Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology



Abstract
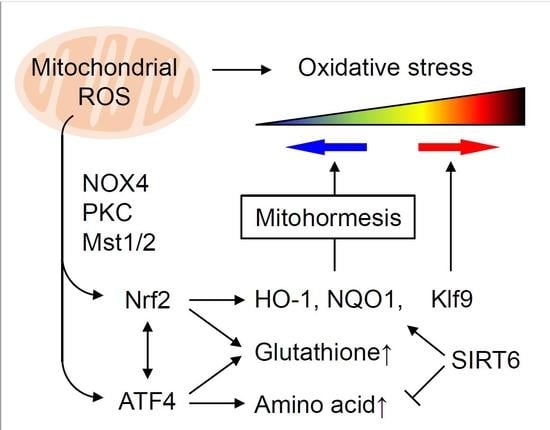
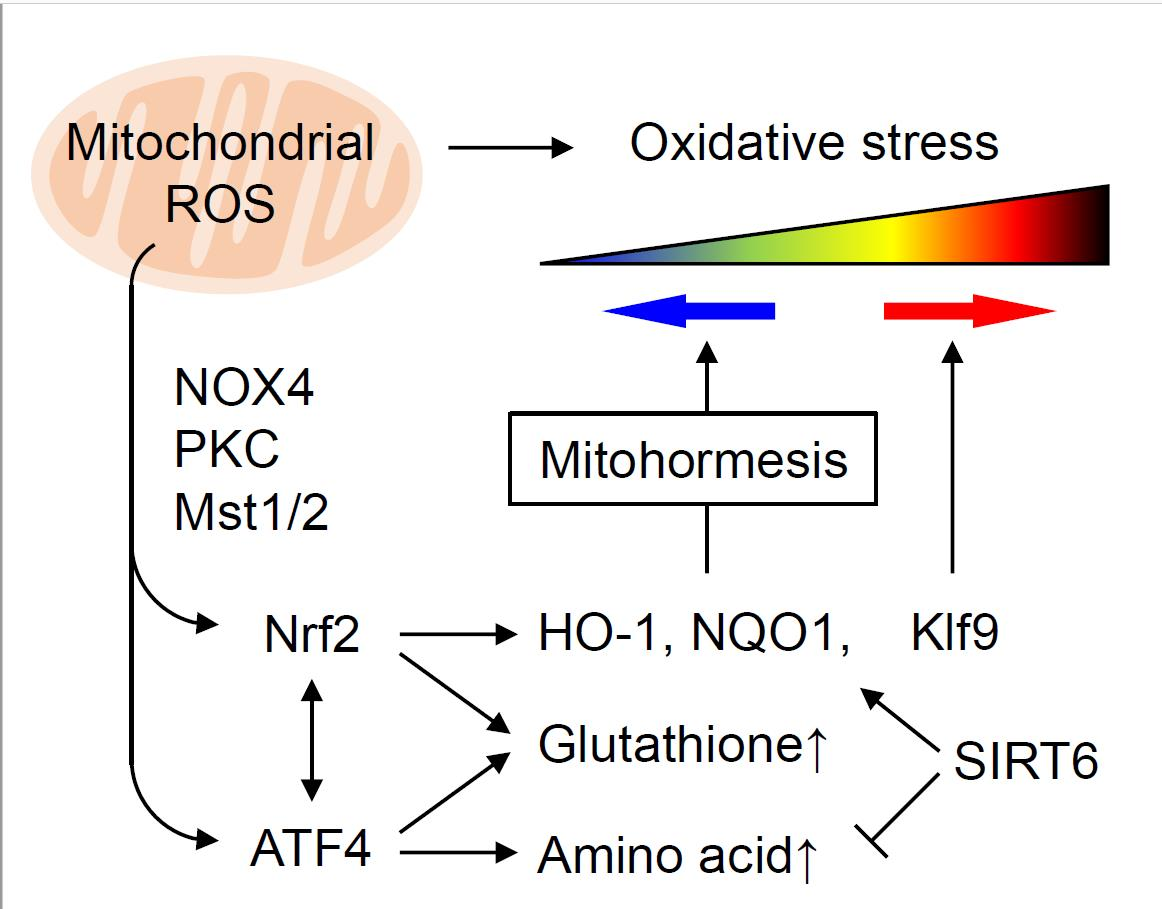{kind=link}
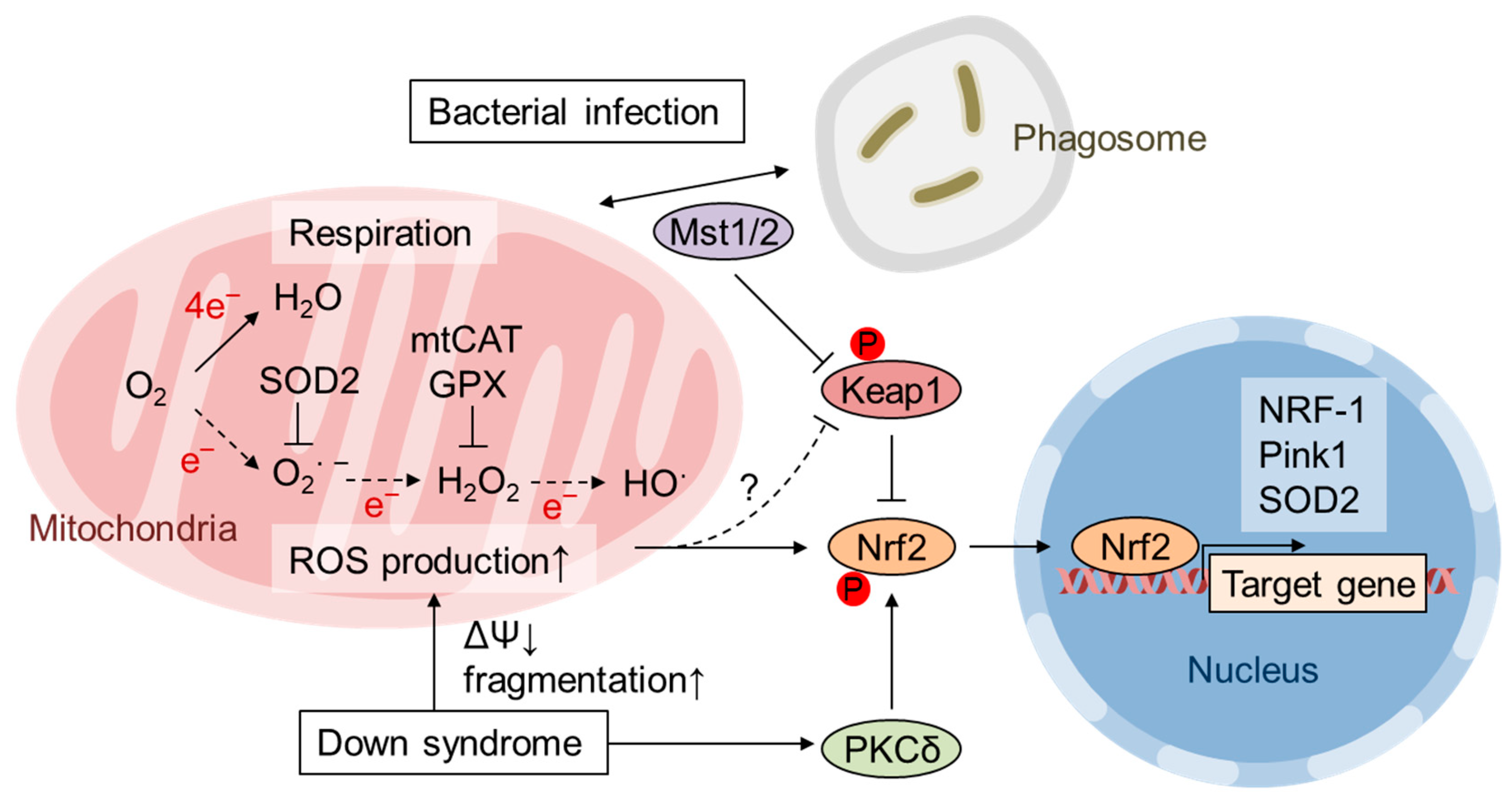{kind=link}
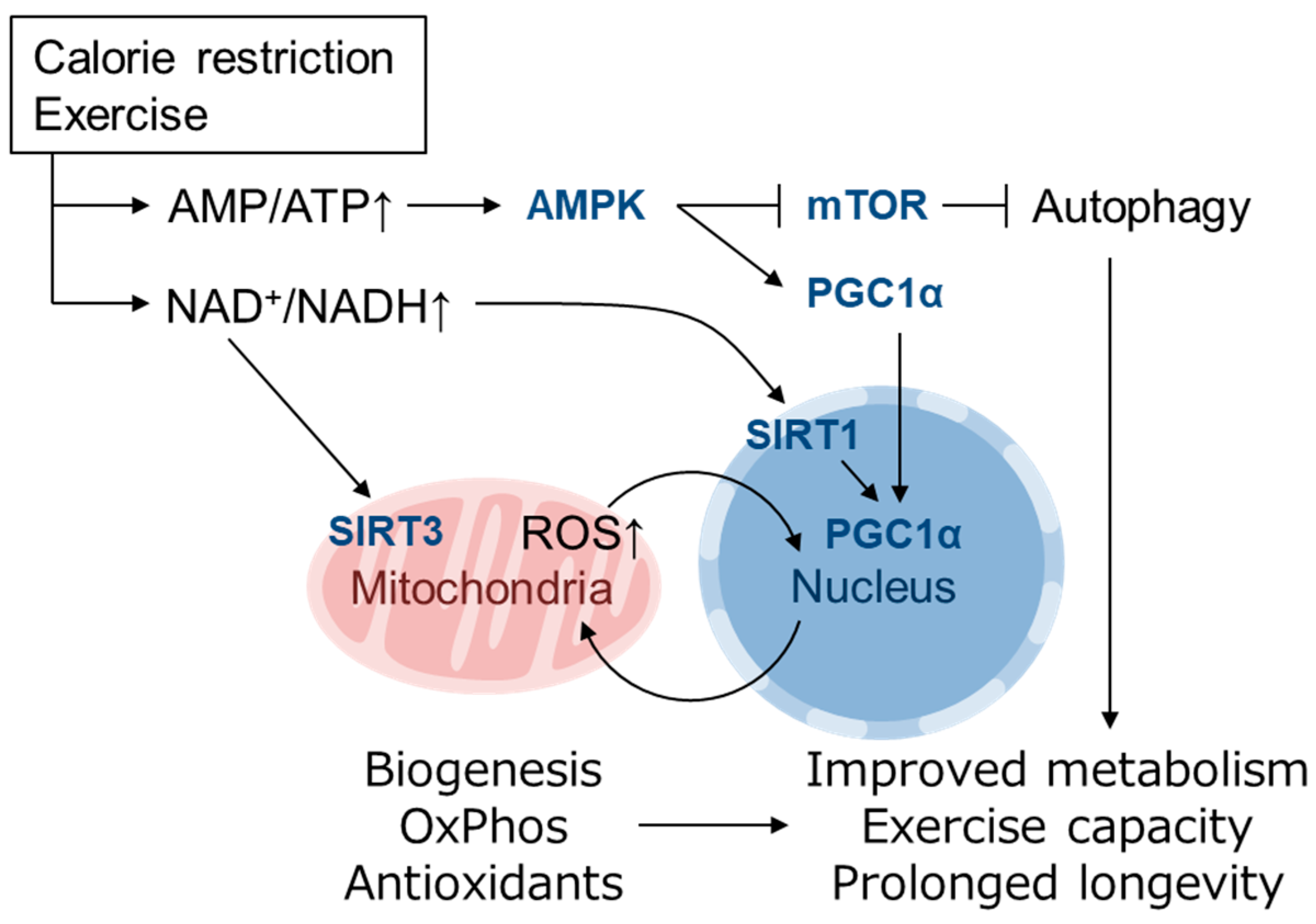{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Nrf2 Contributes to the Homeostasis of Mitochondrial Function
3. Activation of Nrf2 by mtROS
4. Role of mtROS and Nrf2 in Mitohormesis
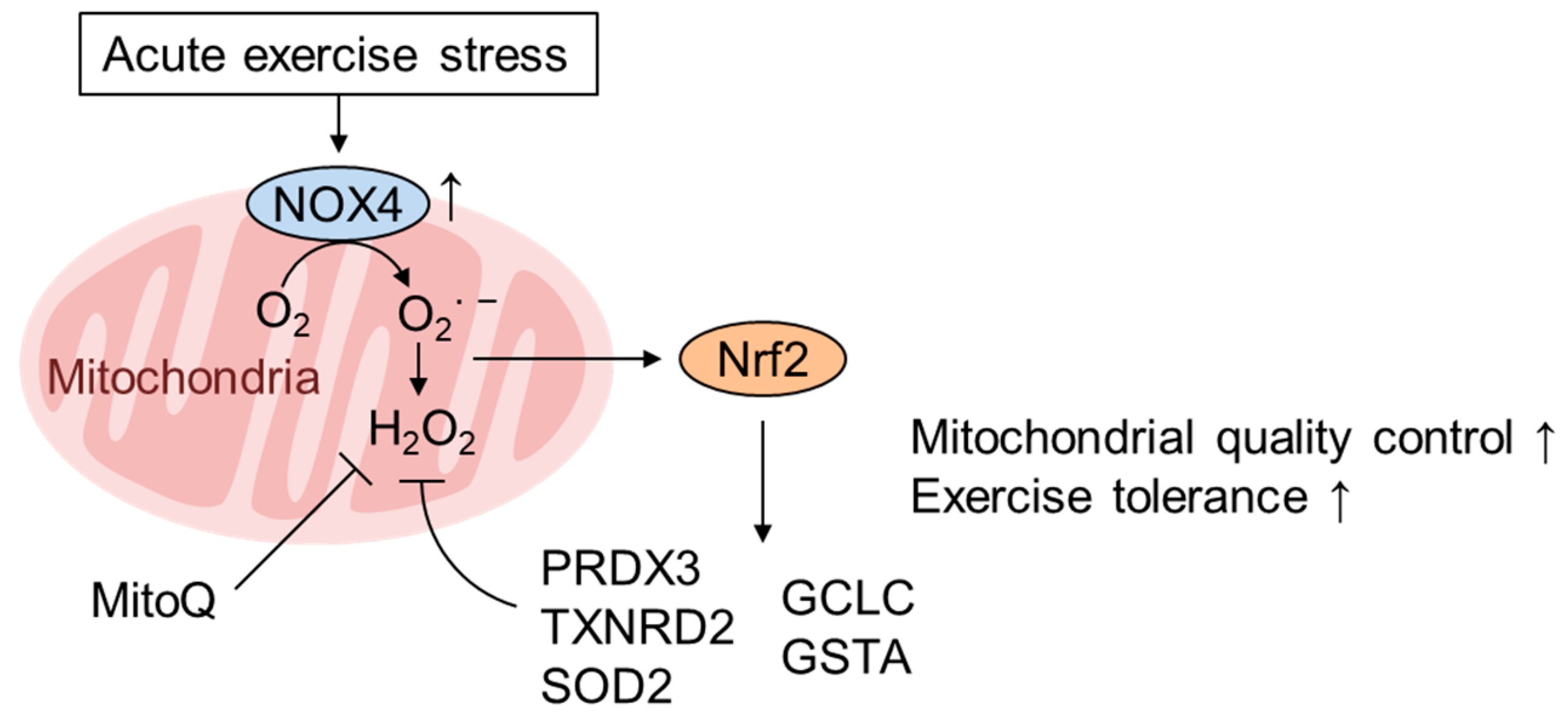
5. Role of mtROS and Nrf2 in Mediating the Healthy Effect of Exercise
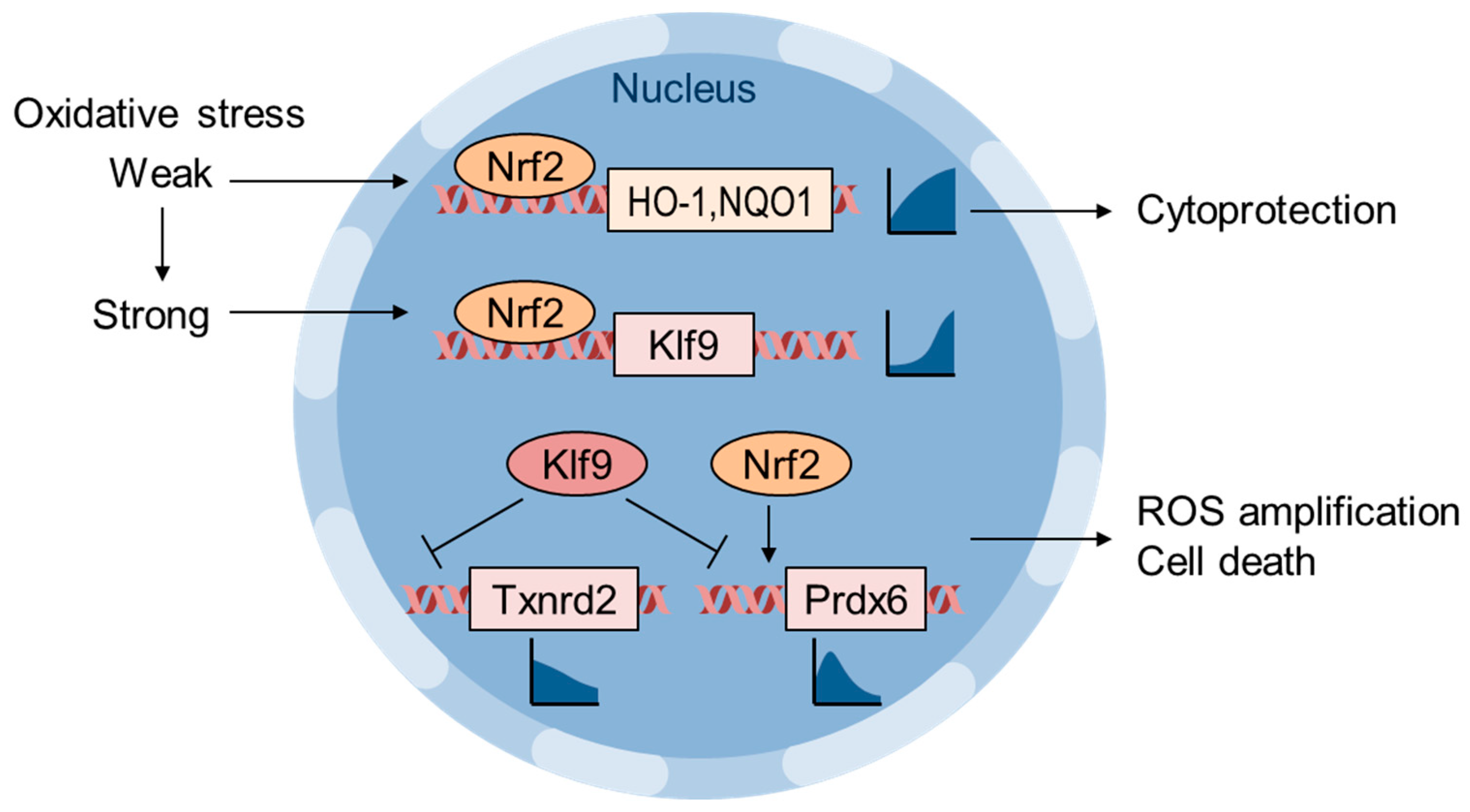
6. Oxidative Stress Thresholding by Nrf2 and Klf9
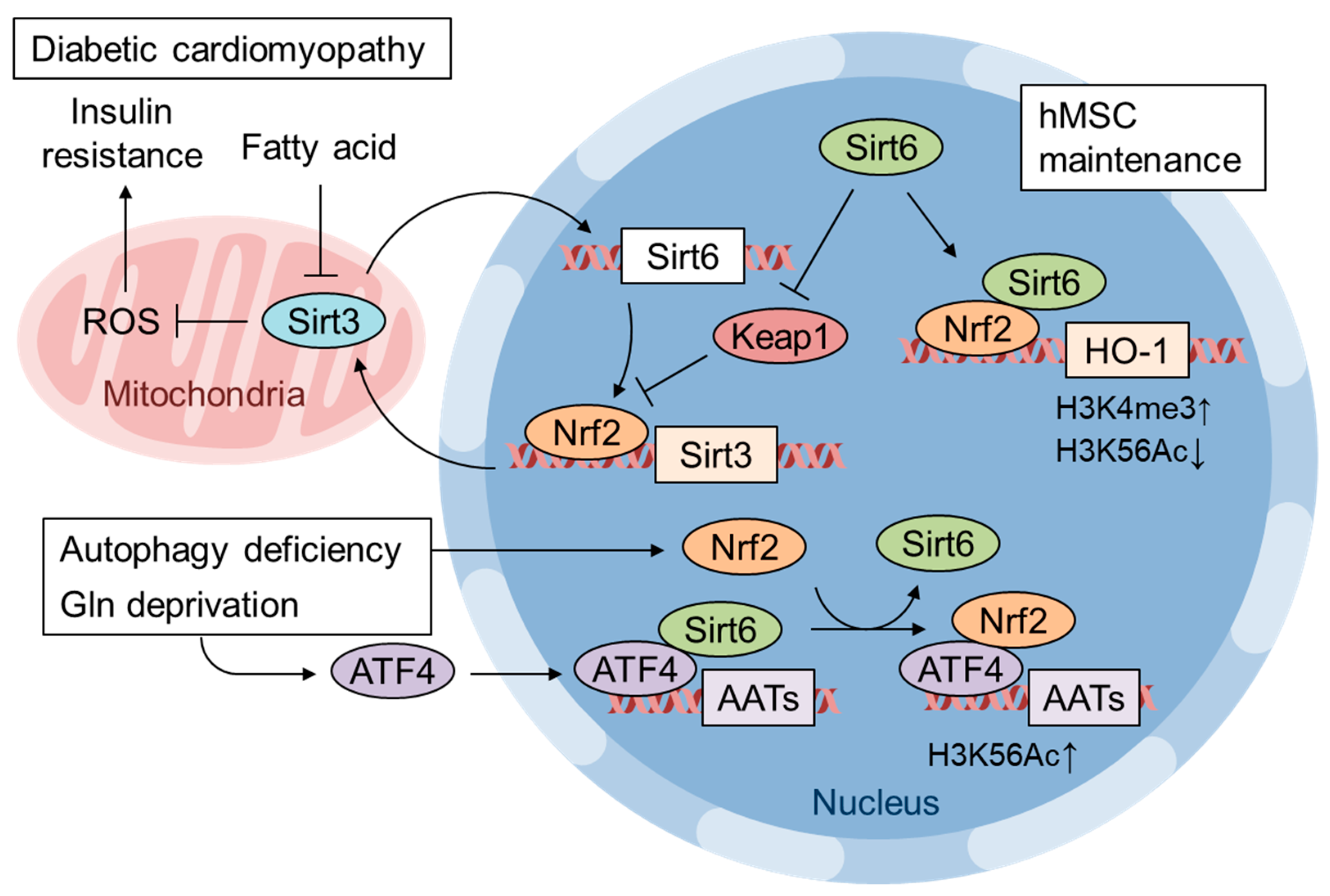
7. Pathological Inactivation of Nrf2 by mtROS
8. Integrated Stress Response (ISR) Is a Major Downstream Pathway of Mitochondrial Dysfunction
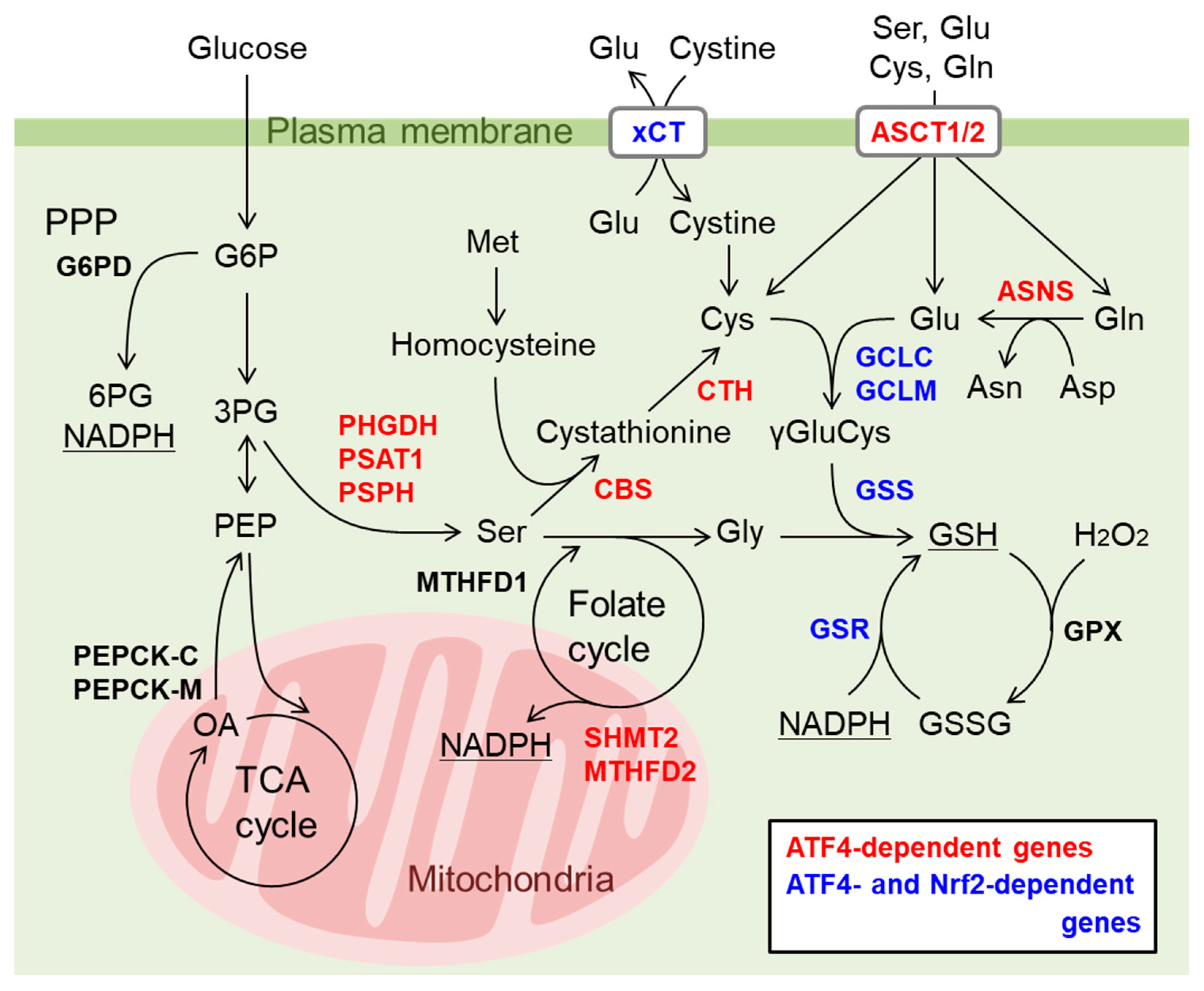
9. Nrf2 and ATF4 Crosstalk in Mitochondrial Retrograde Signaling
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Lane, N. Mitonuclear match: Optimizing fitness and fertility over generations drives ageing within generations. Bioessays 2011, 33, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Li, H.; Forstermann, U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr. Opin. Pharm. 2013, 13, 161–167. [Google Scholar] [CrossRef]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine oxidoreductase-derived reactive species: Physiological and pathological effects. Oxidative Med. Cell. Longev. 2016, 2016, 3527579. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1-Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar] [CrossRef]
- Itoh, K.; Ye, P.; Matsumiya, T.; Tanji, K.; Ozaki, T. Emerging functional cross-talk between the Keap1-Nrf2 system and mitochondria. J. Clin. Biochem. Nutr. 2015, 56, 91–97. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Forman, H.J.; Davies, K.J.; Ursini, F. How do nutritional antioxidants really work: Nucleophilic tone and para-hormesis versus free radical scavenging In Vivo. Free Radic. Biol. Med. 2014, 66, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.S.; McKay, S.E.; Holmbeck, M.A.; Christian, B.E.; Scortea, A.C.; Tsay, A.J.; Newman, L.E.; Shadel, G.S. Mitohormesis in Mice via Sustained Basal Activation of Mitochondrial and Antioxidant Signaling. Cell Metab. 2018, 28, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, E.; Zamponi, N.; Coskun, P.; Quassollo, G.; Lorenzo, A.; Cannas, S.A.; Pigino, G.; Chialvo, D.R.; Gardiner, K.; Busciglio, J.; et al. Nrf2 stabilization prevents critical oxidative damage in Down syndrome cells. Aging Cell 2018, 17, e12812. [Google Scholar] [CrossRef]
- Wang, P.; Geng, J.; Gao, J.; Zhao, H.; Li, J.; Shi, Y.; Yang, B.; Xiao, C.; Linghu, Y.; Sun, X.; et al. Macrophage achieves self-protection against oxidative stress-induced ageing through the Mst-Nrf2 axis. Nat. Commun. 2019, 10, 755. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Carraway, M.S.; Babiker, A.; Suliman, H.B. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 2008, 103, 1232–1240. [Google Scholar] [CrossRef]
- Murata, H.; Takamatsu, H.; Liu, S.; Kataoka, K.; Huh, N.H.; Sakaguchi, M. NRF2 Regulates PINK1 Expression under oxidative stress conditions. PLoS ONE 2015, 10, e0142438. [Google Scholar] [CrossRef]
- Ryoo, I.G.; Kwak, M.K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharm. 2018, 359, 24–33. [Google Scholar] [CrossRef]
