AAPH or Peroxynitrite-Induced Biorelevant Oxidation of Methyl Caffeate Yields a Potent Antitumor Metabolite

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
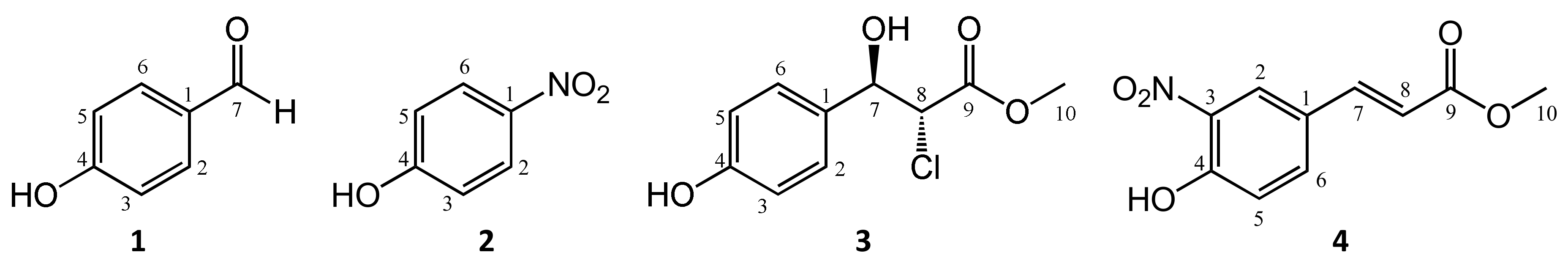
2.1. Continuous-Flow Preparation of Peroxynitrite and Its Reaction with pcm or cm
2.2. Isolation of Products from the Reaction between pcm and Peroxynitrite
2.3. Preparation of Crude Product Mixtures for Bioactivity Screening
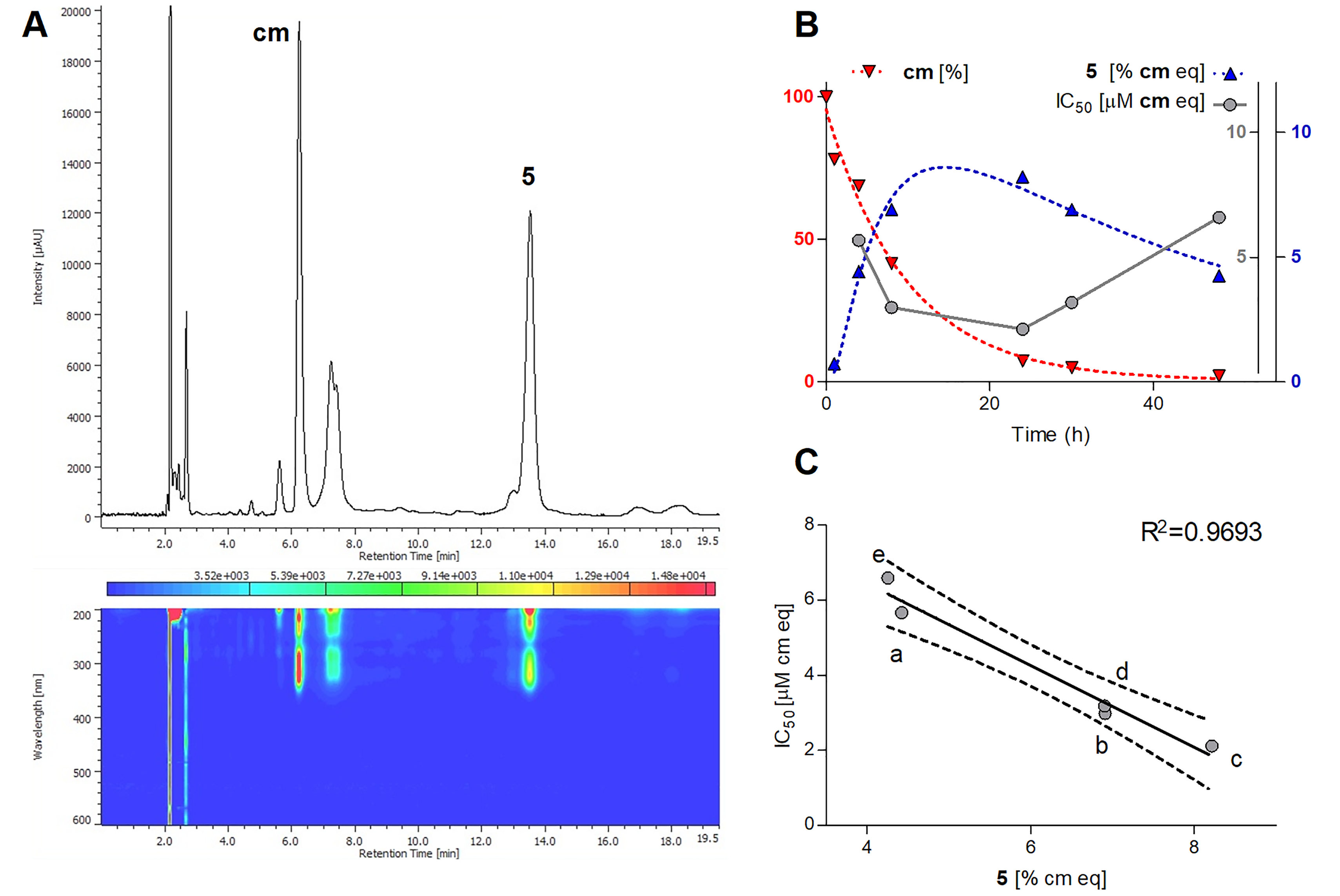
2.4. Longitudinal Study of the Reaction between cm and AAPH
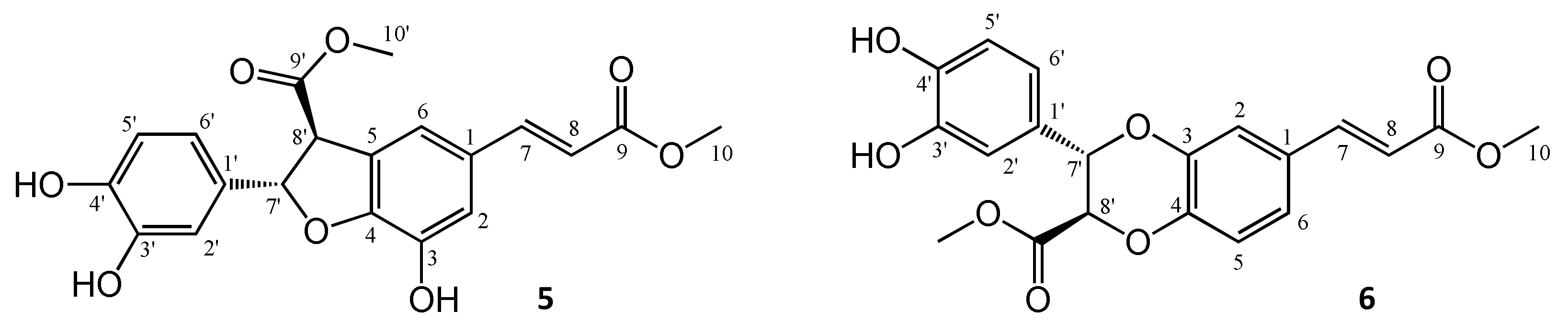
2.5. Isolation of Products from the Reaction between cm and AAPH
2.6. Structure Elucidation
2.7. Cell Lines
2.8. Cell Viability Testing
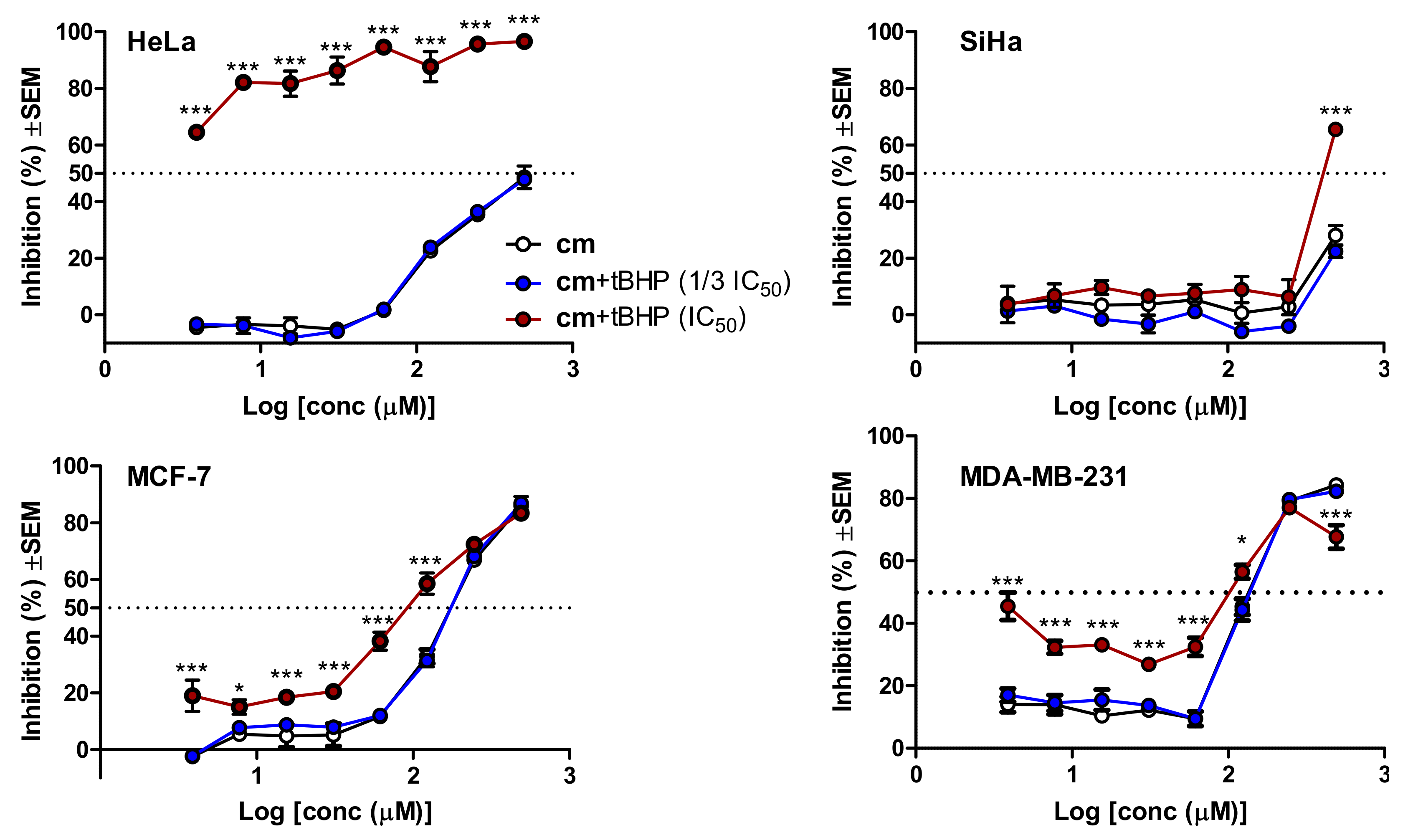
2.9. Testing the Cytotoxicity of cm with or without the Presence of t-BHP-Induced Oxidative Stress
3. Results and Discussion
3.1. In Situ Continuous-Flow Biomimetic Reaction Application of Peroxynitrite as an Oxidative Agent
3.2. Search for Antitumor Metabolites of pcm and cm Oxidized by Peroxynitrite or AAPH
3.3. Bioactivity-Guided Isolation of the Cytotoxic Metabolite of cm
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- El-Seedi, H.R.; El-Said, A.M.A.; Khalifa, S.A.M.; Göransson, U.; Bohlin, L.; Borg-Karlson, A.-K.; Verpoorte, R. Biosynthesis, Natural Sources, Dietary Intake, Pharmacokinetic Properties, and Biological Activities of Hydroxycinnamic Acids. J. Agric. Food Chem. 2012, 60, 10877–10895. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Ho, C.-T. Antioxidant Activities of Caffeic Acid and Its Related Hydroxycinnamic Acid Compounds. J. Agric. Food Chem. 1997, 45, 2374–2378. [Google Scholar] [CrossRef]
