Interface of Phospholipase Activity, Immune Cell Function, and Atherosclerosis




{kind=link}
{kind=link}
Abstract
:1. Introduction
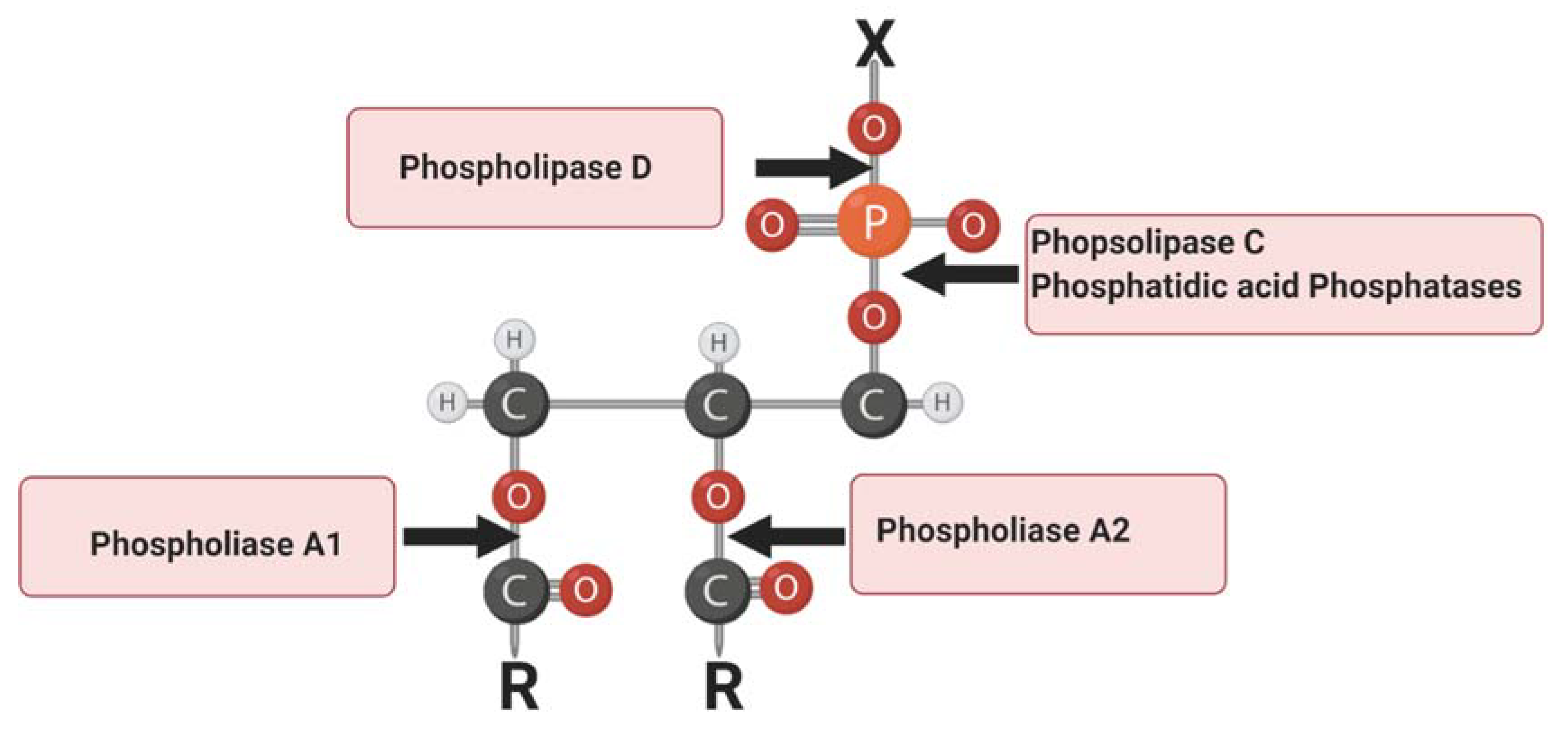
2. Lipoprotein-Associated Phospholipase A2
3. Lipid Phosphate Phosphatases
4. Phospholipase C
5. Phospholipase D
6. Cytosolic Phospholipase A2
7. Lipin 1
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yang, Y.; Lee, M.; Fairn, G.D. Phospholipid subcellular localization and dynamics. J. Biol. Chem. 2018, 293, 6230–6240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. There are three common types of membrane lipids. In Biochemistry, 5th ed.; W H Freeman: New York, NY, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK22361/ (accessed on 30 September 2020).
