Phytoestrogen Agathisflavone Ameliorates Neuroinflammation-Induced by LPS and IL-1β and Protects Neurons in Cocultures of Glia/Neurons

 , , , ,
, , , , 
 and
and
Abstract
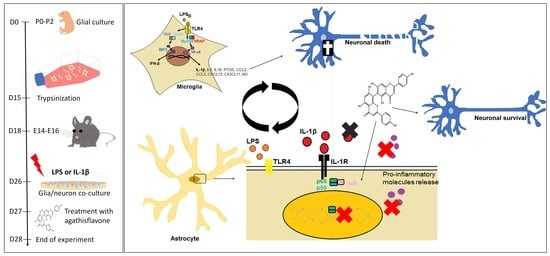
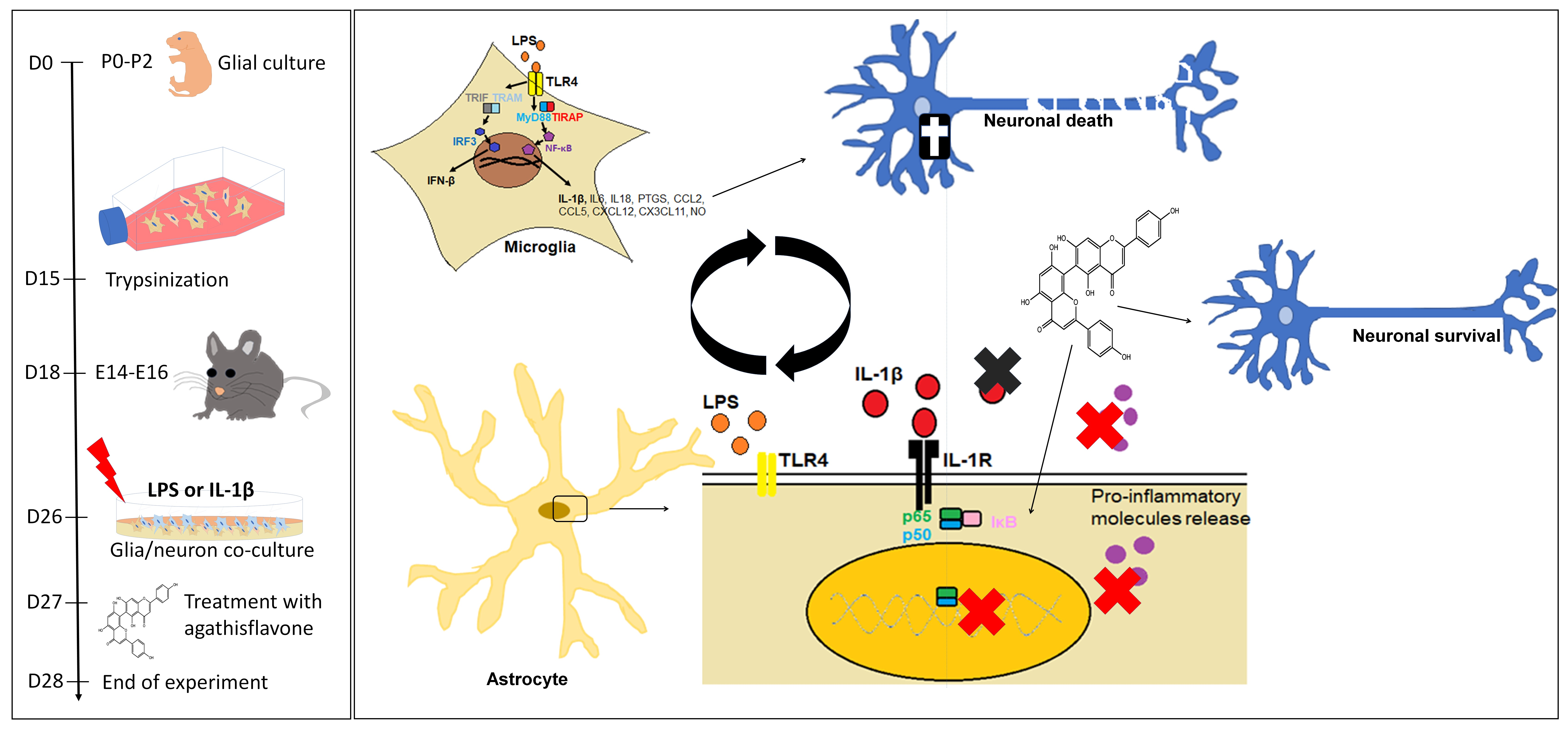{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Neuron–Glial Cell Cocultures
2.2. Agents and Treatments
2.3. Immunocytochemistry
2.4. Bromodeoxyuridine Cell Proliferation Assay
2.5. Fluoro-Jade B (FJ-B) Staining
2.6. RNA Isolation and cDNA Synthesis
2.7. Quantitative PCR (qPCR)
2.8. NO Production
2.9. Statistical Analyses
2.10. Ethics Approval
3. Results
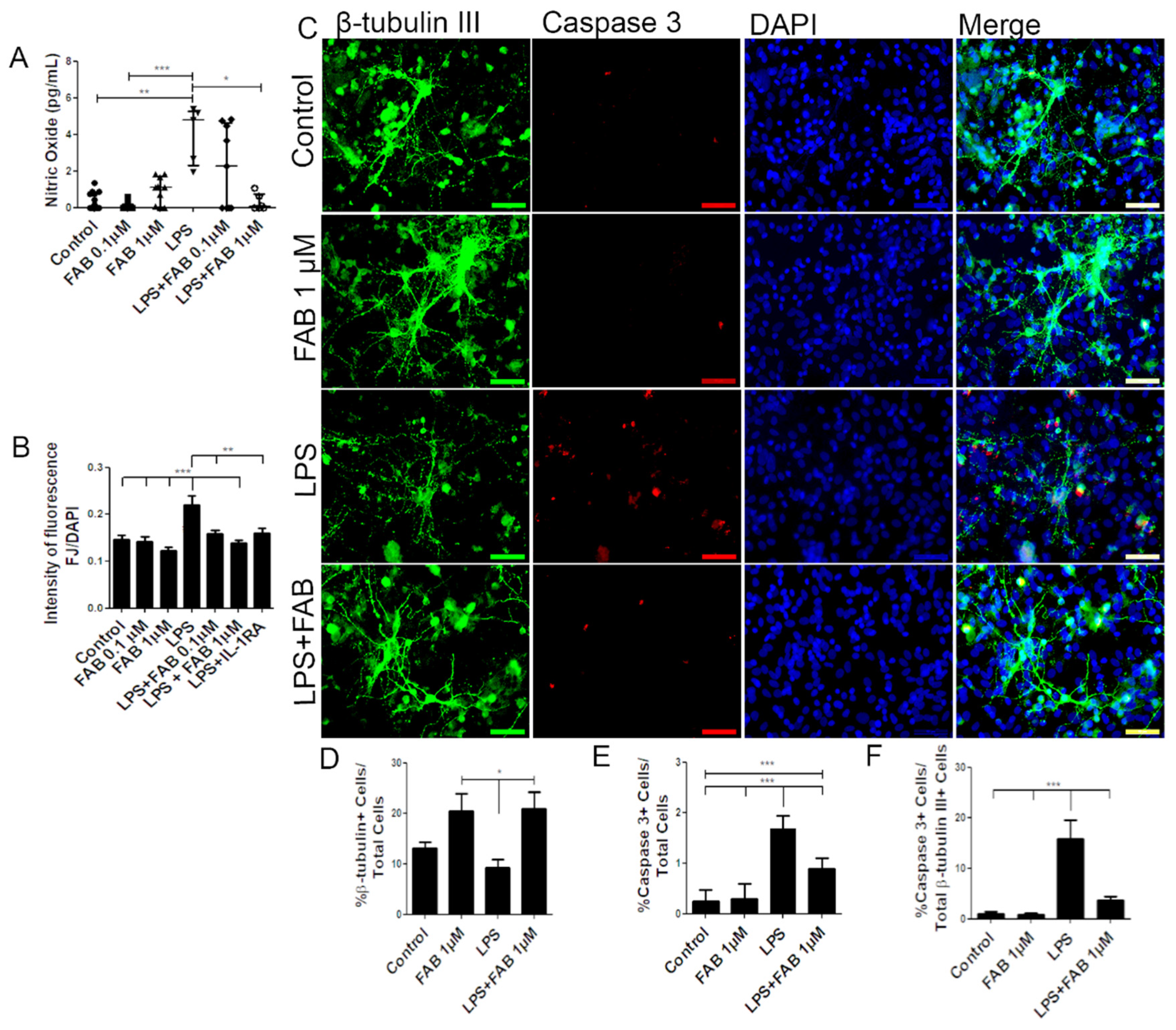
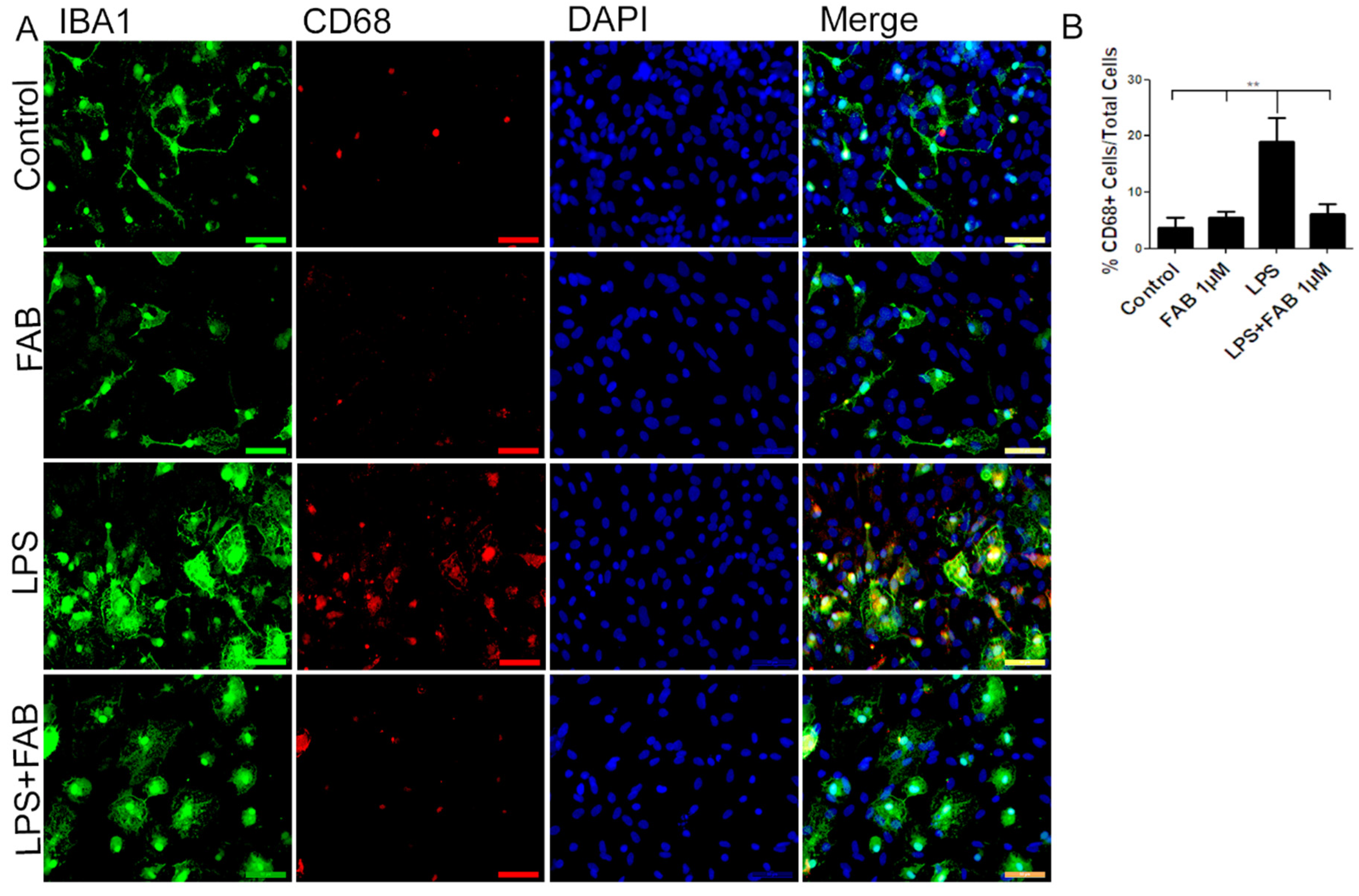
3.1. Agathisflavone (FAB) Protects Against LPS-Induced Neuroinflammatory Damage
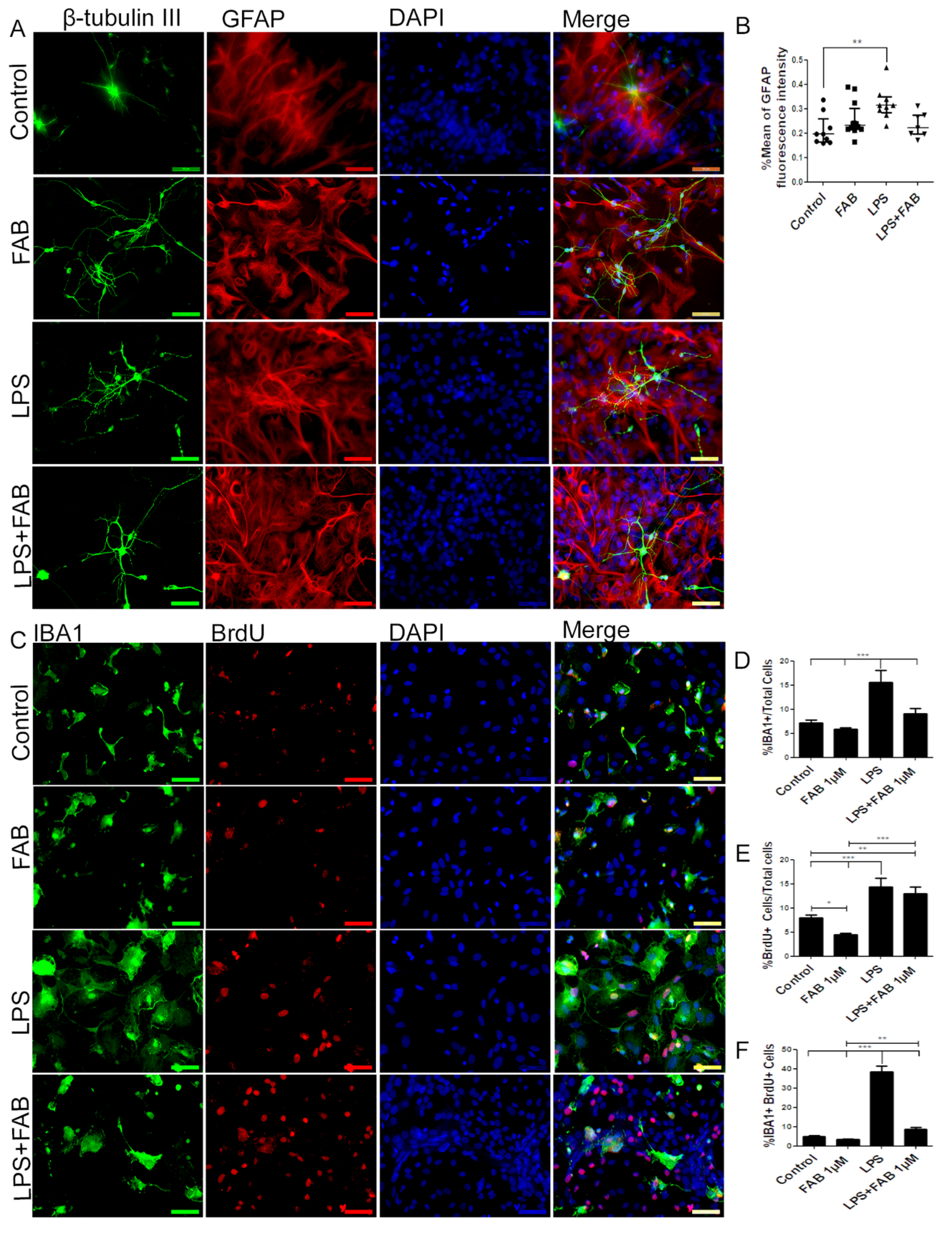
3.2. Agathisflavone (FAB) Preserves Neurons and Decreases Microglial Proliferation after LPS-Induced Neuroinflammatory Damage
3.3. Agathisflavone (FAB) Exerts an Anti-Inflammatory Effect against LPS in Microglia
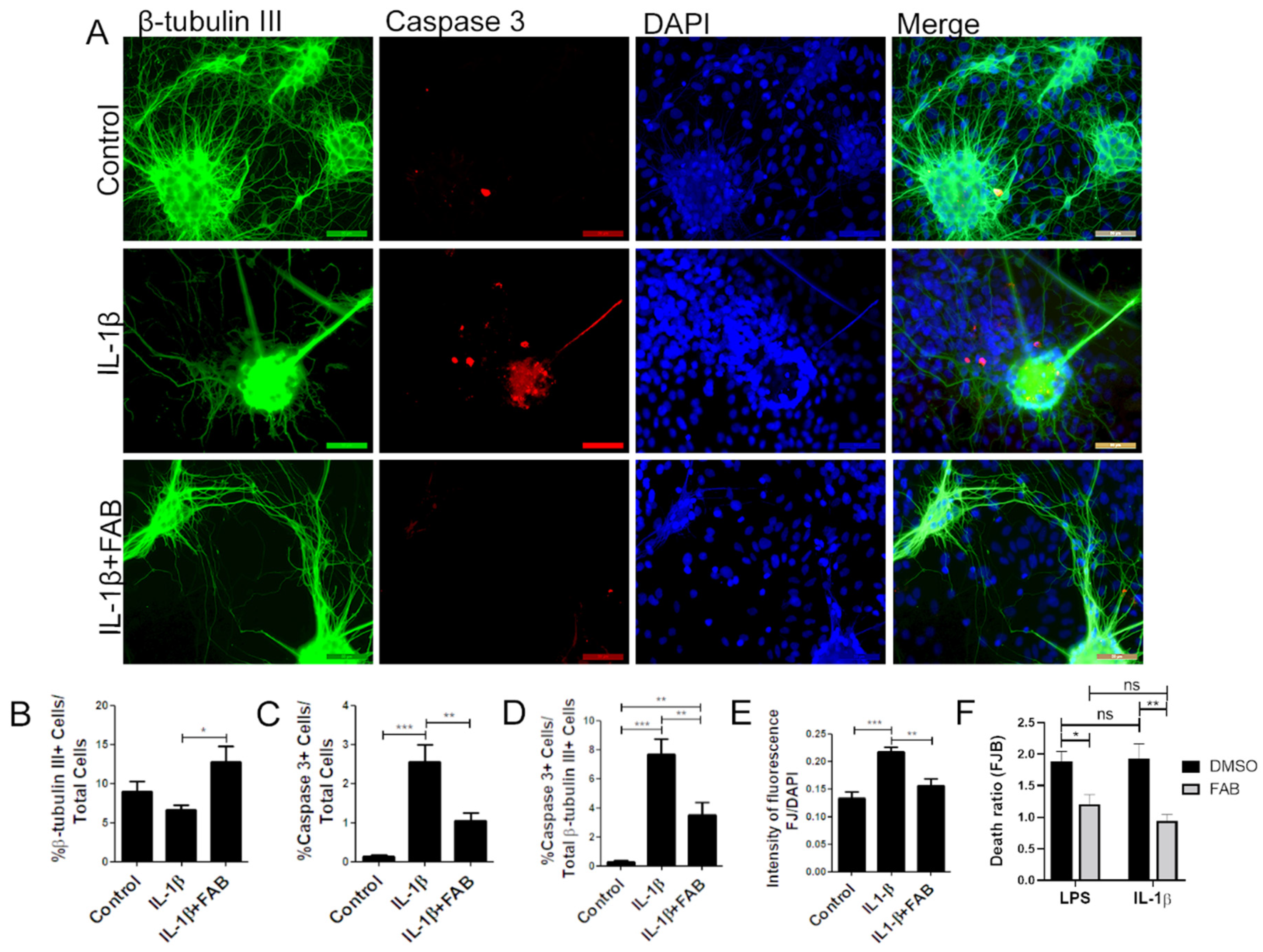
3.4. Agathisflavone (FAB) Protects Against IL-1β-Induced Neuroinflammatory Damage
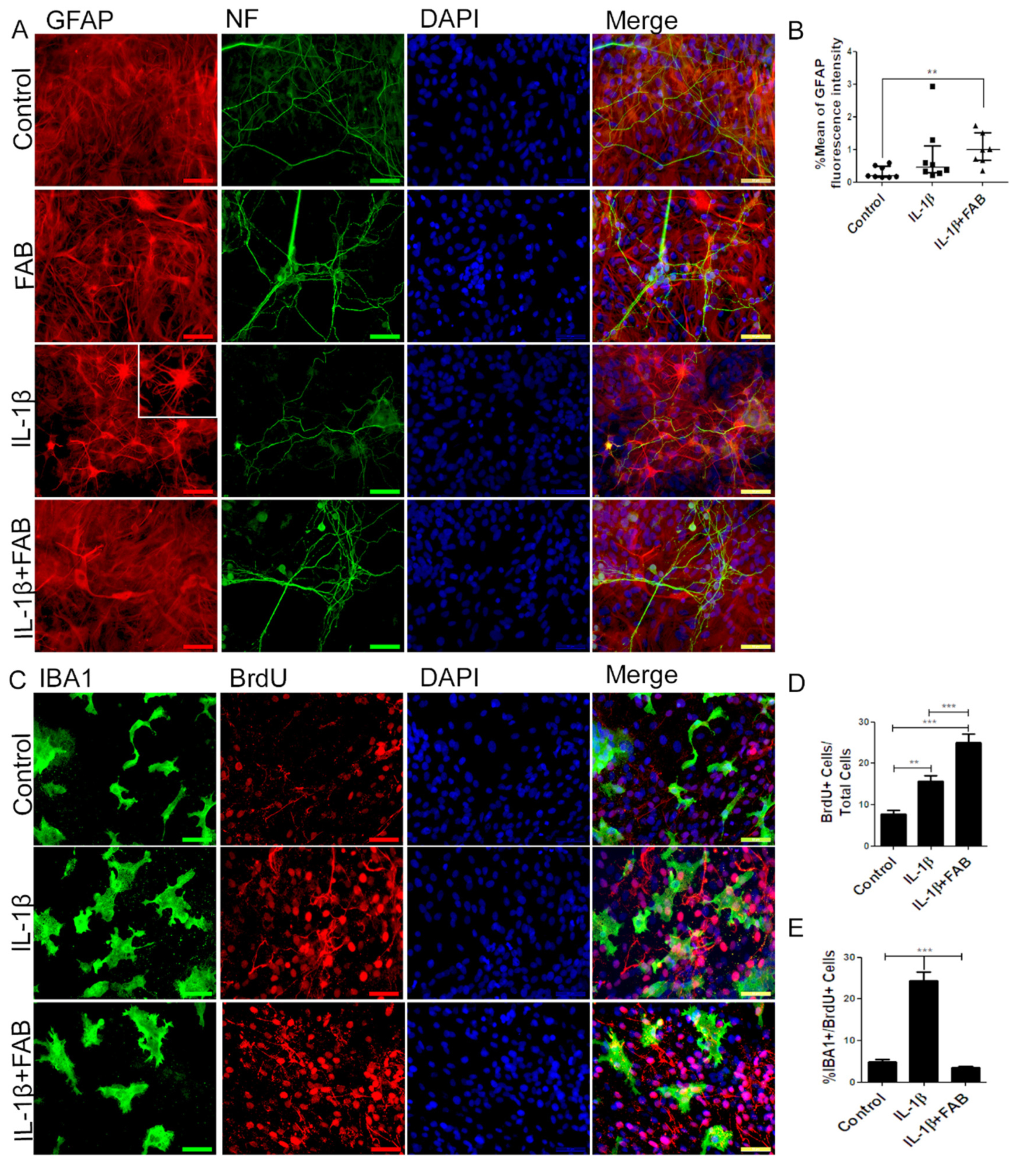
3.5. Agathisflavone (FAB) Preserves Neurons and Astrocytes and Decreases Microglia Proliferation after IL-1β-Induced Neuroinflammatory Damage
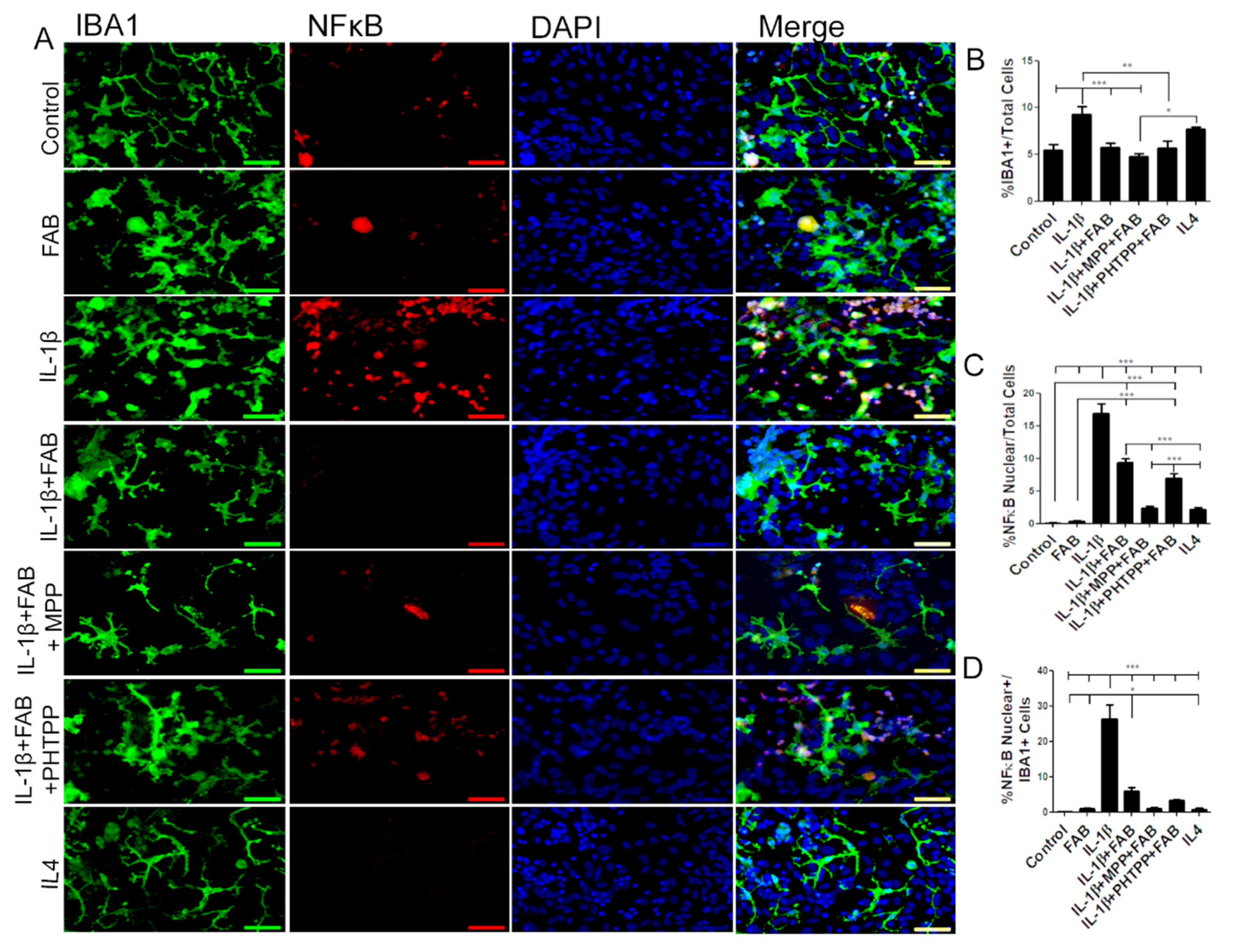
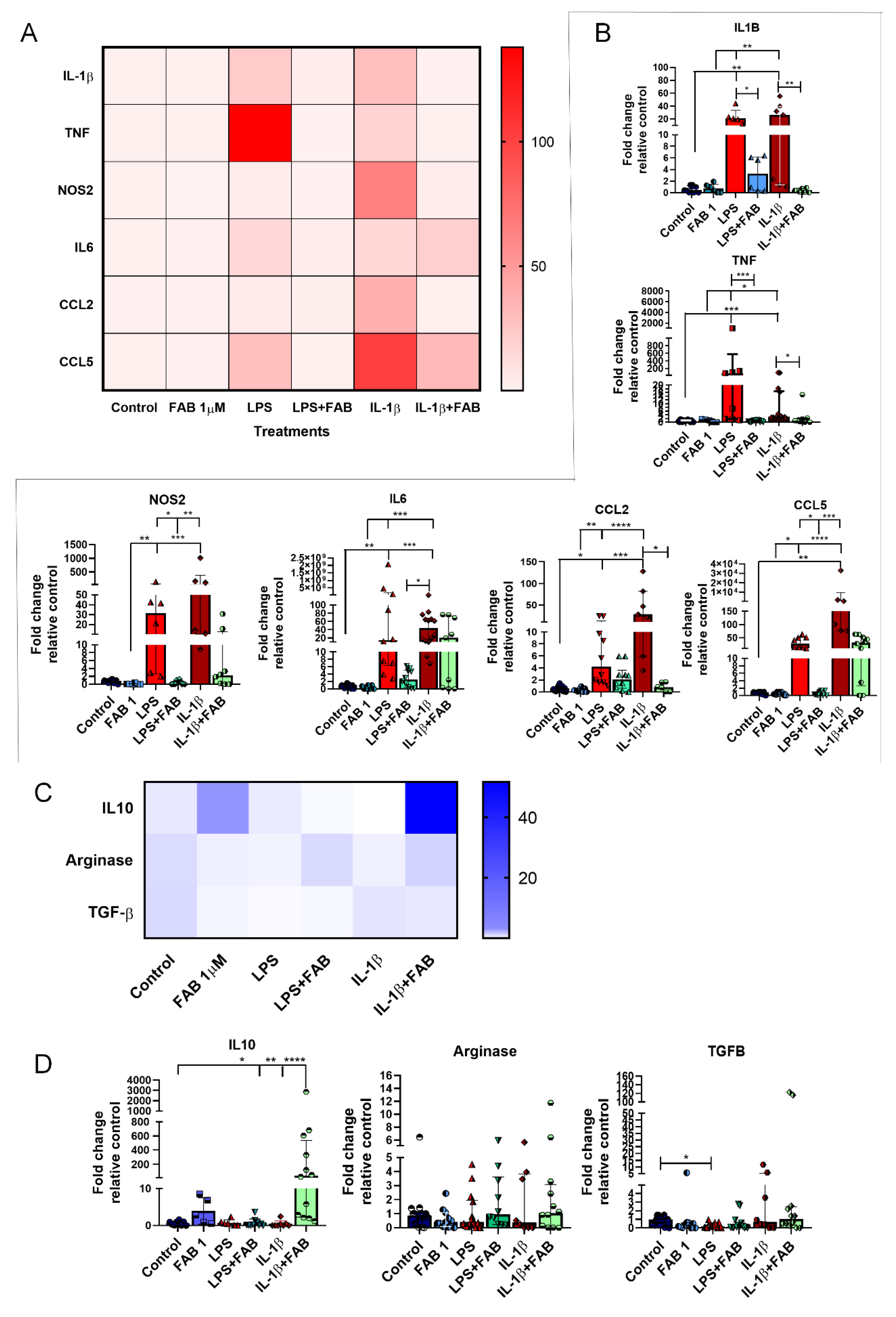
3.6. Agathisflavone (FAB) Positively Impacts Neuroinflammatory Gene Expression after LPS- and IL1-β-Induced Damage
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaur, K.; Gill, J.S.; Bansal, P.K.; Deshmukh, R. Neuroinflammation—A major cause for striatal dopaminergic degeneration in Parkinson’s disease. J. Neurol. Sci. 2017, 381, 308–314. [Google Scholar] [CrossRef] [PubMed]
- González, H.; Elgueta, D.; Montoya, A.; Pacheco, R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J. Neuroimmunol. 2014, 274, 1–13. [Google Scholar] [CrossRef]