- Razzaghi-Asl, N.; Garrido, J.; Khazraei, H.; Borges, F.; Firuzi, O. Antioxidant properties of hydroxycinnamic acids: A review of structure- activity relationships. Curr. Med. Chem. 2013, 20, 4436–4450. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.; Gaspar, A.; Garrido, E.M.; Garrido, J.; Borges, F. Hydroxycinnamic Acid Antioxidants: An Electrochemical Overview. BioMed Res. Int. 2013, 2013, 11. [Google Scholar] [CrossRef] [PubMed]
- Hunyadi, A. The mechanism(s) of action of antioxidants: From scavenging reactive oxygen/nitrogen species to redox signaling and the generation of bioactive secondary metabolites. Med. Res. Rev. 2019, 39, 2505–2533. [Google Scholar] [CrossRef]
- Galloway, W.R.J.D.; Isidro-Llobet, A.; Spring, D.R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 2010, 1, 80–93. [Google Scholar] [CrossRef]
- Kidd, S.L.; Osberger, T.J.; Mateu, N.; Sore, H.F.; Spring, D.R. Recent Applications of Diversity-Oriented Synthesis Toward Novel, 3-Dimensional Fragment Collections. Front. Chem. 2018, 6, 460. [Google Scholar] [CrossRef]
- Yi, S.; Varun, B.V.; Choi, Y.; Park, S.B. A Brief Overview of Two Major Strategies in Diversity-Oriented Synthesis: Build/Couple/Pair and Ring-Distortion. Front. Chem. 2018, 6, 507–515. [Google Scholar] [CrossRef]
- Pavlinov, I.; Gerlach, E.M.; Aldrich, L.N. Next generation diversity-oriented synthesis: A paradigm shift from chemical diversity to biological diversity. Org. Biomol. Chem. 2019, 17, 1608–1623. [Google Scholar] [CrossRef]
