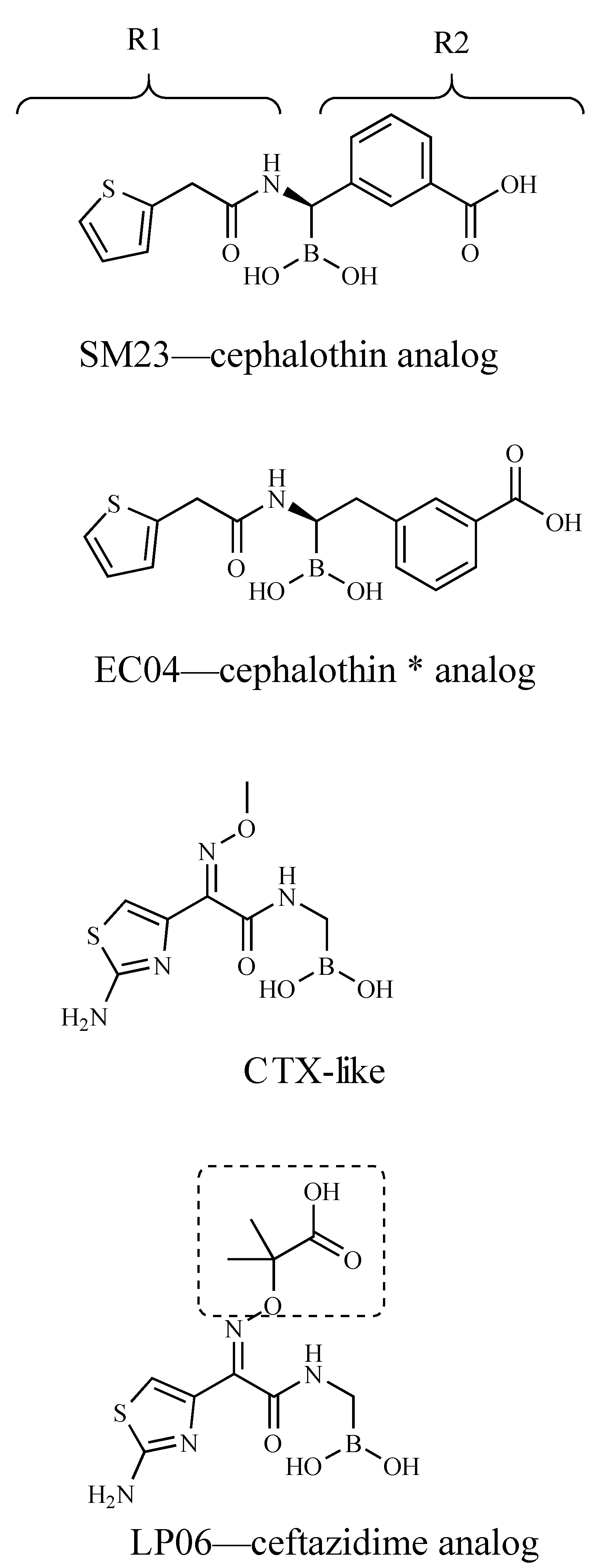
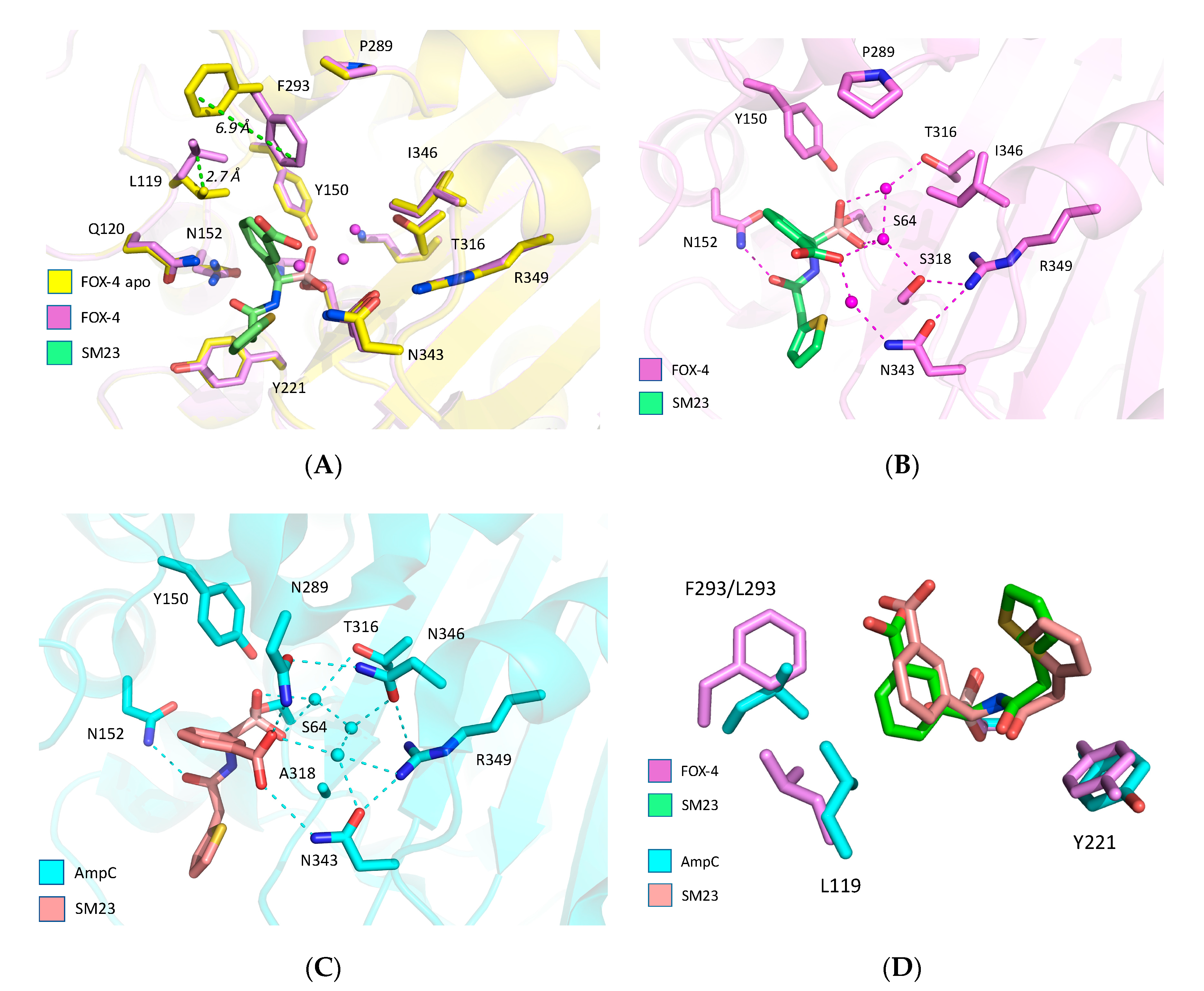
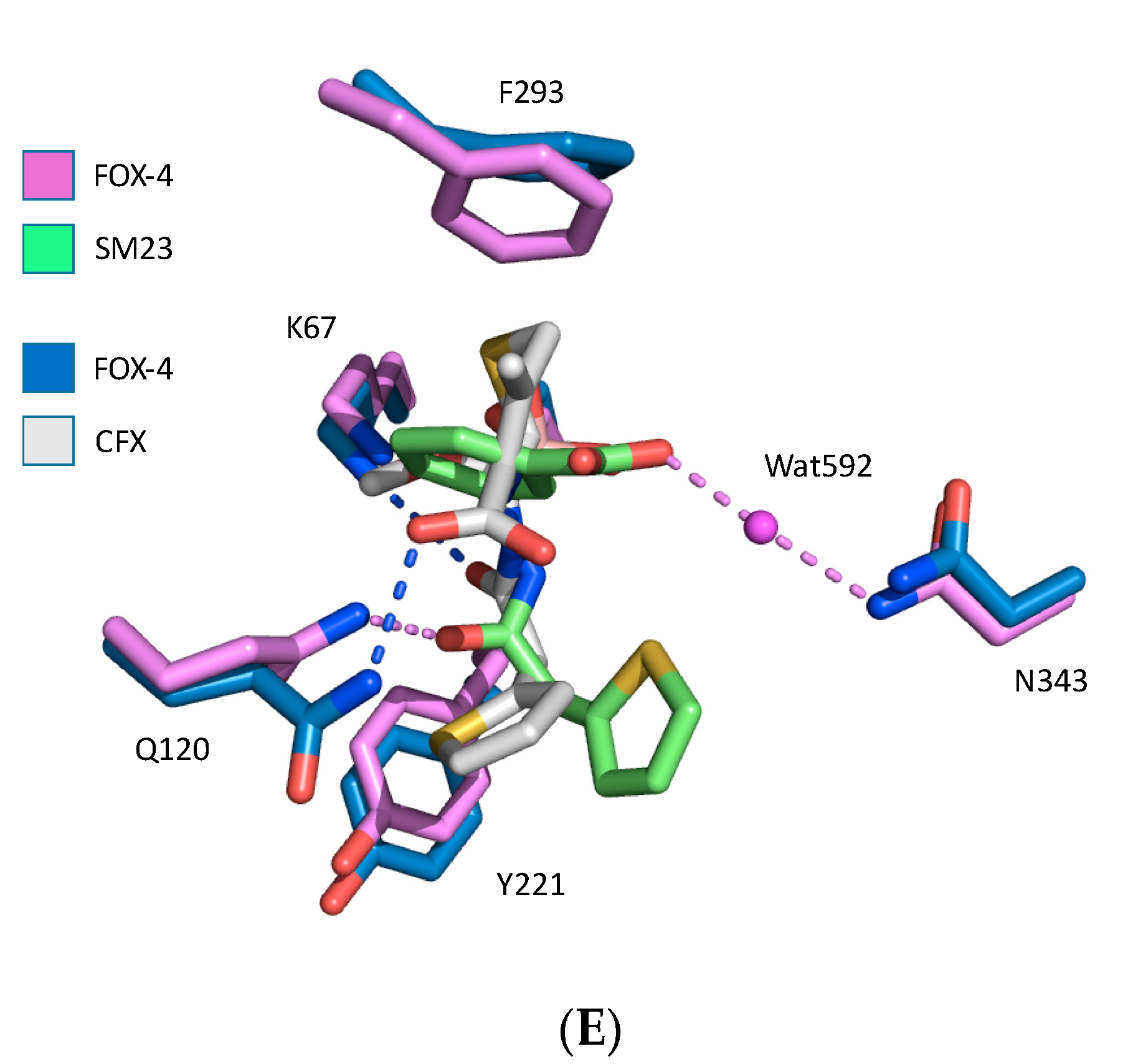
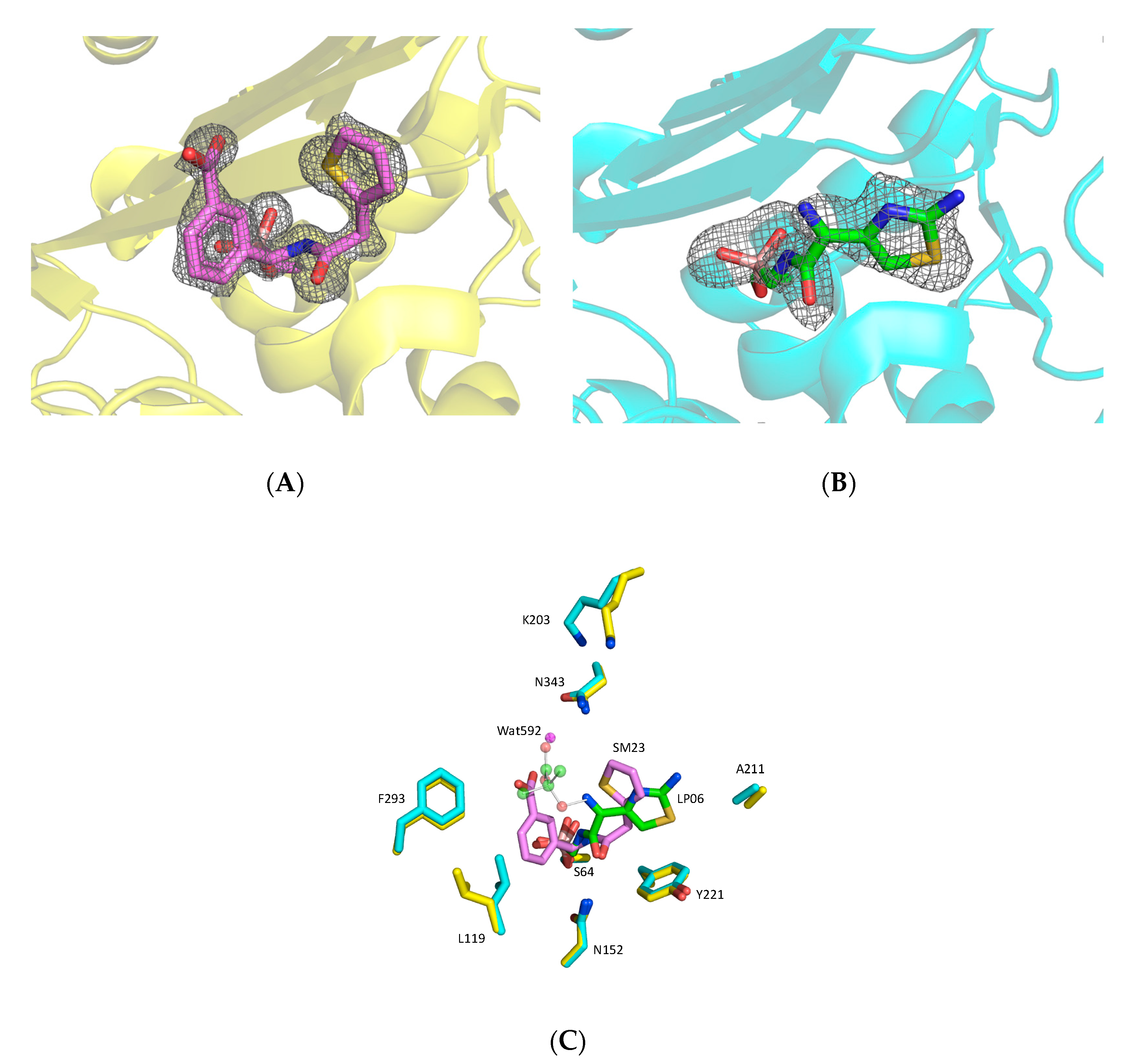
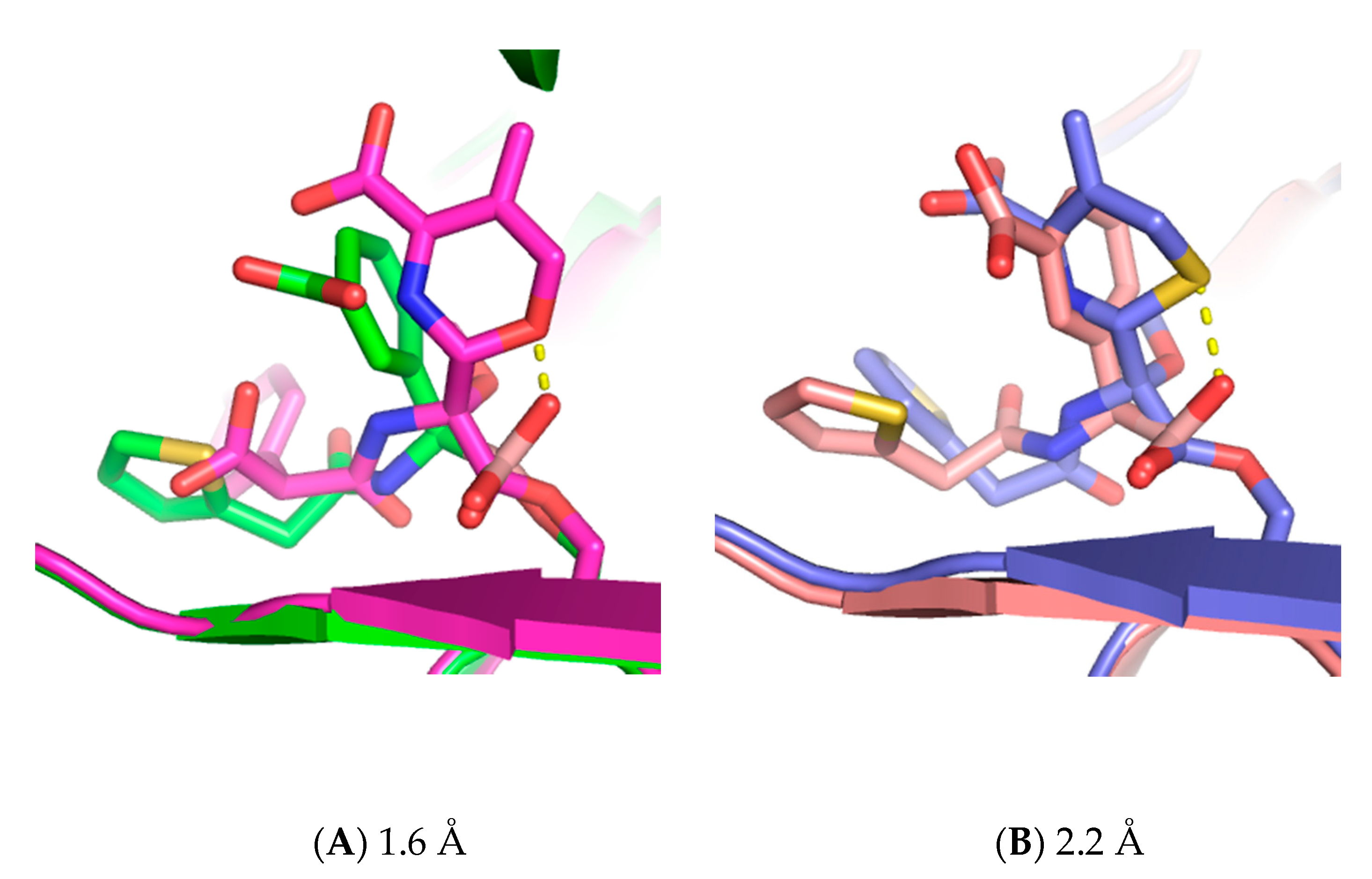
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, Present, and Future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef] [PubMed]
- Drawz, S.M.; Bonomo, R.A. Three Decades of beta-Lactamase Inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef] [PubMed]
- Krajnc, A.; Lang, P.A.; Panduwawala, T.D.; Brem, J.; Schofield, C.J. Will morphing boron-based inhibitors beat the β-lactamases? Curr. Opin. Chem. Biol. 2019, 50, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Papp-Wallace, K.M.; Bonomo, R.A. New β-lactamase Inhibitors in the Clinic. Infect. Dis. Clin. N. Am. 2016, 30, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Caselli, E.; Powers, R.A.; Blasczcak, L.C.; Wu, C.Y.; Prati, F.; Shoichet, B.K. Energetic, structural, and antimicrobial analyses of beta-lactam side chain recognition by beta-lactamases. Chem. Biol. 2001, 8, 17–31. [Google Scholar] [CrossRef][Green Version]
- Morandi, F.; Caselli, E.; Morandi, S.; Focia, P.J.; Blázquez, J.; Shoichet, B.K.; Prati, F. Nanomolar Inhibitors of AmpC β-lactamase. J. Am. Chem. Soc. 2003, 125, 685–695. [Google Scholar] [CrossRef]
- Drawz, S.M.; Papp-Wallace, K.M.; Bonomo, R.A. New beta-Lactamase Inhibitors: A Therapeutic Renaissance in an MDR World. Antimicrob. Agents Chemother. 2014, 58, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.P.; Mitchell, J.M.; Taracila, M.A.; Bonomo, R.A.; Powers, R.A. Exploring the potential of boronic acids as inhibitors of OXA-24/40 β-lactamase. Protein Sci. 2017, 26, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Venturelli, A.; Tondi, D.; Cancian, L.; Morandi, F.; Cannazza, G.; Segatore, B.; Prati, F.; Amicosante, G.; Shoichet, B.K.; Costi, M.P. Optimizing Cell Permeation of an Antibiotic Resistance Inhibitor for Improved Efficacy. J. Med. Chem. 2007, 50, 5644–5654. [Google Scholar] [CrossRef]
- Tondi, D.; Venturelli, A.; Bonnet, R.; Pozzi, C.; Shoichet, B.K.; Costi, M.P. Targeting Class A and C Serine β-lactamases with a Broad-Spectrum Boronic Acid Derivative. J. Med. Chem. 2014, 57, 5449–5458. [Google Scholar] [CrossRef]
- Cendron, L.; Quotadamo, A.; Maso, L.; Bellio, P.; Montanari, M.; Celenza, G.; Venturelli, A.; Costi, M.P.; Tondi, D. X-ray-crystallography deciphers the activity of broad-spectrum boronic acid β-lactamase inhibitors. ACS Med. Chem. Lett. 2019, 10, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Brem, J.; Cain, R.; Cahill, S.; McDonough, M.A.; Clifton, I.J.; Jiménez-Castellanos, J.-C.; Avison, M.B.; Spencer, J.; Fishwick, C.W.G.; Schofield, C.J. Structural basis of metallo-β-lactamase, serine-β-lactamase and penicillin-binding protein inhibition by cyclic boronates. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cahill, S.T.; Cain, R.; Wang, D.Y.; Lohans, C.T.; Wareham, D.W.; Oswin, H.P.; Mohammed, J.; Spencer, J.; Fishwick, C.W.G.; McDonough, M.A.; et al. Cyclic Boronates Inhibit All Classes of β-lactamases. Antimicrob. Agents Chemother. 2017, 61, e02260-16. [Google Scholar] [CrossRef] [PubMed]
- Cahill, S.T.; Tyrrell, J.M.; Navratilova, I.H.; Calvopiña, K.; Robinson, S.W.; Lohans, C.T.; McDonough, M.A.; Cain, R.; Fishwick, C.W.G.; Avison, M.B.; et al. Studies on the inhibition of AmpC and other β-lactamases by cyclic boronates. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Hecker, S.J.; Reddy, K.R.; Totrov, M.; Hirst, G.C.; Lomovskaya, O.; Griffith, D.C.; King, P.; Tsivkovski, R.; Sun, D.; Sabet, M.; et al. Discovery of a Cyclic Boronic Acid β-lactamase Inhibitor (RPX7009) with Utility vs. Class A Serine Carbapenemases. J. Med. Chem. 2015, 58, 3682–3692. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.G.; Lawrence, C.K.; Adam, H.; Schweizer, F.; Zelenitsky, S.; Zhanel, M.; Lagacé-Wiens, P.R.S.; Walkty, A.; Denisuik, A.; Golden, A.; et al. Imipenem-Relebactam and Meropenem-Vaborbactam: Two Novel Carbapenem-β-lactamase Inhibitor Combinations. Drugs 2018, 78, 65–98. [Google Scholar] [CrossRef] [PubMed]
- Bush, K. Game Changers: New β-lactamase Inhibitor Combinations Targeting Antibiotic Resistance in Gram-Negative Bacteria. ACS Infect. Dis. 2018, 4, 84–87. [Google Scholar] [CrossRef]
- Hecker, S.J.; Reddy, K.R.; Lomovskaya, O.; Griffith, D.C.; Rubio-Aparicio, D.; Nelson, K.; Tsivkovski, R.; Sun, D.; Sabet, M.; Tarazi, Z.; et al. Discovery of Cyclic Boronic Acid QPX7728, an Ultra-broad-spectrum Inhibitor of Serine and Metallo Beta-lactamases. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Tsivkovski, R.; Totrov, M.; Lomovskaya, O. Biochemical Characterization of QPX7728, a New Ultra-Broad-Spectrum Beta-lactamase Inhibitor of Serine and Metallo-Beta-Lactamases. Antimicrob. Agents Chemother. 2020, AAC.00130-20. [Google Scholar] [CrossRef]
- Krajnc, A.; Brem, J.; Hinchliffe, P.; Calvopiña, K.; Panduwawala, T.D.; Lang, P.A.; Kamps, J.J.A.G.; Tyrrell, J.M.; Widlake, E.; Saward, B.G.; et al. Bicyclic Boronate VNRX-5133 Inhibits Metallo- and Serine-β-lactamases. J. Med. Chem. 2019, 62, 8544–8556. [Google Scholar] [CrossRef]