- Cherry, A.D.; Suliman, H.B.; Bartz, R.R.; Piantadosi, C.A. Peroxisome proliferator-activated receptor gamma co-activator 1-α as a critical co-activator of the murine hepatic oxidative stress response and mitochondrial biogenesis in Staphylococcus aureus sepsis. J. Biol. Chem. 2014, 289, 41–52. [Google Scholar] [CrossRef]
- Itoh, K.; Tong, K.I.; Yamamoto, M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med. 2004, 36, 1208–1213. [Google Scholar] [CrossRef]
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and non-canonical mechanisms of Nrf2 activation. Pharm. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc. Natl. Acad. Sci. USA 2000, 97, 12475–12480. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.I.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular mechanism of cellular oxidative stress sensing by Keap1. Cell Rep. 2019, 28, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Watai, Y.; Kobayashi, A.; Nagase, H.; Mizukami, M.; McEvoy, J.; Singer, J.D.; Itoh, K.; Yamamoto, M. Subcellular localization and cytoplasmic complex status of endogenous Keap1. Genes Cells 2007, 12, 1163–1178. [Google Scholar] [CrossRef]
- Lo, S.C.; Hannink, M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp. Cell Res. 2008, 314, 1789–1803. [Google Scholar] [CrossRef]
- Itoh, K.; Mimura, J.; Yamamoto, M. Discovery of the negative regulator of Nrf2, Keap1: A historical overview. Antioxid. Redox Signal. 2010, 13, 1665–1678. [Google Scholar] [CrossRef]
- Tsushima, M.; Liu, J.; Hirao, W.; Yamazaki, H.; Tomita, H.; Itoh, K. Emerging evidence for crosstalk between Nrf2 and mitochondria in physiological homeostasis and in heart disease. Arch. Pharm. Res. 2019, 1–11. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef]
- Kasai, S.; Yamazaki, H.; Tanji, K.; Engler, M.J.; Matsumiya, T.; Itoh, K. Role of the ISR-ATF4 pathway and its cross talk with Nrf2 in mitochondrial quality control. J. Clin. Biochem. Nutr. 2019, 64, 1–12. [Google Scholar] [CrossRef]
- Hu, B.; Wu, Y.; Liu, J.; Shen, X.; Tong, F.; Xu, G.; Shen, R. GSK-3beta inhibitor induces expression of Nrf2/TrxR2 signaling pathway to protect against renal ischemia/reperfusion injury in diabetic rats. Kidney Blood Press. Res. 2016, 41, 937–946. [Google Scholar] [CrossRef]
- Miyamoto, N.; Izumi, H.; Miyamoto, R.; Kondo, H.; Tawara, A.; Sasaguri, Y.; Kohno, K. Quercetin induces the expression of peroxiredoxins 3 and 5 via the Nrf2/NRF1 transcription pathway. Invest. Ophthalmol. Vis. Sci. 2011, 52, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-Derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000, 50, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Jovaisaite, V.; Mouchiroud, L.; Auwerx, J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 2014, 217, 137–143. [Google Scholar] [CrossRef]
- Khan, A.U.H.; Allende-Vega, N.; Gitenay, D.; Garaude, J.; Vo, D.N.; Belkhala, S.; Gerbal-Chaloin, S.; Gondeau, C.; Daujat-Chavanieu, M.; Delettre, C.; et al. Mitochondrial Complex I activity signals antioxidant response through ERK5. Sci. Rep. 2018, 8, 7420. [Google Scholar] [CrossRef]
- Indo, H.P.; Hawkins, C.L.; Nakanishi, I.; Matsumoto, K.I.; Matsui, H.; Suenaga, S.; Davies, M.J.; St Clair, D.K.; Ozawa, T.; Majima, H.J. Role of mitochondrial reactive oxygen species in the activation of cellular signals, molecules, and function. Handb. Exp. Pharm. 2017, 240, 439–456. [Google Scholar] [CrossRef]