- Tyagi, E.; Agrawal, R.; Nath, C.; Shukla, R. Influence of LPS-induced neuroinflammation on acetylcholinesterase activity in rat brain. J. Neuroimmunol. 2008, 205, 51–56. [Google Scholar] [CrossRef]
- Davis, B.M.; Salinas-Navarro, M.; Cordeiro, M.F.; Moons, L.; de Groef, L. Characterizing microglia activation: A spatial statistics approach to maximize information extraction. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Chen, T.; Chen, C.; Zhang, Z.; Zou, Y.; Peng, M.; Wang, Y. Toll-like receptor 4 knockout ameliorates neuroinflammation due to lung-brain interaction in mechanically ventilated mice. Brain Behavior Immun. 2016, 56, 42–55. [Google Scholar] [CrossRef]
- Gibbons, H.M.; Dragunow, M. Microglia induce neural cell death via a proximity-dependent mechanism involving nitric oxide. Brain Res. 2006, 1084, 1–15. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Arginase 1+ microglia reduce Aβ plaque deposition during IL-1β-dependent neuroinflammation. J. Neuroinflammation 2015, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.H.; Xiao, Z.B.; Liu, G.Y.; Zhang, X. Development and application of nano-flavor-drug carriers in neurodegenerative diseases. Chin. Chem. Lett. 2017, 28, 1829–1834. [Google Scholar] [CrossRef]
- Beking, K.; Vieira, A. Flavonoid intake and disability-adjusted life years due to Alzheimers and related dementias: A population-based study involving twenty-three developed countries. Public Health Nutr. 2010, 13, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Velagapudi, R.; Ajileye, O.O.; Okorji, U.; Jain, P.; Aderogba, M.A.; Olajide, O.A. Agathisflavone isolated from Anacardium occidentale suppresses SIRT1-mediated neuroinflammation in BV2 microglia and neurotoxicity in APPSwe-transfected SH-SY5Y cells. Phytother. Res. 2018, 32, 1957–1966. [Google Scholar] [CrossRef]