- Saeidnia, S.; Abdollahi, M. Antioxidants: Friends or foe in prevention or treatment of cancer: The debate of the century. Toxicol. Appl. Pharmacol. 2013, 271, 49–63. [Google Scholar] [CrossRef]
- Acharya, A.; Das, I.; Chandhok, D.; Saha, T. Redox regulation in cancer: A double-edged sword with therapeutic potential. Oxidative Med. Cell. Longev. 2010, 3, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Tuveson, D.A. The promise and perils of antioxidants for cancer patients. N. Engl. J. Med. 2014, 371, 177–178. [Google Scholar] [CrossRef] [PubMed]
- Reczek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Fási, L.; Di Meo, F.; Kuo, C.-Y.; Stojkovic Buric, S.; Martins, A.; Kúsz, N.; Béni, Z.; Dékány, M.; Balogh, G.T.; Pesic, M.; et al. Antioxidant-Inspired Drug Discovery: Antitumor Metabolite Is Formed in Situ from a Hydroxycinnamic Acid Derivative upon Free-Radical Scavenging. J. Med. Chem. 2019, 62, 1657–1668. [Google Scholar] [CrossRef]
- Pieters, L.; Van Dyck, S.; Gao, M.; Bai, R.; Hamel, E.; Vlietinck, A.; Lemiere, G. Synthesis and biological evaluation of dihydrobenzofuran lignans and related compounds as potential antitumor agents that inhibit tubulin polymerization. J. Med. Chem. 1999, 42, 5475–5481. [Google Scholar] [CrossRef]
- Apers, S.; Paper, D.; Buergermeister, J.; Baronikova, S.; Van Dyck, S.; Lemiere, G.; Vlietinck, A.; Pieters, L. Antiangiogenic Activity of Synthetic Dihydrobenzofuran Lignans. J. Nat. Prod. 2002, 65, 718–720. [Google Scholar] [CrossRef]
- Yin, S.-Y.; Jian, F.-Y.; Chen, Y.-H.; Chien, S.-C.; Hsieh, M.-C.; Hsiao, P.-W.; Lee, W.-H.; Kuo, Y.-H.; Yang, N.-S. Induction of IL-25 secretion from tumour-associated fibroblasts suppresses mammary tumour metastasis. Nat. Commun. 2016, 7, 11311–11325. [Google Scholar] [CrossRef]