- Robb, E.L.; Gawel, J.M.; Aksentijevic, D.; Cocheme, H.M.; Stewart, T.S.; Shchepinova, M.M.; Qiang, H.; Prime, T.A.; Bright, T.P.; James, A.M.; et al. Selective superoxide generation within mitochondria by the targeted redox cycler MitoParaquat. Free Radic. Biol. Med. 2015, 89, 883–894. [Google Scholar] [CrossRef]
- Imhoff, B.R.; Hansen, J.M. Extracellular redox status regulates Nrf2 activation through mitochondrial reactive oxygen species. Biochem. J. 2009, 424, 491–500. [Google Scholar] [CrossRef]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef]
- Treuting, P.M.; Linford, N.J.; Knoblaugh, S.E.; Emond, M.J.; Morton, J.F.; Martin, G.M.; Rabinovitch, P.S.; Ladiges, W.C. Reduction of age-associated pathology in old mice by overexpression of catalase in mitochondria. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2008, 63, 813–822. [Google Scholar] [CrossRef]
- Lee, H.Y.; Choi, C.S.; Birkenfeld, A.L.; Alves, T.C.; Jornayvaz, F.R.; Jurczak, M.J.; Zhang, D.; Woo, D.K.; Shadel, G.S.; Ladiges, W.; et al. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 2010, 12, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Manczak, M.; Calkins, M.J.; Truong, Q.; Reddy, T.P.; Reddy, A.P.; Shirendeb, U.; Lo, H.H.; Rabinovitch, P.S.; Reddy, P.H. Mitochondria-Targeted catalase reduces abnormal APP processing, amyloid beta production and BACE1 in a mouse model of Alzheimer’s disease: Implications for neuroprotection and lifespan extension. Hum. Mol. Genet. 2012, 21, 2973–2990. [Google Scholar] [CrossRef] [PubMed]
- Umanskaya, A.; Santulli, G.; Xie, W.; Andersson, D.C.; Reiken, S.R.; Marks, A.R. Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc. Natl. Acad. Sci. USA 2014, 111, 15250–15255. [Google Scholar] [CrossRef]
- Geng, J.; Sun, X.; Wang, P.; Zhang, S.; Wang, X.; Wu, H.; Hong, L.; Xie, C.; Li, X.; Zhao, H.; et al. Kinases Mst1 and Mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat. Immunol. 2015, 16, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Vina, J.; Borras, C.; Abdelaziz, K.M.; Garcia-Valles, R.; Gomez-Cabrera, M.C. The free radical theory of aging revisited: The cell signaling disruption theory of aging. Antioxid. Redox Signal. 2013, 19, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Tapia, P.C. Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: “Mitohormesis” for health and vitality. Med. Hypotheses 2006, 66, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef]
- Barcena, C.; Mayoral, P.; Quiros, P.M. Mitohormesis, an antiaging paradigm. Int. Rev. Cell Mol. Biol. 2018, 340, 35–77. [Google Scholar] [CrossRef]
- Quirós, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef]
- Lee, S.-H.; Lee, J.-H.; Lee, H.-Y.; Min, K.-J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, D.A.; Guarente, L. Extrachromosomal rDNA circles—A cause of aging in yeast. Cell 1997, 91, 1033–1042. [Google Scholar] [CrossRef]
- Tissenbaum, H.A.; Guarente, L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 2001, 410, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.G.; Rogina, B.; Lavu, S.; Howitz, K.; Helfand, S.L.; Tatar, M.; Sinclair, D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004, 430, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The role of PGC1α in cancer metabolism and its therapeutic implications. Mol. Cancer 2016, 15, 774–782. [Google Scholar] [CrossRef]
- Qian, X.; Li, X.; Shi, Z.; Bai, X.; Xia, Y.; Zheng, Y.; Xu, D.; Chen, F.; You, Y.; Fang, J.; et al. KDM3A senses oxygen availability to regulate PGC-1α-mediated mitochondrial biogenesis. Mol. Cell 2019, 76, 885–895. [Google Scholar] [CrossRef]