- Powers, R.A.; Caselli, E.; Focia, P.J.; Prati, F.; Shoichet, B.K. Structures of ceftazidime and its transition-state analogue in complex with AmpC beta-lactamase: Implications for resistance mutations and inhibitor design. Biochemistry 2001, 40, 9207–9214. [Google Scholar] [CrossRef]
- Chen, Y.; Shoichet, B.; Bonnet, R. Structure, function, and inhibition along the reaction coordinate of CTX-M beta-lactamases. J. Am. Chem. Soc. 2005, 127, 5423–5434. [Google Scholar] [CrossRef]
- Ness, S.; Martin, R.; Kindler, A.M.; Paetzel, M.; Gold, M.; Jensen, S.E.; Jones, J.B.; Strynadka, N.C. Structure-based design guides the improved efficacy of deacylation transition state analogue inhibitors of TEM-1 beta-Lactamase. Biochemistry 2000, 39, 5312–5321. [Google Scholar] [CrossRef]
- Nguyen, N.Q.; Krishnan, N.P.; Rojas, L.J.; Prati, F.; Caselli, E.; Romagnoli, C.; Bonomo, R.A.; van den Akker, F. Crystal Structures of KPC-2 and SHV-1 β-lactamases in Complex with the Boronic Acid Transition State Analog S02030. Antimicrob. Agents Chemother. 2016, 60, 1760–1766. [Google Scholar] [CrossRef]
- Weston, G.S.; Blázquez, J.; Baquero, F.; Shoichet, B.K. Structure-based enhancement of boronic acid-based inhibitors of AmpC beta-lactamase. J. Med. Chem. 1998, 41, 4577–4586. [Google Scholar] [CrossRef]
- Tondi, D.; Powers, R.A.; Caselli, E.; Negri, M.C.; Blázquez, J.; Costi, M.P.; Shoichet, B.K. Structure-based design and in-parallel synthesis of inhibitors of AmpC beta-lactamase. Chem. Biol. 2001, 8, 593–611. [Google Scholar] [CrossRef]
- Chen, Y.; Minasov, G.; Roth, T.A.; Prati, F.; Shoichet, B.K. The deacylation mechanism of AmpC beta-lactamase at ultrahigh resolution. J. Am. Chem. Soc. 2006, 128, 2970–2976. [Google Scholar] [CrossRef]
