Mitigation of Glucolipotoxicity-Induced Apoptosis, Mitochondrial Dysfunction, and Metabolic Stress by N-Acetyl Cysteine in Pancreatic β-Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Cell Culture and Treatment
2.2.2. Measurement of Apoptosis, DNA Damage, and Caspase Activities
2.2.3. Measurement of Mitochondrial Functions
Measurement of Mitochondrial Membrane Potential MMP (Δψm)
Measurement of Mitochondrial Enzymes and Bioenergetics
2.2.4. Measurement of Activities of Energy Metabolizing Enzymes
2.2.5. SDS-PAGE and Western Blot Analysis
2.2.6. Statistical Analysis
3. Results
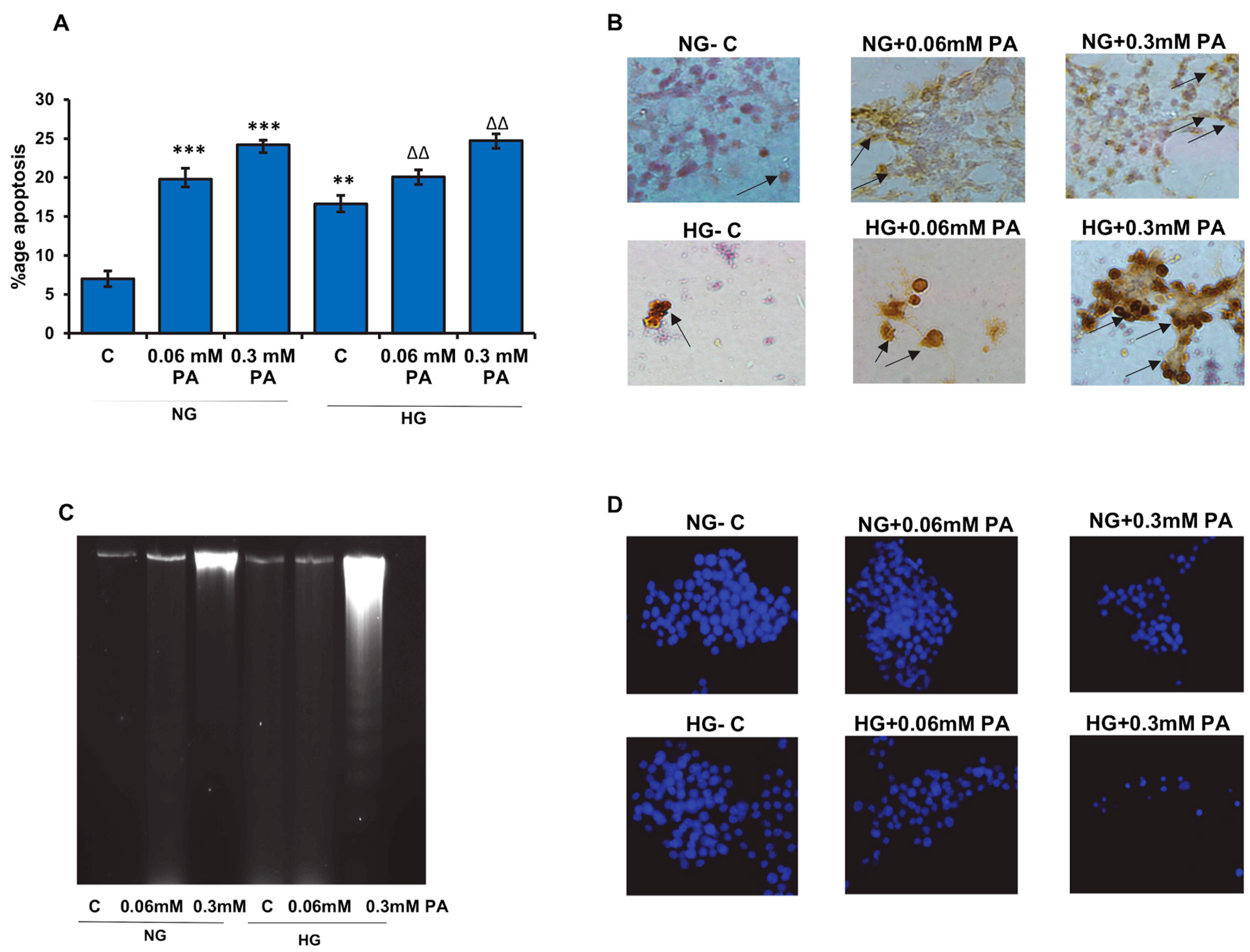
3.1. Effects of High Glucose/High Fatty Acids on Apoptosis
3.2. Effects of High Glucose/High Fatty Acids on Mitochondrial Functions
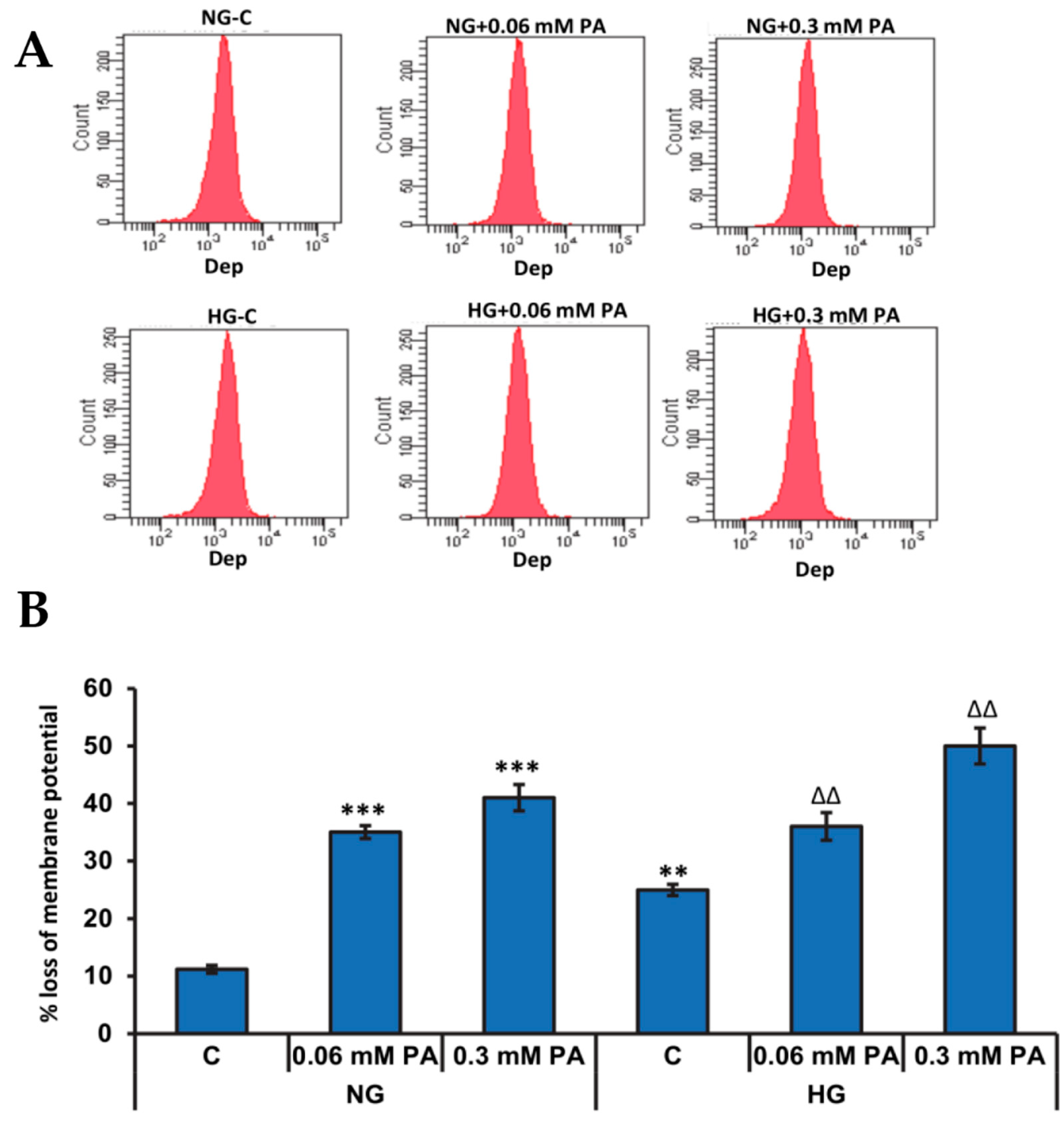
3.2.1. Effects of High Glucose/High Fatty Acids on Mitochondrial Membrane Potential
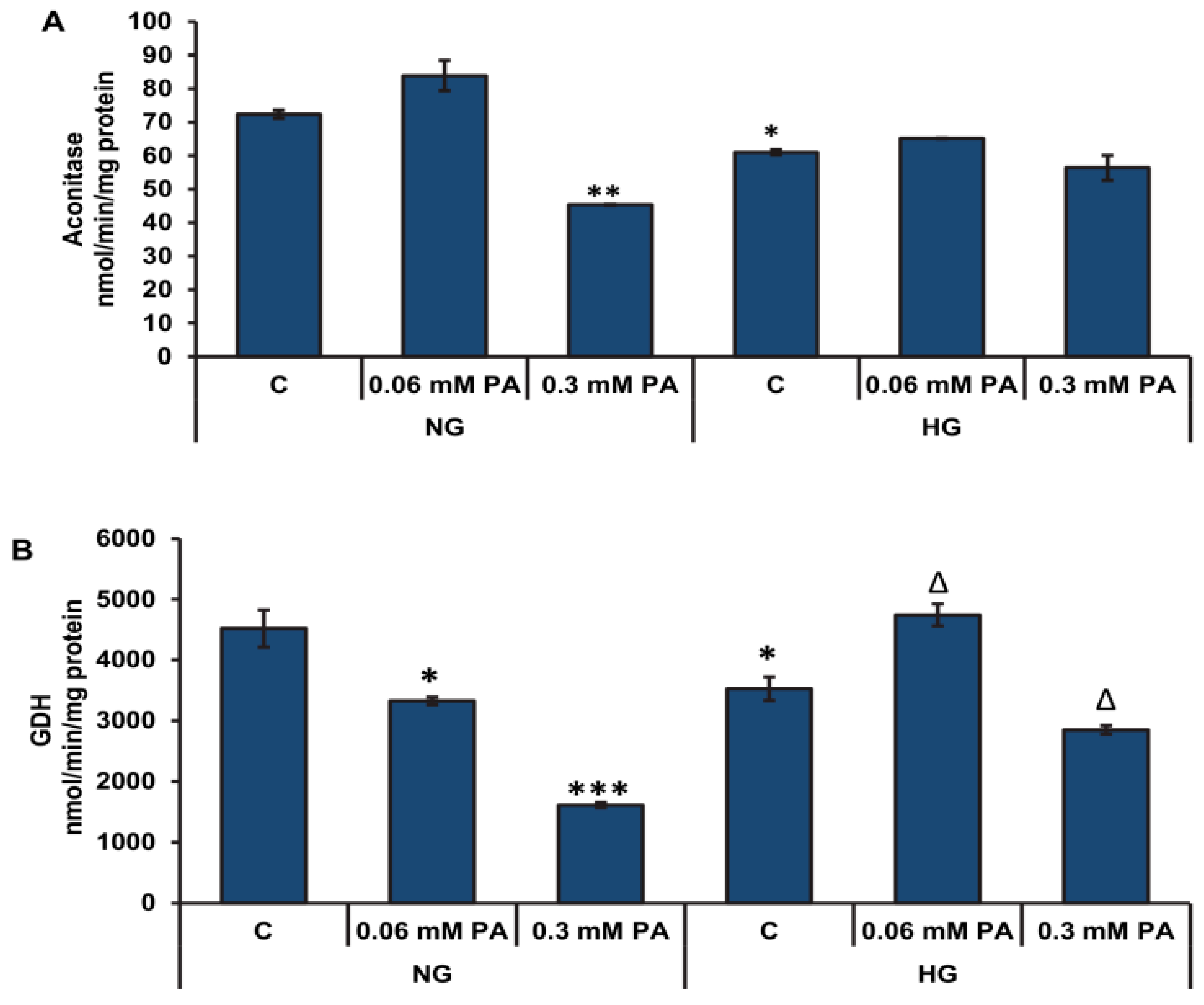
3.2.2. Effects of High Glucose/High Fatty Acids on Mitochondrial Enzymes and Bioenergetics
3.3. Effects of High Glucose/High Palmitic Acid on Cytosolic Energy Metabolizing Enzymes
3.4. Effects of High Glucose/High Palmitic Acid on Expression of Oxidative Stress and Cell Signalling Markers
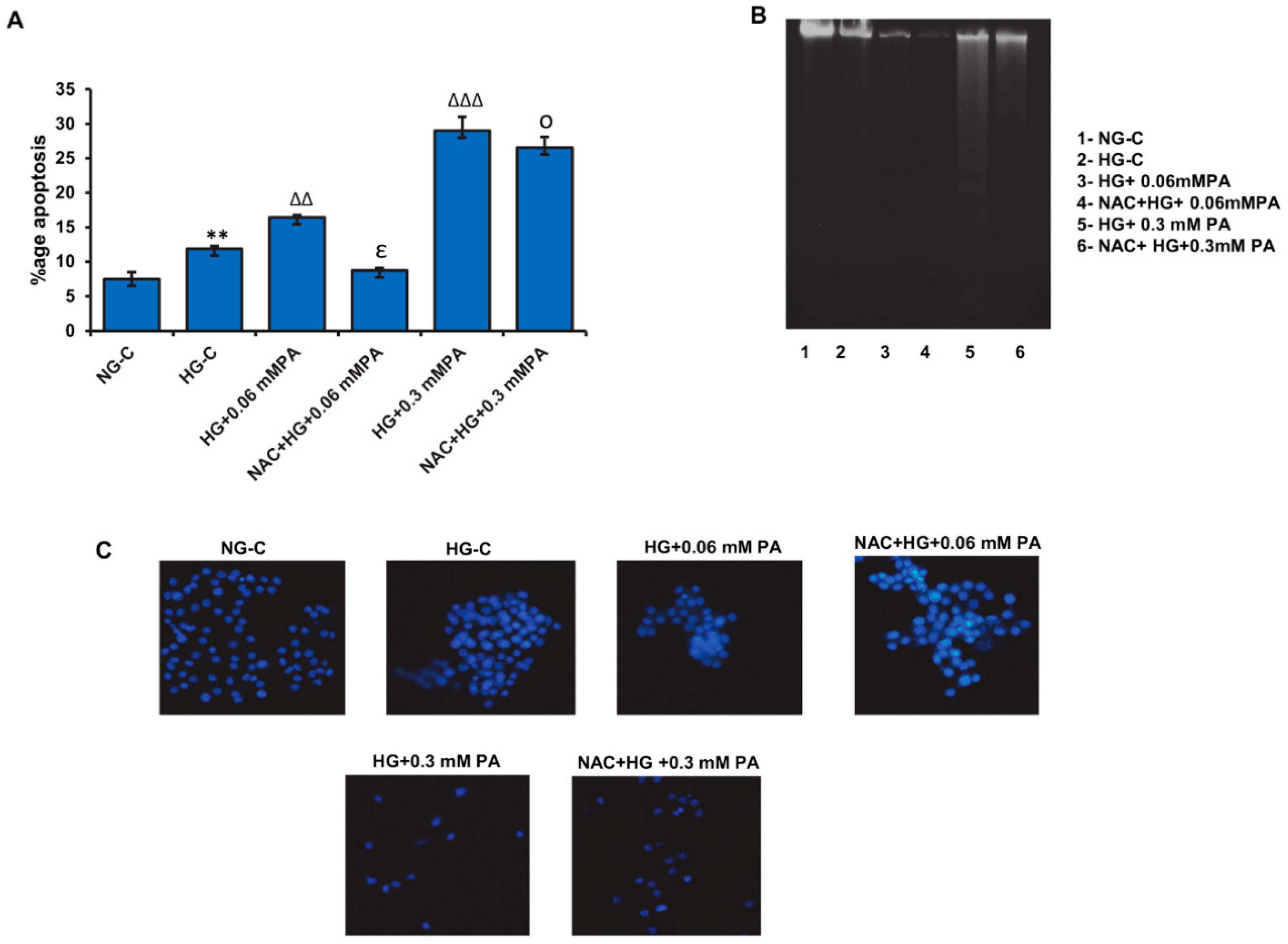
3.5. Effect of NAC Pre-Treatment on Apoptosis in High Glucose/High Palmitic Acid-Treated Cells
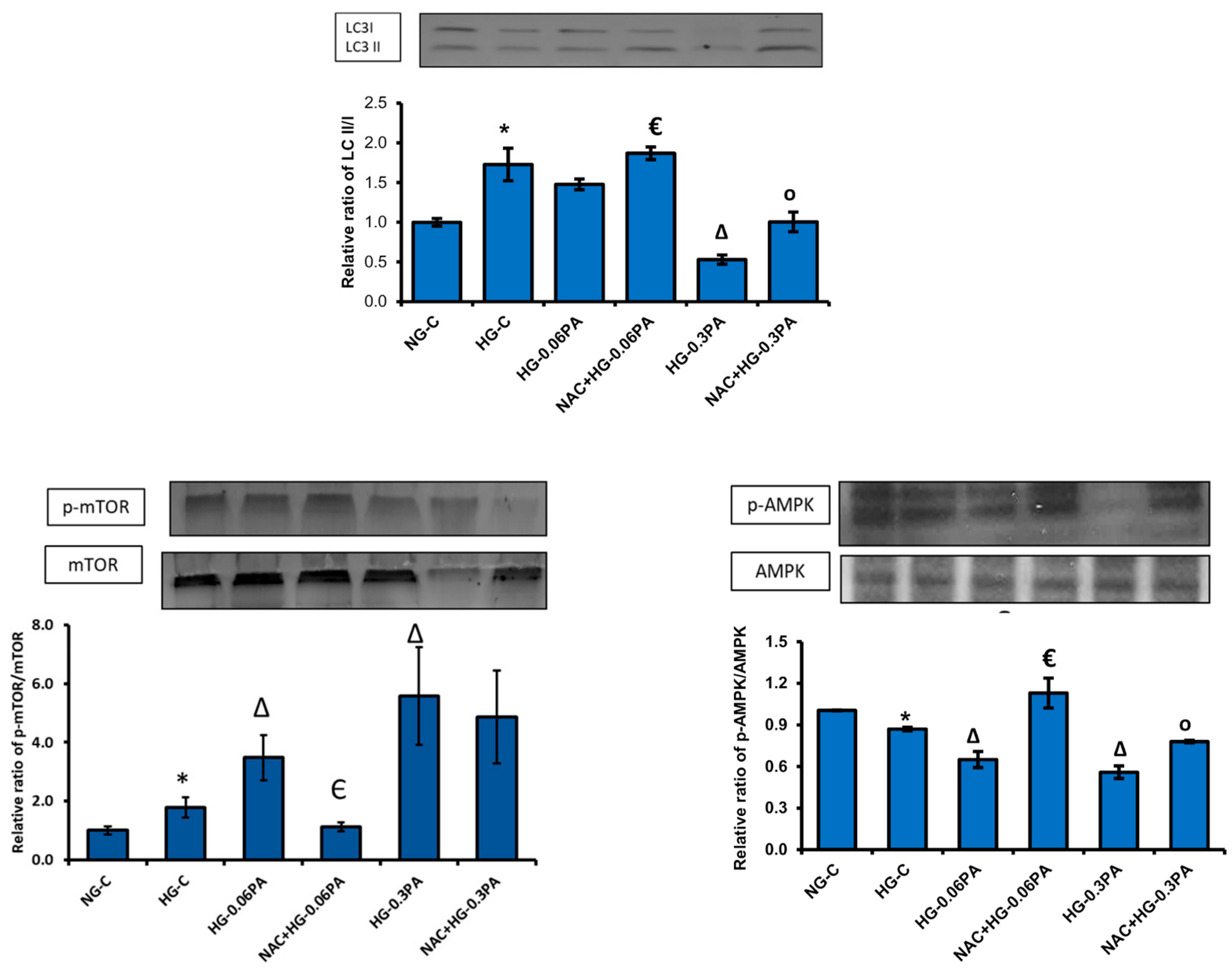