- da Silva, A.B.; Cerqueira Coelho, P.L.; Alves Oliveira Amparo, J.; Alves de Almeida Carneiro, M.M.; Pereira Borges, J.M.; dos Santos Souza, C.; de Fatima Dias Costa, M.; Mecha, M.; Guaza Rodriguez, C.; Amaral da Silva, V.D.; et al. The flavonoid rutin modulates microglial/macrophage activation to a CD150/CD206 M2 phenotype. Chem. Biol. Interact. 2017, 274, 89–99. [Google Scholar] [CrossRef] [PubMed]
- dos Santos Souza, C.; Grangeiro, M.S.; Lima Pereira, E.P.; dos Santos, C.C.; da Silva, A.B.; Sampaio, G.P.; Ribeiro Figueiredo, D.D.; David, J.M.; David, J.P.; da Silva, V.D.A.; et al. Agathisflavone, a flavonoid derived from Poincianella pyramidalis (Tul.), enhances neuronal population and protects against glutamate excitotoxicity. NeuroToxicology 2018, 65, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Serafini, M.; Peluso, I.; Raguzzini, A. Flavonoids as anti-inflammatory agents. Proc. Nutr. Soc. 2010, 69, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, C.; Moccia, S.; Russo, G.L. Anti-inflammatory effects of flavonoids in neurodegenerative disorders. Eur. J. Med. Chem. 2017, 153, 1–11. [Google Scholar] [CrossRef]
- Xu, J.; Hu, C.; Chen, S.; Shen, H.; Jiang, Q.; Huang, P.; Zhao, W. Neuregulin-1 protects mouse cerebellum against oxidative stress and neuroinflammation. Brain Res. 2017, 1670, 32–43. [Google Scholar] [CrossRef]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.B.; Cooper, M.A.; O’Neill, L.A.J.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behavior Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef]
- Mendes, C.C.; Bahia, M.V.; David, J.M.; David, J.P. Constituents of Caesalpinia pyramidalis. Fitoterapia 2000, 71, 205–207. [Google Scholar] [CrossRef]
- Pišlar, A.; Božić, B.; Zidar, N.; Kos, J. Inhibition of cathepsin X reduces the strength of microglial-mediated neuroinflammation. Neuropharmacology 2017, 114, 88–100. [Google Scholar] [CrossRef]