- Latif, A.D.; Gonda, T.; Vagvolgyi, M.; Kusz, N.; Kulmany, A.; Ocsovszki, I.; Zomborszki, Z.P.; Zupko, I. Synthesis and In Vitro Antitumor Activity of Naringenin Oxime and Oxime Ether Derivatives. Int. J. Mol. Sci. 2019, 20, 2184. [Google Scholar] [CrossRef]
- Denicola, A.; Souza, J.M.; Radi, R. Diffusion of peroxynitrite across erythrocyte membranes. Proc. Natl. Acad. Sci. USA 1998, 95, 3566–3571. [Google Scholar] [CrossRef]
- Robinson, K.M.; Beckman, J.S. Synthesis of Peroxynitrite from Nitrite and Hydrogen Peroxide. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2005; Volume 396, pp. 207–214. [Google Scholar]
- Kim, H.; Ralph, J.; Lu, F.; Ralph, S.A.; Boudet, A.-M.; MacKay, J.J.; Sederoff, R.R.; Ito, T.; Kawai, S.; Ohashi, H.; et al. NMR analysis of lignins in CAD-deficient plants. Part 1. Incorporation of hydroxycinnamaldehydes and hydroxybenzaldehydes into lignins. Org. Biomol. Chem. 2003, 1, 268–281. [Google Scholar] [CrossRef]
- Karimi Zarchi, M.A.; Rahmani, F. Regioselective and green synthesis of nitro aromatic compounds using polymer-supported sodium nitrite/KHSO4. J. Appl. Polym. Sci. 2011, 120, 2830–2834. [Google Scholar] [CrossRef]
- Schmidt, B.; Hölter, F.; Berger, R.; Jessel, S. Mizoroki–Heck Reactions with 4-Phenoldiazonium Salts. Adv. Synth. Catal. 2010, 352, 2463–2473. [Google Scholar] [CrossRef]
- Matsuki, K.; Sobukawa, M.; Kawai, A.; Inoue, H.; Takeda, M. Asymmetric Reduction of Aromatic Ketones. II. An Enantioselective Synthesis of Methyl (2R, 3S)-3-(4-Methoxyphenyl)glycidate. Chem. Pharm. Bull. 1993, 41, 643–648. [Google Scholar] [CrossRef]