3.6. Effect of NAC Pre-Treatment on the Expression of Apoptotic and Cell Signalling Markers in High Glucose/High Palmitic Acid-Treated Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Flock, M.R.; Kris-Etherton, P.M. Diverse physiological effects of long-chain saturated fatty acids: Implications for cardiovascular disease. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ryu, T.Y.; Park, J.; Scherer, P.E. Hyperglycemia as a Risk Factor for Cancer Progression. Diabetes Metab. J. 2014, 38, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Savary, S.; Trompier, D.; Andréoletti, P.; Le Borgne, F.; Demarquoy, J.; Lizard, G. Fatty acids—Induced lipotoxicity and inflammation. Curr. Drug Metab. 2012, 13, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Masi, L.N.; Rodrigues, A.C.; Curi, R. Fatty acids regulation of inflammatory and metabolic genes. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 418–424. [Google Scholar] [CrossRef]
- Unger, R.H. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes 1995, 44, 863–870. [Google Scholar] [CrossRef]
- Poitout, V.; Robertson, R.P. Minireview: Secondary β-Cell Failure in Type 2 Diabetes—A Convergence of Glucotoxicity and Lipotoxicity. Endocrinology 2002, 143, 339–342. [Google Scholar] [CrossRef]
- Shimo, N.; Matsuoka, T.; Miyatsuka, T.; Takebe, S.; Tochino, Y.; Takahara, M.; Kaneto, H.; Shimomura, I. Short-term selective alleviation of glucotoxicity and lipotoxicity ameliorates the suppressed expression of key β-cell factors under diabetic conditions. Biochem. Biophys. Res. Commun. 2015, 467, 948–954. [Google Scholar] [CrossRef]
- Gleason, C.E.; Gonzalez, M.; Harmon, J.S.; Robertson, R.P. Determinants of glucose toxicity and its reversibility in the pancreatic islet beta-cell line, HIT-T15. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E997–E1002. [Google Scholar] [CrossRef]