- Zarruk, J.G.; Greenhalgh, A.D.; David, S. Microglia and macrophages differ in their inflammatory profile after permanent brain ischemia. Exp. Neurol. 2017, 301, 120–132. [Google Scholar] [CrossRef]
- Jin, L.; Ding, M.; Oklopcic, A.; Aghdasi, B.; Xiao, L.; Li, Z.; Jevtovic-Todorovic, V.; Li, X. Nanoparticle fullerol alleviates radiculopathy via NLRP3 inflammasome and neuropeptides. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 2049–2059. [Google Scholar] [CrossRef]
- Sun, Y.; Ge, X.; Li, M.; Xu, L.; Shen, Y. Dyrk2 involved in regulating LPS-induced neuronal apoptosis. Int. J. Biol. Macromol. 2017, 104, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.T.; Chen, B.Y.; Zhang, J.Q.; Kuang, F.; Chen, L.W. Lead exposure induced microgliosis and astrogliosis in hippocampus of young mice potentially by triggering TLR4-MyD88-NFκB signaling cascades. Toxicol. Lett. 2015, 239, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Kamphuis, W.; Kooijman, L.; Schetters, S.; Orre, M.; Hol, E.M. Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer’s disease. Biochim. Et Biophys. Acta Mol. Basis Dis. 2016, 1862, 1847–1860. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Behairi, N.; Belkhelfa, M.; Rafa, H.; Labsi, M.; Deghbar, N.; Bouzid, N.; Mesbah-Amroun, H.; Touil-Boukoffa, C. All-trans retinoic acid (ATRA) prevents lipopolysaccharide-induced neuroinflammation, amyloidogenesis and memory impairment in aged rats. J. Neuroimmunol. 2016, 300, 21–29. [Google Scholar] [CrossRef]
- Zhang, F.; Zhong, R.; Li, S.; Fu, Z.; Cheng, C.; Cai, H.; Le, W. Acute hypoxia induced an imbalanced M1/M2 activation of microglia through NF-κB signaling in Alzheimer’s disease mice and wild-type littermates. Front. Aging Neurosci. 2017, 9, 1–12. [Google Scholar] [CrossRef]