- Albeituni, S.; Stiban, J. Roles of ceramides and other sphingolipids in immune cell function and inflammation. Adv. Exp. Med. Biol. 2019, 1161, 169–191. [Google Scholar] [PubMed]
- Gendaszewska-Darmach, E. Lysophosphatidic acids, cyclic phosphatidic acids and autotaxin as promising targets in therapies of cancer and other diseases. Acta Biochim. Pol. 2008, 55, 227–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, D.E. Phospholipid metabolism and cell signalling in eucaryotes. New Compr. Biochem. 1991, 7, 205–240. [Google Scholar]
- Aloulou, A.; Rahier, R.; Arhab, Y.; Noiriel, A.; Abousalham, A. Phospholipases: An Overview. Methods Mol. Biol. 2018, 1835, 69–105. [Google Scholar]
- Gisterå, A.; Hansson, G.K. Immunol. Atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar]
- Boullier, A.; Bird, D.A.; Chang, M.-K.; Dennis, E.A.; Friedman, P.; Gillotte-Taylor, K.; Hörkkö, S.; Palinski, W.; Quehenberger, O.; Shaw, P.; et al. Scavenger receptors, oxidized LDL, and atherosclerosis. Ann. N. Y. Acad. Sci. 2001, 947, 214–222. [Google Scholar] [CrossRef]
- Halliwell, B.; Cross, C.E. Oxygen-derived species: Their relation to human disease and environmental stress. Environ. Health Perspect. 1994, 102 (Suppl. 10), 5–12. [Google Scholar]
- Serafini, M.; Del Rio, D. Understanding the association between dietary antioxidants, redox status and disease: Is the total antioxidant capacity the right tool? Redox Rep. 2004, 9, 145–152. [Google Scholar] [CrossRef]
- Hazell, L.J.; Stocker, R. Oxidation of low-density lipoprotein with hypochlorite causes transformation of the lipoprotein into a high-uptake form for macrophages. Biochem. J. 1993, 290, 165–172. [Google Scholar] [CrossRef]
- Stocker, R.; Keaney, J.F., Jr. New insights on oxidative stress in the artery wall. J. Thromb. Haemost. 2005, 3, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.; Ley, K. Immunity and inflammation in atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Heinz, J.; Marinello, M.; Fredman, G. Pro-resolution therapeutics for cardiovascular diseases. Prostaglandins Other Lipid Mediat. 2017, 132, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Schlegel, M.P.; Afonso, M.S.; Brown, E.J.; Rahman, K.; Weinstock, A.; Sansbury, B.E.; Corr, E.M.; Van Solingen, C.; Koelwyn, G.J.; et al. Regulatory T cells license macrophage pro-resolving functions during atherosclerosis regression. Circ. Res. 2020, 127, 335–353. [Google Scholar] [CrossRef]
- O’Donnell, V.B.; Rossjohn, J.; Wakelam, M.J. Phospholipid signaling in innate immune cells. J. Clin. Investig. 2018, 128, 2670–2679. [Google Scholar] [CrossRef]
- Tjoelker, L.W.; Wilder, C.; Eberhardt, C.; Stafforinit, D.M.; Dietsch, G.; Schimpf, B.; Hooper, S.; Le Trong, H.; Cousens, L.S.; Zimmerman, G.A.; et al. Anti-inflammatory properties of a platelet-activating factor acetylhydrolase. Nature 1995, 374, 549–553. [Google Scholar] [CrossRef]
- Min, J.H.; Jain, M.K.; Wilder, C.; Paul, L.; Apitz-Castro, R.; Aspleaf, D.C.; Gelb, M.H. Membrane-bound plasma platelet activating factor acetylhydrolase acts on substrate in the aqueous phase. Biochemistry 1999, 38, 12935–12942. [Google Scholar] [CrossRef]
- Stafforini, D.M.; McIntyre, T.M.; E Carter, M.; Prescott, S.M. Human plasma platelet-activating factor acetylhydrolase. Association with lipoprotein particles and role in the degradation of platelet-activating factor. J. Biol. Chem. 1987, 262, 4215–4222. [Google Scholar]