- Unger, R.H.; Grundy, S. Hyperglycemia as an inducer as well as a consequence of impaired islet cell function and insulin resistance: Implications for the management of diabetes. Diabetologia 1985, 28, 119–121. [Google Scholar]
- El-Assaad, W.; Buteau, J.; Peyot, M.-L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef]
- Prentki, M.; Vischer, S.; Glennon, M.C.; Regazzi, R.; Deeney, J.T.; Corkey, B.E. Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion. J. Biol. Chem. 1992, 267, 5802–5810. [Google Scholar] [PubMed]
- Alnahdi, A.; John, A.; Raza, H. Augmentation of Glucotoxicity, Oxidative Stress, Apoptosis and Mitochondrial Dysfunction in HepG2 Cells by Palmitic Acid. Nutrients 2019, 11, 1979. [Google Scholar] [CrossRef] [PubMed]
- Alnahdi, A.; John, A.; Raza, H. N-acetyl cysteine attenuates oxidative stress and GSH-dependent redox imbalance caused by high glucose/high palmitic acid treatment in pancreatic Rin-5F cells. PLoS ONE 2019, 14, e0226696. [Google Scholar] [CrossRef] [PubMed]
- Prause, M.; Christensen, D.P.; Billestrup, N.; Mandrup-Poulsen, T. JNK1 protects against glucolipotoxicity-mediated beta-cell apoptosis. PLoS ONE 2014, 9, e87067. [Google Scholar] [CrossRef] [PubMed]
- Joshi-Barve, S.; Barve, S.S.; Amancherla, K.; Gobejishvili, L.; Hill, D.; Cave, M.; Hote, P.; McClain, C.J. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 2007, 46, 823–830. [Google Scholar] [CrossRef]
- Raza, H.; John, A. Implications of altered glutathione metabolism in aspirin-induced oxidative stress and mitochondrial dysfunction in HepG2 cells. PLoS ONE 2012, 7, e36325. [Google Scholar] [CrossRef]