- Lamming, D.W. Inhibition of the mechanistic target of rapamycin (mTOR)-rapamycin and beyond. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]
- Warburton, D.E.; Nicol, C.W.; Bredin, S.S. Health benefits of physical activity: The evidence. CMAJ 2006, 174, 801–809. [Google Scholar] [CrossRef]
- Lanza, I.R.; Short, D.K.; Short, K.R.; Raghavakaimal, S.; Basu, R.; Joyner, M.J.; McConnell, J.P.; Nair, K.S. Endurance exercise as a countermeasure for aging. Diabetes 2008, 57, 2933–2942. [Google Scholar] [CrossRef]
- Vargas-Ortiz, K.; Perez-Vazquez, V.; Macias-Cervantes, M.H. Exercise and sirtuins: A way to mitochondrial health in skeletal muscle. Int. J. Mol. Sci. 2019, 20, 2717. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Hoj, P.B.; Hoogenraad, N.J. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem. 1996, 240, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Munch, C.; Harper, J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 2016, 534, 710–713. [Google Scholar] [CrossRef]
- Munch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018, 16, 81. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Steinbaugh, M.J.; Hourihan, J.M.; Ewald, C.Y.; Isik, M. SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans. Free Radic. Biol. Med. 2015, 88, 290–301. [Google Scholar] [CrossRef]
- Pulliam, D.A.; Deepa, S.S.; Liu, Y.; Hill, S.; Lin, A.L.; Bhattacharya, A.; Shi, Y.; Sloane, L.; Viscomi, C.; Zeviani, M.; et al. Complex IV-deficient Surf1(-/-) mice initiate mitochondrial stress responses. Biochem. J. 2014, 462, 359–371. [Google Scholar] [CrossRef]
- Wedatilake, Y.; Brown, R.M.; McFarland, R.; Yaplito-Lee, J.; Morris, A.A.; Champion, M.; Jardine, P.E.; Clarke, A.; Thorburn, D.R.; Taylor, R.W.; et al. SURF1 deficiency: A multi-centre natural history study. Orphanet J. Rare Dis. 2013, 8, 96. [Google Scholar] [CrossRef]
- Dell’agnello, C.; Leo, S.; Agostino, A.; Szabadkai, G.; Tiveron, C.; Zulian, A.; Prelle, A.; Roubertoux, P.; Rizzuto, R.; Zeviani, M. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 2007, 16, 431–444. [Google Scholar] [CrossRef]
- Memme, J.M.; Erlich, A.T.; Hood, D.A.; Phukan, G. Exercise and mitochondrial health. J. Physiol. 2019. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Salvador-Pascual, A.; Cabo, H.; Ferrando, B.; Vina, J. Redox modulation of mitochondriogenesis in exercise. Does antioxidant supplementation blunt the benefits of exercise training? Free Radic. Biol. Med. 2015, 86, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K.; Oberbach, A.; Kloting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Bluher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Hancock, M.; Hafstad, A.D.; Nabeebaccus, A.A.; Catibog, N.; Logan, A.; Smyrnias, I.; Hansen, S.S.; Lanner, J.; Schroder, K.; Murphy, M.P.; et al. Myocardial NADPH oxidase-4 regulates the physiological response to acute exercise. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Done, A.J.; Traustadottir, T. Nrf2 mediates redox adaptations to exercise. Redox Biol. 2016, 10, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, V.R.; Kannan, S.; Sadhaasivam, K.; Gounder, S.S.; Davidson, C.J.; Boeheme, C.; Hoidal, J.R.; Wang, L.; Rajasekaran, N.S. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic. Biol. Med. 2012, 52, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Kirber, M.T.; Xiao, H.; Yang, Y.; Keaney, J.F., Jr. Regulation of ROS signal transduction by NADPH oxidase 4 localization. J. Cell Biol. 2008, 181, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Casciaro, F.; Beretti, F.; Zavatti, M.; McCubrey, J.A.; Ratti, S.; Marmiroli, S.; Follo, M.Y.; Maraldi, T. Nuclear Nox4 interaction with prelamin A is associated with nuclear redox control of stem cell aging. Aging 2018, 10, 2911–2934. [Google Scholar] [CrossRef]