- Caselli, E.; Romagnoli, C.; Powers, R.A.; Taracila, M.A.; Bouza, A.A.; Swanson, H.C.; Smolen, K.A.; Fini, F.; Wallar, B.J.; Bonomo, R.A.; et al. Inhibition of Acinetobacter-Derived Cephalosporinase: Exploring the Carboxylate Recognition Site Using Novel β-lactamase Inhibitors. ACS Infect. Dis. 2018, 4, 337–348. [Google Scholar] [CrossRef]
- Bouza, A.A.; Swanson, H.C.; Smolen, K.A.; VanDine, A.L.; Taracila, M.A.; Romagnoli, C.; Caselli, E.; Prati, F.; Bonomo, R.A.; Powers, R.A.; et al. Structure-Based Analysis of Boronic Acids as Inhibitors of Acinetobacter-Derived Cephalosporinase-7, a Unique Class C β-lactamase. ACS Infect. Dis. 2018, 4, 325–336. [Google Scholar] [CrossRef]
- Cejas, D.; Fernández Canigia, L.; Quinteros, M.; Giovanakis, M.; Vay, C.; Lascialandare, S.; Mutti, D.; Pagniez, G.; Almuzara, M.; Gutkind, G.; et al. Plasmid-Encoded AmpC (pAmpC) in Enterobacteriaceae: Epidemiology of microorganisms and resistance markers. Rev. Argent. Microbiol. 2012, 44, 182–186. [Google Scholar]
- Jacoby, G.A. AmpC-Lactamases. Clin. Microbiol. Rev. 2009, 22, 161–182. [Google Scholar] [CrossRef] [PubMed]
- Bou, G.; Oliver, A.; Ojeda, M.; Monzón, C.; Martínez-Beltrán, J. Molecular characterization of FOX-4, a new AmpC-type plasmid-mediated beta-lactamase from an Escherichia coli strain isolated in Spain. Antimicrob. Agents Chemother. 2000, 44, 2549–2553. [Google Scholar] [CrossRef][Green Version]
- Lefurgy, S.T.; Malashkevich, V.N.; Aguilan, J.T.; Nieves, E.; Mundorff, E.C.; Biju, B.; Noel, M.A.; Toro, R.; Baiwir, D.; Papp-Wallace, K.M.; et al. Analysis of the Structure and Function of FOX-4 Cephamycinase. Antimicrob. Agents Chemother. 