- Raza, H.; John, A.; Benedict, S. Acetylsalicylic acid-induced oxidative stress, cell cycle arrest, apoptosis and mitochondrial dysfunction in human hepatoma HepG2 cells. Eur. J. Pharmacol. 2011, 668, 15–24. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar]
- Raza, H.; John, A.; Shafarin, J. Potentiation of LPS-Induced Apoptotic Cell Death in Human Hepatoma HepG2 Cells by Aspirin via ROS and Mitochondrial Dysfunction: Protection by N-Acetyl Cysteine. PLoS ONE 2016, 11, e0159750. [Google Scholar] [CrossRef]
- Al-Nahdi, A.M.T.; John, A.; Raza, H. Cytoprotective Effects of N-Acetylcysteine on Streptozotocin- Induced Oxidative Stress and Apoptosis in RIN-5F Pancreatic β-Cells. Cell. Physiol. Biochem. 2018, 51, 201–216. [Google Scholar] [CrossRef]
- Nahdi, A.M.T.A.; John, A.; Raza, H. Elucidation of Molecular Mechanisms of Streptozotocin-Induced Oxidative Stress, Apoptosis, and Mitochondrial Dysfunction in Rin-5F Pancreatic β-Cells. Oxid. Med. Cell. Longev. 2017, 2017, 7054272. [Google Scholar] [CrossRef] [PubMed]
- Schmid, I.; Uittenbogaart, C.; Jamieson, B.D. Live-cell assay for detection of apoptosis by dual-laser flow cytometry using Hoechst 33342 and 7-amino-actinomycin D. Nat. Protoc. 2007, 2, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Sibirtsev, V.S.; Garabadzhiu, A.V.; Ivanov, S.D. Spectral Properties of Bisbenzimidazole Dyes upon Interaction with DNA. Russ. J. Bioorg. Chem. C/C Bioorganicheskaia Khimiia 1997, 23, 857–865. [Google Scholar]