- Tselepis, A.D.; Chapman, M.D. Inflammation, bioactive lipids and atherosclerosis: Potential roles of a lipoprotein-associated phospholipase A2, platelet activating factor-acetylhydrolase. Atheroscler. Suppl. 2002, 3, 57–68. [Google Scholar] [CrossRef]
- Khakpour, H.; Frishman, W.H. Lipoprotein-associated phospholipase A2: An independent predictor of cardiovascular risk and a novel target for immunomodulation therapy. Cardiol. Rev. 2009, 17, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.D.; Navab, M.; Hama, S.Y.; Sevanian, A.; Prescott, S.M.; Stafforini, D.M.; McIntyre, T.M.; Du, B.N.; Fogelman, A.M.; A Berliner, J. Effect of platelet activating factor-acetylhydrolase on the formation and action of minimally oxidized low density lipoprotein. J. Clin. Investig. 1995, 95, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Stafforini, D.M.; McIntyre, T.M.; A Zimmerman, G.; Prescott, S.M. Platelet-activating factor acetylhydrolases. J. Biol. Chem. 1997, 272, 17895–17898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marathe, G.K.; Pandit, C.; Lakshmikanth, C.L.; Chaithra, V.H.; Jacob, S.P.; D’Souza, C.J.M. To hydrolyze or not to hydrolyze: The dilemma of platelet-activating factor acetylhydrolase. J. Lipid Res. 2014, 55, 1847–1854. [Google Scholar] [CrossRef] [Green Version]
- Maiolino, G.; Bisogni, V.; Rossitto, G.; Rossi, G.P. Lipoprotein-associated phospholipase A2 prognostic role in atherosclerotic complications. World J. Cardiol. 2015, 7, 609–620. [Google Scholar] [CrossRef]
- Yamada, Y.; Yoshida, H.; Ichihara, S.; Imaizumi, T.; Satoh, K.; Yokota, M. Correlations between plasma platelet-activating factor acetylhydrolase (PAF-AH) activity and PAF-AH genotype, age, and atherosclerosis in a Japanese population. Atherosclerosis 2000, 150, 209–216. [Google Scholar] [CrossRef]
- Unno, N.; Nakamura, T.; Kaneko, H.; Uchiyama, T.; Yamamoto, N.; Sugatani, J.; Miwa, M. Plasma platelet-activating factor acetylhydrolase deficiency is associated with atherosclerotic occlusive disease in japan. J. Vasc. Surg. 2000, 32, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Tsaoussis, V.; Vakirtzi-Lemonias, C. The mouse plasma PAF acetylhydrolase: II. It consists of two enzymes both associated with the HDL. J. Lipid Mediat. Cell Signal. 1994, 9, 317–331. [Google Scholar]
- Singh, U.; Zhong, S.; Xiong, M.; Li, T.-B.; Sniderman, A.; Teng, B.-B. Increased plasma non-esterified fatty acids and platelet-activating factor acetylhydrolase are associated with susceptibility to atherosclerosis in mice. Clin. Sci. 2004, 106, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Chroni, A.; Mavri-Vavayanni, M. Characterization of a platelet activating factor acetylhydrolase from rat adipocyte. Life Sci. 2000, 67, 2807–2825. [Google Scholar] [CrossRef]
- Elstad, M.R.; Stafforini, D.M.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A. Platelet-activating factor acetylhydrolase increases during macrophage differentiation. A novel mechanism that regulates accumulation of platelet-activating factor. J. Biol. Chem. 1989, 264, 8467–8470. [Google Scholar] [PubMed]
- Stafforini, D.M.; Tjoelker, L.W.; McCormick, S.P.; Vaitkus, D.; McIntyre, T.M.; Gray, P.W.; Young, S.G.; Prescott, S.M. Molecular basis of the interaction between plasma platelet-activating factor acetylhydrolase and low density lipoprotein. J. Biol. Chem. 1999, 274, 7018–7024. [Google Scholar] [CrossRef] [Green Version]