- Andy, S.N.; Chan, C.K.; Kadir, H.A. Deoxyelephantopin from Elephantopus scaber modulates neuroinflammatory response through MAPKs and PI3K/Akt-dependent NF-κB signaling pathways in LPS-stimulated BV-2 microglial cells. J. Funct. Foods 2017, 38, 221–231. [Google Scholar] [CrossRef]
- Kempuraj, D.; Tagen, M.; Iliopoulou, B.P.; Clemons, A.; Vasiadi, M.; Boucher, W.; House, M.; Wolfberg, A.; Theoharides, T.C. Luteolin inhibits myelin basic protein-induced human mast cell activation and mast cell-dependent stimulation of Jurkat T cells. Br. J. Pharmacol. 2008, 155, 1076–1084. [Google Scholar] [CrossRef]
- Wang, Y.X.; Ren, Q.; Yan, Z.Y.; Wang, W.; Zhao, L.; Bai, M.; Wang, X.B.; Huang, X.X.; Song, S.J. Flavonoids and their derivatives with β-amyloid aggregation inhibitory activity from the leaves and twigs of Pithecellobium clypearia Benth. Bioorganic Med. Chem. Lett. 2017, 27, 4823–4827. [Google Scholar] [CrossRef]
- Kumaran, A.; Ho, C.C.; Hwang, L.S. Protective effect of Nelumbo nucifera extracts on beta amyloid protein induced apoptosis in PC12 cells, in vitro model of Alzheimer’s disease. J. Food Drug Anal. 2016, 26, 1–10. [Google Scholar] [CrossRef]
- Kim, B.W.; Koppula, S.; Kim, J.W.; Lim, H.W.; Hwang, J.W.; Kim, I.S.; Park, P.J.; Choi, D.K. Modulation of LPS-stimulated neuroinflammation in BV-2 microglia by Gastrodia elata: 4-Hydroxybenzyl alcohol is the bioactive candidate. J. Ethnopharmacol. 2012, 139, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Kitoh, Y.; Tsukada, M.; Miki, K.; Koyama, K.; Juliawaty, L.D.; Hakim, E.H.; Takahashi, K.; Kinoshita, K. Inhibitory activities of biflavonoids against amyloid-β peptide 42 cytotoxicity in PC-12 cells. Bioorganic Med. Chem. Lett. 2015, 25, 2831–2833. [Google Scholar] [CrossRef] [PubMed]
- Thapa, A.; Woo, E.-R.; Chi, E.Y.; Sharoar, M.G.; Jin, H.-G.; Shin, S.Y.; Park, I.-S. Biflavonoids are superior to monoflavonoids in inhibiting amyloid-beta toxicity and fibrillogenesis via accumulation of nontoxic oligomer-like structures. Biochemistry 2011, 50, 2445–2455. [Google Scholar] [CrossRef]