- Green, M.R.; Sambrook, J.; Sambrook, J. Molecular Cloning: A Laboratory Manual, 4th ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2012; ISBN 978-1-936113-41-5. [Google Scholar]
- Al-Belooshi, T.; John, A.; Tariq, S.; Al-Otaiba, A.; Raza, H. Increased mitochondrial stress and modulation of mitochondrial respiratory enzyme activities in acetaminophen-induced toxicity in mouse macrophage cells. Food Chem. Toxicol. 2010, 48, 2624–2632. [Google Scholar] [CrossRef]
- Raza, H.; John, A.; Shafarin, J. NAC attenuates LPS-induced toxicity in aspirin-sensitized mouse macrophages via suppression of oxidative stress and mitochondrial dysfunction. PLoS ONE 2014, 9, e103379. [Google Scholar] [CrossRef]
- Birch-Machin, M.A.; Turnbull, D.M. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 2001, 65, 97–117. [Google Scholar]
- Raza, H.; John, A.; Howarth, F.C. Increased oxidative stress and mitochondrial dysfunction in zucker diabetic rat liver and brain. Cell. Physiol. Biochem. 2015, 35, 1241–1251. [Google Scholar] [CrossRef]
- McElroy, W.D. Properties of the Reaction Utilizing Adenosinetriphosphate for Bioluminescence. J. Biol. Chem. 1951, 191, 547–557. [Google Scholar]
- Raza, H.; John, A. Streptozotocin-induced cytotoxicity, oxidative stress and mitochondrial dysfunction in human hepatoma HepG2 cells. Int. J. Mol. Sci. 2012, 13, 5751–5767. [Google Scholar] [CrossRef]
- Briaud, I.; Harmon, J.S.; Kelpe, C.L.; Segu, V.B.; Poitout, V. Lipotoxicity of the pancreatic beta-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes 2001, 50, 315–321. [Google Scholar] [CrossRef]