- de la Mare, P.B.D.; Wilson, M.A.; Rosser, M.J. The kinetics and mechanisms of additions to olefinic substances. Part XI. Stereochemistry of addition of chlorine acetate and of chlorine to some unsaturated compounds. J. Chem. Soc. Perkin Trans. 2 1973, 1480–1490. [Google Scholar] [CrossRef]
- Inoue, H.; Matsuki, K.; Oh-Ishi, T. A New Enantioselective Synthesis of (2R, 3S)-3-(4-Methoxyphenyl)glycidic Ester via the Enzymatic Hydrolysis of erythro-N-Acetyl-β-(4-methoxyphenyl)serine. Chem. Pharm. Bull. 1993, 41, 1521–1523. [Google Scholar] [CrossRef]
- Cabon, O.; Buisson, D.; Larcheveque, M.; Azerad, R. The microbial reduction of 2-chloro-3-oxoesters. Tetrahedron Asymmetry 1995, 6, 2199–2210. [Google Scholar] [CrossRef]
- Rubbo, H.; Radi, R. Protein and lipid nitration: Role in redox signaling and injury. Biochim. Biophys. Acta (BBA) Gen. Subj. 2008, 1780, 1318–1324. [Google Scholar] [CrossRef]
- Matsumoto, K.; Takahashi, H.; Miyake, Y.; Fukuyama, Y. Convenient syntheses of neurotrophic americanol A and isoamericanol A by HRP catalyzed oxidative coupling of caffeic acid. Tetrahedron Lett. 1999, 40, 3185–3186. [Google Scholar] [CrossRef]
- Halliwell, B. Cell culture, oxidative stress, and antioxidants: Avoiding pitfalls. Biomed. J. 2014, 37, 99–105. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5849. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, D.; Asaduzzaman, M.; Young, F. Real time monitoring and quantification of reactive oxygen species in breast cancer cell line MCF-7 by 2’,7’-dichlorofluorescin diacetate (DCFDA) assay. J. Pharmacol. Toxicol. Methods 2018, 94, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 [95% C.I.] (µM) | |||