2016, 60, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Drawz, S.M.; Taracila, M.; Caselli, E.; Prati, F.; Bonomo, R.A. Exploring sequence requirements for C3/C4 carboxylate recognition in the Pseudomonas aeruginosa cephalosporinase: Insights into plasticity of the AmpC β-lactamase: C3/C4 Carboxylate Recognition in P. aeruginosa AmpC. Protein Sci. 2011, 20, 941–958. [Google Scholar] [CrossRef] [PubMed]
- Mallo, S.; Pérez-Llarena, F.J.; Kerff, F.; Soares, N.C.; Galleni, M.; Bou, G. A tripeptide deletion in the R2 loop of the class C beta-lactamase enzyme FOX-4 impairs cefoxitin hydrolysis and slightly increases susceptibility to beta-lactamase inhibitors. J. Antimicrob. Chemother. 2010, 65, 1187–1194. [Google Scholar] [CrossRef]
- Patel, J.B.; Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017; ISBN 978-1-56238-804-1. [Google Scholar]
- Michaelis, L.; Menten, M. Die Kinetik der Invertinwirkung. Biochemische Zeitschrift 1913, 49, 333–369. [Google Scholar]
- Crompton, I.E.; Cuthbert, B.K.; Lowe, G.; Waley, S.G. Beta-lactamase inhibitors. The inhibition of serine beta-lactamases by specific boronic acids. Biochem. J. 1988, 251, 453–459. [Google Scholar]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP 4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1997; Volume 276, pp. 307–326. ISBN 978-0-12-182177-7. [Google Scholar]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Adams, P.D.; Grosse-Kunstleve, R.W.; Hung, L.W.; Ioerger, T.R.; McCoy, A.J.; Moriarty, N.W.; Read, R.J.; Sacchettini, J.C.; Sauter, N.K.; Terwilliger, T.C. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 1948–1954. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Kabsch, W. A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. A 1976, 32, 922–923. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2256–2268. [Google Scholar] [CrossRef]