- Gao, C.-L.; Zhu, C.; Zhao, Y.-P.; Chen, X.-H.; Ji, C.-B.; Zhang, C.-M.; Zhu, J.-G.; Xia, Z.-K.; Tong, M.-L.; Guo, X.-R. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3-L1 adipocytes. Mol. Cell. Endocrinol. 2010, 320, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Park, E.-J.; Lee, A.Y.; Park, S.; Kim, J.-H.; Cho, M.-H. Multiple pathways are involved in palmitic acid-induced toxicity. Food Chem. Toxicol. 2014, 67, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.; Affourtit, C. Novel insights into pancreatic β-cell glucolipotoxicity from real-time functional analysis of mitochondrial energy metabolism in INS-1E insulinoma cells. Biochem. J. 2013, 456, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Wojtczak, L.; Wieckowski, M.R. The mechanisms of fatty acid-induced proton permeability of the inner mitochondrial membrane. J. Bioenerg. Biomembr. 1999, 31, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Rashid, M.A.; Jang, M.; Kim, Y.; Won, H.; Lee, J.; Woo, J.; Kim, Y.S.; Murphy, M.P.; Ali, L.; et al. Mitochondria-targeted antioxidants protect pancreatic β-cells against oxidative stress and improve insulin secretion in glucotoxicity and glucolipotoxicity. Cell. Physiol. Biochem. 2011, 28, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef]
- Penzo, D.; Tagliapietra, C.; Colonna, R.; Petronilli, V.; Bernardi, P. Effects of fatty acids on mitochondria: Implications for cell death. Biochim. Biophys. Acta 2002, 1555, 160–165. [Google Scholar] [CrossRef]
- Mastorodemos, V.; Zaganas, I.; Spanaki, C.; Bessa, M.; Plaitakis, A. Molecular basis of human glutamate dehydrogenase regulation under changing energy demands. J. Neurosci. Res. 2005, 79, 65–73. [Google Scholar] [CrossRef]