- Szot, P.; Franklin, A.; Figlewicz, D.P.; Beuca, T.P.; Bullock, K.; Hansen, K.; Banks, W.A.; Raskind, M.A.; Peskind, E.R. Multiple lipopolysaccharide (LPS) injections alter interleukin 6 (IL-6), IL-7, IL-10 and IL-6 and IL-7 receptor mRNA in CNS and spleen. Neuroscience 2017, 355, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Lopes Andrade, A.W.; Dias Ribeiro Figueiredo, D.; TorequlIslam, M.; Viana Nunes, A.M.; da ConceiçãoMachado, K.; da Conceição Machado, K.; Uddin, S.J.; Ahmed Shilpi, J.; Rouf, R.; de Carvalho Melo-Cavalcante, A.A.; et al. Toxicological evaluation of the biflavonoid, agathisflavone in albino Swiss mice. Biomed. Pharmacother. 2019, 110, 68–73. [Google Scholar] [CrossRef]
- Sam, S.K.; Ji, Y.L.; Yoo, K.C.; Sun, S.S.; Ju, S.K.; Su, J.J.; Yong, N.H.; Kun, H.S.; Byung, H.H. Neuroprotective effects of naturally occurring biflavonoids. Bioorganic Med. Chem. Lett. 2005, 15, 3588–3591. [Google Scholar]
- D’Amelio, M.; Cavallucci, V.; Middei, S.; Marchetti, C.; Pacioni, S.; Ferri, A.; Diamantini, A.; de Zio, D.; Carrara, P.; Battistini, L.; et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2010, 14, 69–76. [Google Scholar] [CrossRef]
- Paulsen, B.S.; Souza, C.S.; Chicaybam, L.; Bonamino, M.H.; Bahia, M.; Costa, S.L.; Borges, H.L.; Rehen, S.K. Agathisflavone Enhances Retinoic Acid-Induced Neurogenesis and Its Receptors α and β in Pluripotent Stem Cells. Stem Cells Dev. 2011, 20, 1711–1721. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Kobayashi, K.; Imagama, S.; Ohgomori, T.; Hirano, K.; Uchimura, K.; Sakamoto, K.; Hirakawa, A.; Takeuchi, H.; Suzumura, A.; Ishiguro, N.; et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 2013, 4, e525–e529. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim. Et Biophys. Acta Bioenerg. 2001, 1504, 46–57. [Google Scholar] [CrossRef]