- Beadle, B.M.; Trehan, I.; Focia, P.J.; Shoichet, B.K. Structural milestones in the reaction pathway of an amide hydrolase: Substrate, acyl, and product complexes of cephalothin with AmpC beta-lactamase. Structure 2002, 10, 413–424. [Google Scholar] [CrossRef]
- Beadle, B.M.; Shoichet, B.K. Structural basis for imipenem inhibition of class C beta-lactamases. Antimicrob. Agents Chemother. 2002, 46, 3978–3980. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | WT | |
|---|---|---|
| Ligand | SM23 cephalothin (CEF) analog | LP06 ceftazidime (CAZ) analog |
| PDB entry | 5CHJ | 5CHM |
| Data Collection | ||
| Resolution range (Å) | 20.0–1.4 | 20.0–1.9 |
| Wavelength (Å) | 0.979 | 0.979 |
| Space group | P21 | P21 |
| Unit cell dimensions (Å) | a = 74.29 | a = 54.85 |
| b = 57.16 | b = 57.09 | |
| c = 83.23 | c = 56.75 | |
| α = γ = 90° | α = γ = 90° | |
| β = 91.67° | β = 96.45° | |
| Observed reflections | 539,219 | 94,986 |
| Unique reflections | 147,034 | 27,189 |
| Completeness (%) a | 97.8 (96.5) | 98.8 (98.0) |
| I/σ | 9.9 (2.6) | 7.6 (2.8) |
| Rmerge (I) b | 0.067 (0.448) | 0.094 (0.422) |
| Structure Refinement | ||
| Rcryst (%) c | 0.163 | 0.170 |
| Rfree (%) c | 0.192 | 0.228 |
| Protein non-hydrogen atoms | 5511 | 2722 |
| Water molecules | 1009 | 237 |
| Average B-factor (Å2) | 9.5 | 15.6 |
| RMS Deviations from Ideal Value | ||
| Bonds (Å) | 0.011 | 0.007 |
| Angles (°) | 1.46 | 1.09 |
| Torsion angles (°) | 13.5 | 13.5 |
| Overall coordinate error | ||
| (Maximum likelihood) | 0.11 | 0.22 |
| Ramachandran Statistics (%) | ||
| (For non-Gly/Pro residues) | ||
| Most favorable | 98.1 | 97.8 |
| Additional allowed | 1.9 | 2.2 |
| Strain | CAZ # Only | CAZ/SM23 (CEF-Like) | CAZ/EC04 (CEF *-Like) | CAZ/CTX-Like | CAZ/LP06 (CAZ-Like) |