- Broniarek, I.; Koziel, A.; Jarmuszkiewicz, W. The effect of chronic exposure to high palmitic acid concentrations on the aerobic metabolism of human endothelial EA.hy926 cells. Pflugers Arch. 2016, 468, 1541–1554. [Google Scholar] [CrossRef]
- Zhang, Z.; Liew, C.W.; Handy, D.E.; Zhang, Y.; Leopold, J.A.; Hu, J.; Guo, L.; Kulkarni, R.N.; Loscalzo, J.; Stanton, R.C. High glucose inhibits glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and β-cell apoptosis. FASEB J. 2010, 24, 1497–1505. [Google Scholar] [CrossRef]
- Gonzalez, C.D.; Lee, M.-S.; Marchetti, P.; Pietropaolo, M.; Towns, R.; Vaccaro, M.I.; Watada, H.; Wiley, J.W. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy 2011, 7, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Karantza-Wadsworth, V. Role and regulation of autophagy in cancer. Biochim. Biophys. Acta 2009, 1793, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Volpe, C.M.O.; Villar-Delfino, P.H.; dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Mir, S.U.R.; George, N.M.; Zahoor, L.; Harms, R.; Guinn, Z.; Sarvetnick, N.E. Inhibition of Autophagic Turnover in β-Cells by Fatty Acids and Glucose Leads to Apoptotic Cell Death. J. Biol. Chem. 2015, 290, 6071–6085. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Shanware, N.P.; Bray, K.; Abraham, R.T. The PI3K, Metabolic, and Autophagy Networks: Interactive Partners in Cellular Health and Disease. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 89–106. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Stienstra, R.; Haim, Y.; Riahi, Y.; Netea, M.; Rudich, A.; Leibowitz, G. Autophagy in adipose tissue and the beta cell: Implications for obesity and diabetes. Diabetologia 2014, 57, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Lasram, M.M.; Dhouib, I.B.; Annabi, A.; El Fazaa, S.; Gharbi, N. A review on the possible molecular mechanism of action of N-acetylcysteine against insulin resistance and type-2 diabetes development. Clin. Biochem. 2015, 48, 1200–1208. [Google Scholar] [CrossRef] [PubMed]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alnahdi, A.; John, A.; Raza, H. Mitigation of Glucolipotoxicity-Induced Apoptosis, Mitochondrial Dysfunction, and Metabolic Stress by N-Acetyl Cysteine in Pancreatic β-Cells. Biomolecules 2020, 10, 239. https://doi.org/10.3390/biom10020239
Alnahdi A, John A, Raza H. Mitigation of Glucolipotoxicity-Induced Apoptosis, Mitochondrial Dysfunction, and Metabolic Stress by N-Acetyl Cysteine in Pancreatic β-Cells. Biomolecules. 2020; 10(2):239. https://doi.org/10.3390/biom10020239
Chicago/Turabian StyleAlnahdi, Arwa, Annie John, and Haider Raza. 2020. "Mitigation of Glucolipotoxicity-Induced Apoptosis, Mitochondrial Dysfunction, and Metabolic Stress by N-Acetyl Cysteine in Pancreatic β-Cells" Biomolecules 10, no. 2: 239. https://doi.org/10.3390/biom10020239
APA StyleAlnahdi, A., John, A., & Raza, H. (2020). Mitigation of Glucolipotoxicity-Induced Apoptosis, Mitochondrial Dysfunction, and Metabolic Stress by N-Acetyl Cysteine in Pancreatic β-Cells. Biomolecules, 10(2), 239. https://doi.org/10.3390/biom10020239