- Rosenson, R.S. Lp-PLA(2) and risk of atherosclerotic vascular disease. Lancet 2010, 375, 1498–1500. [Google Scholar] [CrossRef]
- Macphee, C.H.; Moores, K.E.; Boyd, H.F.; Dhanak, D.; Ife, R.J.; Leach, C.A.; Leake, D.S.; Milliner, K.J.; Patterson, R.A.; Suckling, K.E.; et al. Lipoprotein-associated phospholipase A2, platelet-activating factor acetylhydrolase, generates two bioactive products during the oxidation of low-density lipoprotein: Use of a novel inhibitor. Biochem. J. 1999, 338 Pt 2, 479–487. [Google Scholar] [CrossRef]
- Karakas, M.; Koenig, W. Lp-Pla2 Inhib. -Atheroscler. Panacea? Pharmaceuticals 2010, 3, 1360–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, K.L.; Dennis, I.F.; Challis, I.R.; Osborn, D.P.; Macphee, C.H.; Leake, D.S.; Arends, M.J.; Mitchinson, M.J. Inhibition of lipoprotein-associated phospholipase A2 diminishes the death-inducing effects of oxidised LDL on human monocyte-macrophages. FEBS Lett. 2001, 505, 357–363. [Google Scholar] [CrossRef] [Green Version]
- Quinn, M.T.; Parthasarathy, S.; Steinberg, D. Lysophosphatidylcholine: A chemotactic factor for human monocytes and its potential role in atherogenesis. Proc. Natl. Acad. Sci. USA 1988, 85, 2805–2809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavi, S.; McConnell, J.P.; Rihal, C.S.; Prasad, A.; Mathew, V.; Lerman, L.O.; Lerman, L.O. Local production of lipoprotein-associated phospholipase A2 and lysophosphatidylcholine in the coronary circulation: Association with early coronary atherosclerosis and endothelial dysfunction in humans. Circulation 2007, 115, 2715–2721. [Google Scholar] [CrossRef] [Green Version]
- Hazen, S.L. Oxidized phospholipids as endogenous pattern recognition ligands in innate immunity. J. Biol. Chem. 2008, 283, 15527–15531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilensky, R.L.; Macphee, C.H. Lipoprotein-Assoc. Phospholipase A(2) Atherosclerosis. Curr. Opin. Lipidol. 2009, 20, 415–420. [Google Scholar] [CrossRef]
- Zalewski, A.; Macphee, C. Role of lipoprotein-associated phospholipase A2 in atherosclerosis: Biology, epidemiology, and possible therapeutic target. Arter. Thromb. Vasc. Biol. 2005, 25, 923–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häkkinen, T.; Luoma, J.S.; Hiltunen, M.O.; Macphee, C.H.; Milliner, K.J.; Patel, L.; Rice, S.Q.; Tew, D.G.; Karkola, K.; Ylä-Herttuala, S. Lipoprotein-associated phospholipase A(2), platelet-activating factor acetylhydrolase, is expressed by macrophages in human and rabbit atherosclerotic lesions. Arter. Thromb. Vasc. Biol. 1999, 19, 2909–2917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolodgie, F.D.; Burke, A.P.; Taye, A.; Liu, W.H.; Sudhir, K.; Virmani, R. Lipoprotein-associated phospholipase A2 is highly expressed in macrophages of coronary lesions prone to rupture. Circulation 2004, 110, 246–247. [Google Scholar]
- Stewart, R.A.; White, H.D. The role of lipoprotein-associated phospholipase a2 as a marker and potential therapeutic target in atherosclerosis. Curr. Atheroscler. Rep. 2011, 13, 132–137. [Google Scholar] [CrossRef]