|---|---|---|---|---|
| HeLa | SiHa | MCF-7 | MDA-MB-231 | |
| cm | 450 [396.7–551.2] | >500 | 175.4 [162.3–189.7] | 139.3 [116.5–166.6] |
| 5 | 1.1 [1.0–1.2] | >30 | 1.1 [0.9–1.4] | 3.9 [3.1–4.9] |
| cisplatin | 11.7 [10.3–13.1] | 13.6 [12.6–14.7] | 5.2 [4.6–5.8] | 25.8 [24.4–27.4] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fási, L.; Latif, A.D.; Zupkó, I.; Lévai, S.; Dékány, M.; Béni, Z.; Könczöl, Á.; Balogh, G.T.; Hunyadi, A. AAPH or Peroxynitrite-Induced Biorelevant Oxidation of Methyl Caffeate Yields a Potent Antitumor Metabolite. Biomolecules 2020, 10, 1537. https://doi.org/10.3390/biom10111537
Fási L, Latif AD, Zupkó I, Lévai S, Dékány M, Béni Z, Könczöl Á, Balogh GT, Hunyadi A. AAPH or Peroxynitrite-Induced Biorelevant Oxidation of Methyl Caffeate Yields a Potent Antitumor Metabolite. Biomolecules. 2020; 10(11):1537. https://doi.org/10.3390/biom10111537
Chicago/Turabian StyleFási, Laura, Ahmed Dhahir Latif, István Zupkó, Sándor Lévai, Miklós Dékány, Zoltán Béni, Árpád Könczöl, György Tibor Balogh, and Attila Hunyadi. 2020. "AAPH or Peroxynitrite-Induced Biorelevant Oxidation of Methyl Caffeate Yields a Potent Antitumor Metabolite" Biomolecules 10, no. 11: 1537. https://doi.org/10.3390/biom10111537
APA StyleFási, L., Latif, A. D., Zupkó, I., Lévai, S., Dékány, M., Béni, Z., Könczöl, Á., Balogh, G. T., & Hunyadi, A. (2020). AAPH or Peroxynitrite-Induced Biorelevant Oxidation of Methyl Caffeate Yields a Potent Antitumor Metabolite. Biomolecules, 10(11), 1537. https://doi.org/10.3390/biom10111537