- Fricker, M.; Oliva-Martin, M.J.; Brown, G.C. Primary phagocytosis of viable neurons by microglia activated with LPS or Aβ is dependent on calreticulin/LRP phagocytic signalling. J. Neuroinflammation 2012, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Langley, M.R.; Harischandra, D.S.; Neal, M.L.; Jin, H.; Anantharam, V.; Joseph, J.; Brenza, T.; Narasimhan, B.; Kanthasamy, A.; et al. Mitoapocynin Treatment Protects Against Neuroinflammation and Dopaminergic Neurodegeneration in a Preclinical Animal Model of Parkinson’s Disease. J. Neuroimmune Pharmacol. 2016, 11, 259–278. [Google Scholar] [CrossRef] [PubMed]
- Víteček, J.; Lojek, A.; Valacchi, G.; Kubala, L. Arginine-based inhibitors of nitric oxide synthase: Therapeutic potential and challenges. Mediat. Inflamm. 2012, 2012, 1–22. [Google Scholar] [CrossRef]
- Wong, V.; Wai, C.; Lerner, E. Nitric oxide inhibition strategies. Future Sci. Oa 2015, 1, 1–5. [Google Scholar] [CrossRef]
- Costa, S.L.; Silva, V.D.A.; dos Santos Souza, C.; Santos, C.C.; Paris, I.; Muñoz, P.; Segura-Aguilar, J. Impact of Plant-Derived Flavonoids on Neurodegenerative Diseases. Neurotox. Res. 2016, 30, 41–52. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Olabarria, M.; Noristani, H.N.; Yeh, C.Y.; Rodriguez, J.J. Astrocytes in Alzheimer’s Disease. Neurotherapeutics 2010, 7, 399–412. [Google Scholar] [CrossRef]
- Gorina, R.; Font-Nieves, M.; Márquez-Kisinousky, L.; Santalucia, T.; Planas, A.M. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia 2011, 59, 242–255. [Google Scholar] [CrossRef]
- Masgrau, R.; Guaza, C.; Ransohoff, R.M.; Galea, E. Should We Stop Saying ‘Glia’ and ‘Neuroinflammation’? Trends Mol. Med. 2017, 23, 486–500. [Google Scholar] [CrossRef]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Owens, T. Toll-like receptors in neurodegeneration. Curr. Top. Microbiol. Immunol. 2009, 336, 105–120. [Google Scholar] [PubMed]