- Kolodgie, F.D.; Burke, A.; Skorija, K.S.; Ladich, E.; Kutys, R.; Makuria, A.T.; Virmani, R. Lipoprotein-Associated Phospholipase A 2 Protein Expression in the Natural Progression of Human Coronary Atherosclerosis. Arter. Thromb. Vasc. Biol. 2006, 26, 2523–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sairam, S.G.; Sola, S.; Barooah, A.; Javvaji, S.K.; Jaipuria, J.; Venkateshan, V.; Chelli, J.; Sanjeevi, C.B. The role of Lp-PLA2 and biochemistry parameters as potential biomarkers of coronary artery disease in Asian South-Indians: A case-control study. Cardiovasc. Diagn. Ther. 2017, 7, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Sakka, S.; Siahanidou, T.; Voyatzis, C.; Pervanidou, P.; Kaminioti, C.; Lazopoulou, N.; Kanaka-Gantenbein, C.; Chrousos, G.P.; Papassotiriou, I. Elevated circulating levels of lipoprotein-associated phospholipase A2 in obese children. Clin. Chem. Lab. Med. 2015, 53, 1119–1125. [Google Scholar] [CrossRef]
- Donato, L.J.; Meeusen, J.W.; Callanan, H.; Saenger, A.K.; Jaffe, A.S. Advantages of the lipoprotein-associated phospholipase A2 activity assay. Clin. Biochem. 2016, 49, 172–175. [Google Scholar] [CrossRef]
- Dennis, E.A. Introduction to Thematic Review Series: Phospholipases: Central Role in Lipid Signaling and Disease. J. Lipid Res. 2015, 56, 1245–1247. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Benesch, M.G.K.; Brindley, D.N. Lipid phosphate phosphatases and their roles in mammalian physiology and pathology. J. Lipid Res. 2015, 56, 2048–2060. [Google Scholar] [CrossRef] [Green Version]
- Brindley, D.N.; Pilquil, C. Lipid phosphate phosphatases and signaling. J. Lipid Res. 2008, 50, S225–S230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntyre, T.M.; Pontsler, A.V.; Silva, A.R.; St Hilaire, A. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc. Natl. Acad. Sci. USA 2003, 100, 131–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabbai, S.; Moreno-Fernández, R.D.; Zambrana-Infantes, E.; Nieto-Quero, A.; Chun, J.; García-Fernández, M.; Estivill-Torrús, G.; De Fonseca, F.R.; Santín, L.J.; Gil Oliveira, T.; et al. Effects of the LPA1 Receptor Deficiency and Stress on the Hippocampal LPA Species in Mice. Front. Mol. Neurosci. 2019, 12, 146. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.-W.; Mutoh, T.; Lin, M.-E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; et al. LPA Receptors: Subtypes and Biological Actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef] [Green Version]
- Loveridge, C.; Tonelli, F.; Leclercq, T.; Lim, K.G.; Long, J.S.; Berdyshev, E.; Tate, R.J.; Natarajan, V.; Pitson, S.M.; Pyne, N.J.; et al. The Sphingosine Kinase 1 Inhibitor 2-(p-Hydroxyanilino)-4-(p-chlorophenyl)thiazole Induces Proteasomal Degradation of Sphingosine Kinase 1 in Mammalian Cells. J. Biol. Chem. 2010, 285, 38841–38852. [Google Scholar] [CrossRef] [Green Version]
- Sobel, K.; Menyhart, K.; Killer, N.; Renault, B.; Bauer, Y.; Studer, R.; Steiner, B.; Bolli, M.H.; Nayler, O.; Gatfield, J. Sphingosine 1-Phosphate (S1P) Receptor Agonists Mediate Pro-fibrotic Responses in Normal Human Lung Fibroblasts via S1P2 and S1P3 Receptors and Smad-independent Signaling. J. Biol. Chem. 2013, 288, 14839–14851. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Yuan, Y.; Lin, W.; Zhong, H.; Xu, K.; Qi, X. Roles of sphingosine-1-phosphate signaling in cancer. Cancer Cell Int. 2019, 19, 295. [Google Scholar] [CrossRef]
- Zhang, Q.X.; Carlos, S.P.; Jay, D.; Luc, G.B.; David, N.B. Identification of structurally important domains of lipid phosphate phosphatase-1: Implications for its sites of action. Biochem. J. 2000, 345 Pt 2, 181–184. [Google Scholar] [CrossRef]
- Humtsoe, J.O.; Bowling, R.A.; Feng, S.; Wary, K.K. Murine lipid phosphate phosphohydrolase-3 acts as a cell-associated integrin ligand. Biochem. Biophys. Res. Commun. 2005, 335, 906–919. [Google Scholar] [CrossRef]