- Ago, T.; Kuroda, J.; Pain, J.; Fu, C.; Li, H.; Sadoshima, J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 2010, 106, 1253–1264. [Google Scholar] [CrossRef]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef]
- Vendrov, A.E.; Vendrov, K.C.; Smith, A.; Yuan, J.; Sumida, A.; Robidoux, J.; Runge, M.S.; Madamanchi, N.R. NOX4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid. Redox Signal. 2015, 23, 1389–1409. [Google Scholar] [CrossRef]
- Langbein, H.; Shahid, A.; Hofmann, A.; Mittag, J.; Bornstein, S.R.; Morawietz, H.; Brunssen, C. NADPH oxidase 4 mediates the protective effects of physical activity against obesity-induced vascular dysfunction. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef]
- Merry, T.L.; Ristow, M. Nuclear factor erythroid-derived 2-like 2 (NFE2L2, Nrf2) mediates exercise-induced mitochondrial biogenesis and the anti-oxidant response in mice. J. Physiol. 2016, 594, 5195–5207. [Google Scholar] [CrossRef]
- Yamada, M.; Iwata, M.; Warabi, E.; Oishi, H.; Lira, V.A.; Okutsu, M. p62/SQSTM1 and Nrf2 are essential for exercise-mediated enhancement of antioxidant protein expression in oxidative muscle. FASEB J. 2019, 33, 8022–8032. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Han, E.; Bui, C.B.; Shin, W.; Lee, J.; Lee, S.; Choi, Y.B.; Lee, A.H.; Lee, K.H.; Park, C.; et al. Assurance of mitochondrial integrity and mammalian longevity by the p62-Keap1-Nrf2-Nqo1 cascade. EMBO Rep. 2012, 13, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Simmons, A.N.; Kajino-Sakamoto, R.; Tsuji, Y.; Ninomiya-Tsuji, J. TAK1 Regulates the Nrf2 antioxidant system through modulating p62/SQSTM1. Antioxid. Redox Signal. 2016, 25, 953–964. [Google Scholar] [CrossRef]
- Kajino-Sakamoto, R.; Omori, E.; Nighot, P.K.; Blikslager, A.T.; Matsumoto, K.; Ninomiya-Tsuji, J. TGF-β-activated kinase 1 signaling maintains intestinal integrity by preventing accumulation of reactive oxygen species in the intestinal epithelium. J. Immunol. 2010, 185, 4729–4737. [Google Scholar] [CrossRef]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi-Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 amplifies oxidative stress via induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Sulforaphane-Induced Klf9/Prdx6 axis acts as a molecular switch to control redox signaling and determines fate of cells. Cells 2019, 8, 1159. [Google Scholar] [CrossRef]
- Cheng, X.; Chapple, S.J.; Patel, B.; Puszyk, W.; Sugden, D.; Yin, X.; Mayr, M.; Siow, R.C.; Mann, G.E. Gestational diabetes mellitus impairs Nrf2-mediated adaptive antioxidant defenses and redox signaling in fetal endothelial cells in utero. Diabetes 2013, 62, 4088–4097. [Google Scholar] [CrossRef]
- Smith, R.A.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 2010, 1201, 96–103. [Google Scholar] [CrossRef]
- Rabbani, P.S.; Soares, M.A.; Hameedi, S.G.; Kadle, R.L.; Mubasher, A.; Kowzun, M.; Ceradini, D.J. Dysregulation of Nrf2/Keap1 redox pathway in diabetes affects multipotency of stromal cells. Diabetes 2019, 68, 141–155. [Google Scholar] [CrossRef]