- Vegeto, E.; Bonincontro, C.; Pollio, G.; Sala, A.; Viappiani, S.; Nardi, F.; Brusadelli, A.; Viviani, B.; Ciana, P.; Maggi, A. Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J. Neurosci. 2001, 21, 1809–1818. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Almeida, M.M.A.; Souza, C.d.S.; Dourado, N.S.; da Silva, A.B.; Ferreira, R.S.; David, J.M.; David, J.P.; Costa, M.d.F.D.; da Silva, V.D.A.; Butt, A.M.; et al. Phytoestrogen Agathisflavone Ameliorates Neuroinflammation-Induced by LPS and IL-1β and Protects Neurons in Cocultures of Glia/Neurons. Biomolecules 2020, 10, 562. https://doi.org/10.3390/biom10040562
de Almeida MMA, Souza CdS, Dourado NS, da Silva AB, Ferreira RS, David JM, David JP, Costa MdFD, da Silva VDA, Butt AM, et al. Phytoestrogen Agathisflavone Ameliorates Neuroinflammation-Induced by LPS and IL-1β and Protects Neurons in Cocultures of Glia/Neurons. Biomolecules. 2020; 10(4):562. https://doi.org/10.3390/biom10040562
Chicago/Turabian Stylede Almeida, Monique Marylin Alves, Cleide dos Santos Souza, Naiara Silva Dourado, Alessandra Bispo da Silva, Rafael Short Ferreira, Jorge Mauricio David, Juceni Pereira David, Maria de Fátima Dias Costa, Victor Diógenes Amaral da Silva, Arthur Morgan Butt, and et al. 2020. "Phytoestrogen Agathisflavone Ameliorates Neuroinflammation-Induced by LPS and IL-1β and Protects Neurons in Cocultures of Glia/Neurons" Biomolecules 10, no. 4: 562. https://doi.org/10.3390/biom10040562
APA Stylede Almeida, M. M. A., Souza, C. d. S., Dourado, N. S., da Silva, A. B., Ferreira, R. S., David, J. M., David, J. P., Costa, M. d. F. D., da Silva, V. D. A., Butt, A. M., & Costa, S. L. (2020). Phytoestrogen Agathisflavone Ameliorates Neuroinflammation-Induced by LPS and IL-1β and Protects Neurons in Cocultures of Glia/Neurons. Biomolecules, 10(4), 562. https://doi.org/10.3390/biom10040562