- Humtsoe, J.O.; Feng, S.; Thakker, G.D.; Yang, J.; Hong, J.; Wary, K.K. Regulation of cell–cell interactions by phosphatidic acid phosphatase 2b/VCIP. EMBO J. 2003, 22, 1539–1554. [Google Scholar] [CrossRef] [Green Version]
- Tanyi, J.L.; Hasegawa, Y.; LaPushin, R.; Morris, A.J.; Wolf, J.K.; Berchuck, A.; Lu, K.; Smith, D.I.; Kalli, K.; Hartmann, L.C.; et al. Role of decreased levels of lipid phosphate phosphatase-1 in accumulation of lysophosphatidic acid in ovarian cancer. Clin. Cancer Res. 2003, 9, 3534–3545. [Google Scholar] [PubMed]
- Smyth, S.S.; Sciorra, V.A.; Sigal, Y.J.; Pamuklar, Z.; Wang, Z.; Xu, Y.; Prestwich, G.D.; Morris, A.J. Lipid Phosphate Phosphatases Regulate Lysophosphatidic Acid Production and Signaling in Platelets. J. Biol. Chem. 2003, 278, 43214–43223. [Google Scholar] [CrossRef] [Green Version]
- Panchatcharam, M.; Salous, A.K.; Brandon, J.; Miriyala, S.; Wheeler, J.; Patil, P.; Sunkara, M.; Morris, A.J.; Escalante-Alcalde, D.; Smyth, S.S. Mice with Targeted Inactivation of Ppap2b in Endothelial and Hematopoietic Cells Display Enhanced Vascular Inflammation and Permeability. Arter. Thromb. Vasc. Biol. 2014, 34, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Martínez, E.; Fernández-Ulibarri, I.; Lázaro-Diéguez, F.; Johannes, L.; Pyne, S.; Sarri, E.; Egea, G. Lipid phosphate phosphatase 3 participates in transport carrier formation and protein trafficking in the early secretory pathway. J. Cell Sci. 2013, 126, 2641–2655. [Google Scholar] [CrossRef] [Green Version]
- Sciorra, V.A.; Morris, A.J. Sequential Actions of Phospholipase D and Phosphatidic Acid Phosphohydrolase 2b Generate Diglyceride in Mammalian Cells. Mol. Biol. Cell 1999, 10, 3863–3876. [Google Scholar] [CrossRef] [Green Version]
- Schunkert, H.; Cardiogenics, J.F.C.; König, I.R.; Kathiresan, S.; Reilly, M.P.; Assimes, T.L.; Holm, H.; Preuss, M.; Stewart, A.F.R.; Barbalic, M.; et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 2011, 43, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Mueller, P.A.; Yang, L.; Ubele, M.; Mao, G.; Brandon, J.; Vandra, J.; Nichols, T.C.; Escalante-Alcalde, D.; Morris, A.J.; Smyth, S.S. Coronary Artery Disease Risk-Associated Plpp3 gene and its product lipid phosphate phosphatase 3 regulate experimental atheroscerosis. Arter. Thromb. Vasc. Biol. 2019, 39, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Wirtwein, M.; Olle, M.; Marketa, S.; Michal, H.; Krzysztof, N.; Marcin, G.; Wojciech, S. Relationship between selected DNA polymorphisms and coronary artery disease complications. Int. J. Cardiol. 2017, 228, 814–820. [Google Scholar] [CrossRef]
- Reschen, M.E.; Gaulton, K.J.; Lin, D.; Soilleux, E.J.; Morris, A.J.; Smyth, S.S.; O’Callaghan, C.A. Lipid-Induced Epigenomic Changes in Human Macrophages Identify a Coronary Artery Disease-Associated Variant that Regulates PPAP2B Expression through Altered C/EBP-Beta Binding. PLoS Genet. 2015, 11, e1005061. [Google Scholar] [CrossRef]
- Siess, W.; Zangl, K.J.; Essler, M.; Bauer, M.; Brandl, R.; Corrinth, C.; Bittman, R.; Tigyi, G.; Aepfelbacher, M. Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proc. Natl. Acad. Sci. USA 1999, 96, 6931–6936. [Google Scholar] [CrossRef] [Green Version]