- Zuo, H.; Wang, S.; Feng, J.; Liu, X. BRD4 contributes to high-glucose-induced podocyte injury by modulating Keap1/Nrf2/ARE signaling. Biochimie 2019, 165, 100–107. [Google Scholar] [CrossRef]
- Cheng, X.; Siow, R.C.; Mann, G.E. Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: A role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid. Redox Signal. 2011, 14, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Bohmann, D. BET-Ting on Nrf2: How Nrf2 signaling can influence the therapeutic activities of BET protein inhibitors. Bioessays 2018, 40, e1800007. [Google Scholar] [CrossRef] [PubMed]
- Anzovino, A.; Chiang, S.; Brown, B.E.; Hawkins, C.L.; Richardson, D.R.; Huang, M.L. Molecular alterations in a mouse cardiac model of friedreich ataxia: An Impaired Nrf2 response mediated via upregulation of Keap1 and activation of the Gsk3beta axis. Am. J. Pathol. 2017, 187, 2858–2875. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef]
- Pan, H.; Guan, D.; Liu, X.; Li, J.; Wang, L.; Wu, J.; Zhou, J.; Zhang, W.; Ren, R.; Zhang, W.; et al. SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell Res. 2016, 26, 190–205. [Google Scholar] [CrossRef]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef]
- Kanwal, A.; Pillai, V.B.; Samant, S.; Gupta, M.; Gupta, M.P. The nuclear and mitochondrial sirtuins, Sirt6 and Sirt3, regulate each other’s activity and protect the heart from developing obesity-mediated diabetic cardiomyopathy. FASEB J. 2019, 33, 10872–10888. [Google Scholar] [CrossRef]
- Wen, J.J.; Porter, C.; Garg, N.J. Inhibition of NFE2L2-antioxidant response element pathway by mitochondrial reactive oxygen species contributes to development of cardiomyopathy and left ventricular dysfunction in chagas disease. Antioxid. Redox Signal. 2017, 27, 550–566. [Google Scholar] [CrossRef] [PubMed]
- Paupe, V.; Dassa, E.P.; Goncalves, S.; Auchere, F.; Lonn, M.; Holmgren, A.; Rustin, P. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS ONE 2009, 4, e4253. [Google Scholar] [CrossRef] [PubMed]
- D’Oria, V.; Petrini, S.; Travaglini, L.; Priori, C.; Piermarini, E.; Petrillo, S.; Carletti, B.; Bertini, E.; Piemonte, F. Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2-related factor (Nrf2) in cultured motor neurons. Int. J. Mol. Sci. 2013, 14, 7853–7865. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Schoenfeld, R.A.; Hayashi, G.; Napoli, E.; Akiyama, T.; Iodi Carstens, M.; Carstens, E.E.; Pook, M.A.; Cortopassi, G.A. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid. Redox Signal. 2013, 19, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-Inducer prevents mitochondrial defects and oxidative stress in Friedreich’s ataxia models. Front. Cell. Neurosci. 2018, 12, 188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yang, X.; Yuan, F.; Zhang, L.; Wang, Y.; Wang, L.; Mao, Z.; Luo, J.; Zhang, H.; Zhu, W.G.; et al. Increased amino acid uptake supports autophagy-deficient cell survival upon glutamine deprivation. Cell Rep. 2018, 23, 3006–3020. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Baird, T.D.; Wek, R.C. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv. Nutr. 2012, 3, 307–321. [Google Scholar] [CrossRef]
- Iwawaki, T.; Akai, R.; Toyoshima, T.; Takeda, N.; Ishikawa, T.O.; Yamamura, K.I. Transgenic mouse model for imaging of ATF4 translational activation-related cellular stress responses in vivo. Sci. Rep. 2017, 7, 46230. [Google Scholar] [CrossRef]