|---|---|---|---|---|---|
| E. coli blaFOX-4 | >128 | 32 | 64 | 64 | 16 |
| E. coli TG1 pBGS18-blaFOX-4 | 128 | 4 | 16 | 16 | 1 |
| BATSI | FOX-4 1 IC50 (μM) | E. coli AmpC 2 IC50 (μM) | PDC-3 3 IC50 (μM) |
|---|---|---|---|
| SM23 (CEF-like) | 0.032 ± 0.003 | 0.001 | 0.004 |
| EC04 (CEF *-like) | 0.42 ± 0.02 | n.d. | 0.22 |
| CTX-like | 0.26 ± 0.02 | 0.31 4 | 0.17 |
| LP06 (CAZ-like) | 0.11 ± 0.02 | 0.020 4 | 0.004 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lefurgy, S.T.; Caselli, E.; Taracila, M.A.; Malashkevich, V.N.; Biju, B.; Papp-Wallace, K.M.; Bonanno, J.B.; Prati, F.; Almo, S.C.; Bonomo, R.A. Structures of FOX-4 Cephamycinase in Complex with Transition-State Analog Inhibitors. Biomolecules 2020, 10, 671. https://doi.org/10.3390/biom10050671
Lefurgy ST, Caselli E, Taracila MA, Malashkevich VN, Biju B, Papp-Wallace KM, Bonanno JB, Prati F, Almo SC, Bonomo RA. Structures of FOX-4 Cephamycinase in Complex with Transition-State Analog Inhibitors. Biomolecules. 2020; 10(5):671. https://doi.org/10.3390/biom10050671
Chicago/Turabian StyleLefurgy, Scott T., Emilia Caselli, Magdalena A. Taracila, Vladimir N. Malashkevich, Beena Biju, Krisztina M. Papp-Wallace, Jeffrey B. Bonanno, Fabio Prati, Steven C. Almo, and Robert A. Bonomo. 2020. "Structures of FOX-4 Cephamycinase in Complex with Transition-State Analog Inhibitors" Biomolecules 10, no. 5: 671. https://doi.org/10.3390/biom10050671
APA StyleLefurgy, S. T., Caselli, E., Taracila, M. A., Malashkevich, V. N., Biju, B., Papp-Wallace, K. M., Bonanno, J. B., Prati, F., Almo, S. C., & Bonomo, R. A. (2020). Structures of FOX-4 Cephamycinase in Complex with Transition-State Analog Inhibitors. Biomolecules, 10(5), 671. https://doi.org/10.3390/biom10050671