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent advances in mitochondrial disease. Annu. Rev. Genom. Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef]
- Fujita, Y.; Ito, M.; Nozawa, Y.; Yoneda, M.; Oshida, Y.; Tanaka, M. CHOP (C/EBP homologous protein) and ASNS (asparagine synthetase) induction in cybrid cells harboring MELAS and NARP mitochondrial DNA mutations. Mitochondrion 2007, 7, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Mjosund, K.P.; Wanrooij, S.; Lappalainen, I.; Ylikallio, E.; Jalanko, A.; Spelbrink, J.N.; Paetau, A.; Suomalainen, A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 17687–17692. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-Omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Jeong, Y.T.; Kim, S.H.; Jung, H.S.; Park, K.S.; Lee, H.Y.; Lee, M.S. Metformin-Induced inhibition of the mitochondrial respiratory chain increases FGF21 expression via ATF4 activation. Biochem. Biophys. Res. Commun. 2013, 440, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.F.; Chen, M.S.; Chou, Y.C.; Ueng, Y.F.; Yin, P.H.; Yeh, T.S.; Lee, H.C. Mitochondrial dysfunction enhances cisplatin resistance in human gastric cancer cells via the ROS-activated GCN2-eIF2α-ATF4-xCT pathway. Oncotarget 2016, 7, 74132–74151. [Google Scholar] [CrossRef]
- Baker, B.M.; Nargund, A.M.; Sun, T.; Haynes, C.M. Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet. 2012, 8, e1002760. [Google Scholar] [CrossRef]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.-H.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial dysfunction is signaled to the integrated stress response by OMA1, DELE1 and HRI. bioRxiv 2019. [Google Scholar] [CrossRef]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease—The long and short of it. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Chen, P.H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef]
- Afonyushkin, T.; Oskolkova, O.V.; Philippova, M.; Resink, T.J.; Erne, P.; Binder, B.R.; Bochkov, V.N. Oxidized phospholipids regulate expression of ATF4 and VEGF in endothelial cells via NRF2-dependent mechanism: Novel point of convergence between electrophilic and unfolded protein stress pathways. Arter. Thromb. Vasc. Biol. 2010, 30, 1007–1013. [Google Scholar] [CrossRef]
- Miyamoto, N.; Izumi, H.; Miyamoto, R.; Bin, H.; Kondo, H.; Tawara, A.; Sasaguri, Y.; Kohno, K. Transcriptional regulation of activating transcription factor 4 under oxidative stress in retinal pigment epithelial ARPE-19/HPV-16 cells. Invest. Ophthalmol. Vis. Sci. 2011, 52, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- He, C.H.; Gong, P.; Hu, B.; Stewart, D.; Choi, M.E.; Choi, A.M.; Alam, J. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J. Biol. Chem. 2001, 276, 20858–20865. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Mimura, J.; Okada, T.; Sato, H.; Liu, T.; Maruyama, A.; Ohyama, C.; Itoh, K. Nrf2-and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol. Cell. Biol. 2014, 34, 3421–3434. [Google Scholar] [CrossRef] [PubMed]
- Mimura, J.; Inose-Maruyama, A.; Taniuchi, S.; Kosaka, K.; Yoshida, H.; Yamazaki, H.; Kasai, S.; Harada, N.; Kaufman, R.J.; Oyadomari, S.; et al. Concomitant Nrf2-and ATF4-activation by carnosic acid cooperatively induces expression of cytoprotective genes. Int. J. Mol. Sci. 2019, 20, 1706. [Google Scholar] [CrossRef] [PubMed]
- Zong, Z.-H.; Du, Z.-X.; Li, N.; Li, C.; Zhang, Q.; Liu, B.-Q.; Guan, Y.; Wang, H.-Q. Implication of Nrf2 and ATF4 in differential induction of CHOP by proteasome inhibition in thyroid cancer cells. Biochim. Biophys. Acta 2012, 1823, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Coleman, V.; Sa-Nguanmoo, P.; Koenig, J.; Schulz, T.J.; Grune, T.; Klaus, S.; Kipp, A.P.; Ost, M. Partial involvement of Nrf2 in skeletal muscle mitohormesis as an adaptive response to mitochondrial uncoupling. Sci. Rep. 2018, 8, 2446. [Google Scholar] [CrossRef]
- Klaus, S.; Rudolph, B.; Dohrmann, C.; Wehr, R. Expression of uncoupling protein 1 in skeletal muscle decreases muscle energy efficiency and affects thermoregulation and substrate oxidation. Physiol. Genom. 2005, 21, 193–200. [Google Scholar] [CrossRef][Green Version]
- Gates, A.C.; Bernal-Mizrachi, C.; Chinault, S.L.; Feng, C.; Schneider, J.G.; Coleman, T.; Malone, J.P.; Townsend, R.R.; Chakravarthy, M.V.; Semenkovich, C.F. Respiratory uncoupling in skeletal muscle delays death and diminishes age-related disease. Cell Metab. 2007, 6, 497–505. [Google Scholar] [CrossRef]
- Adjeitey, C.N.-K.; Mailloux, R.J.; deKemp, R.A.; Harper, M.-E. Mitochondrial uncoupling in skeletal muscle by UCP1 augments energy expenditure and glutathione content while mitigating ROS production. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E405–E415. [Google Scholar] [CrossRef]
- Keipert, S.; Ost, M.; Chadt, A.; Voigt, A.; Ayala, V.; Portero-Otin, M.; Pamplona, R.; Al-Hasani, H.; Klaus, S. Skeletal muscle uncoupling-induced longevity in mice is linked to increased substrate metabolism and induction of the endogenous antioxidant defense system. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E495–E506. [Google Scholar] [CrossRef] [PubMed]
- Ost, M.; Keipert, S.; van Schothorst, E.M.; Donner, V.; van der Stelt, I.; Kipp, A.P.; Petzke, K.J.; Jove, M.; Pamplona, R.; Portero-Otin, M.; et al. Muscle mitohormesis promotes cellular survival via serine/glycine pathway flux. FASEB J. 2015, 29, 1314–1328. [Google Scholar] [CrossRef]
- Klaus, S.; Ost, M. Mitochondrial uncoupling and longevity—A role for mitokines? Exp. Gerontol. 2019, 130, 110796. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, T.; Hinoi, E.; Jung, D.Y.; Kajimura, D.; Ferron, M.; Seo, J.; Graff, J.M.; Kim, J.K.; Karsenty, G. The transcription factor ATF4 regulates glucose metabolism in mice through its expression in osteoblasts. J. Clin. Investig. 2009, 119, 2807–2817. [Google Scholar] [CrossRef]
- Mendez-Lucas, A.; Hyrossova, P.; Novellasdemunt, L.; Vinals, F.; Perales, J.C. Mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) is a pro-survival, endoplasmic reticulum (ER) stress response gene involved in tumor cell adaptation to nutrient availability. J. Biol. Chem. 2014, 289, 22090–22102. [Google Scholar] [CrossRef]
- Potter, J.D. The failure of cancer chemoprevention. Carcinogenesis 2014, 35, 974–982. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. https://doi.org/10.3390/biom10020320
Kasai S, Shimizu S, Tatara Y, Mimura J, Itoh K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules. 2020; 10(2):320. https://doi.org/10.3390/biom10020320
Chicago/Turabian StyleKasai, Shuya, Sunao Shimizu, Yota Tatara, Junsei Mimura, and Ken Itoh. 2020. "Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology" Biomolecules 10, no. 2: 320. https://doi.org/10.3390/biom10020320
APA StyleKasai, S., Shimizu, S., Tatara, Y., Mimura, J., & Itoh, K. (2020). Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules, 10(2), 320. https://doi.org/10.3390/biom10020320