- Escalante-Alcalde, D.; Hernandez, L.; Le Stunff, H.; Maeda, R.; Lee, H.S.; Sciorra, V.A.; Daar, I.; Spiegel, S.; Morris, A.J.; Stewart, C.L. The lipid phosphatase LPP3 regulates extra-embryonic vasculogenesis and axis patterning. Development 2003, 130, 4623–4637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busnelli, M.; Manzini, S.; Hilvo, M.; Parolini, C.; Ganzetti, G.S.; Dellera, F.; Ekroos, K.; Jänis, M.; Escalante-Alcalde, D.; Sirtori, C.R.; et al. Liver-specific deletion of the Plpp3 gene alters plasma lipid composition and worsens atherosclerosis in apoE−/− mice. Sci. Rep. 2017, 7, 44503. [Google Scholar] [CrossRef]
- Gresset, A.; Sondek, J.; Harden, T.K. The Phospholipase C Isozymes and Their Regulation. Subcell. Biochem. 2012, 58, 61–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Fukami, K. Regulation and physiological functions of mammalian phospholipase C. J. Biochem. 2017, 89, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocco, L.; Follo, M.Y.; Manzoli, L.; Suh, P.-G. Phosphoinositide-specific phospholipase C in health and disease. J. Lipid Res. 2015, 56, 1853–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebecchi, M.J.; Pentyala, S.N. Structure, Function, and Control of Phosphoinositide-Specific Phospholipase C. Physiol. Rev. 2000, 80, 1291–1335. [Google Scholar] [CrossRef] [PubMed]
- Cecchetti, S.; Spadaro, F.; Gessani, S.; Podo, F.; Fantuzzi, L. Phospholipases: At the crossroads of the immune system and the pathogenesis of HIV-1 infection. J. Leukoc. Biol. 2016, 101, 53–75. [Google Scholar] [CrossRef]
- Bi, K.; Tanaka, Y.; Coudronniere, N.; Sugie, K.; Hong, S.; Van Stipdonk, M.J.B.; Altman, A. Antigen-induced translocation of PKC-θ to membrane rafts is required for T cell activation. Nat. Immunol. 2001, 2, 556–563. [Google Scholar] [CrossRef]
- Schulze-Luehrmann, J.; Ghosh, S. Antigen-receptor signaling to nuclear factor kappa B. Immunity 2006, 25, 701–715. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-U.; Kim, L.-K.; Choi, J.-M. Revisiting the Concept of Targeting NFAT to Control T Cell Immunity and Autoimmune Diseases. Front. Immunol. 2018, 9, 2747. [Google Scholar] [CrossRef] [Green Version]
- Chuck, M.I.; Zhu, M.; Shen, S.; Zhang, W. The role of the LAT-PLC-gamma1 interaction in T regulatory cell function. J. Immunol. 2010, 184, 2476–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, C.M.; Claudia, B.; Carlos, C.B.; Alice, T.F.; Edgar, J.P. PLCγ2 and PKC are important to myeloid lineage commitment triggered by M-SCF and G-CSF. J. Cell. Biochem. 2014, 115, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Luft, T.; Rodionova, E.; Maraskovsky, E.; Kirsch, M.; Hess, M.; Buchholtz, C.; Goerner, M.; Schnurr, M.; Skoda, R.; Ho, A. Adaptive functional differentiation of dendritic cells: Integrating the network of extra- and intracellular signals. Blood 2006, 107, 4763–4769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohdanowicz, M.; Schlam, D.; Hermansson, M.; Rizzuti, D.; Fairn, G.D.; Ueyama, T.; Somerharju, P.; Du, G.; Grinstein, S. Phosphatidic acid is required for the constitutive ruffling and macropinocytosis of phagocytes. Mol. Biol. Cell 2013, 24, 1700–1712. [Google Scholar] [CrossRef]
- Yurdagul, A.J.; Doran, A.C.; Cai, B.; Fredman, G.; Tabas, I.A. Mechanisms and Consequences of Defective Efferocytosis in Atherosclerosis. Front. Cardiovasc. Med. 2018, 4, 86. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Liu, B.; Wang, P.; Dong, X.; Fernandez-Hernando, C.; Li, Z.; Hla, T.; Li, Z.; Claffey, K.; Smith, J.D.; et al. Phospholipase C β3 deficiency leads to macrophage hypersensitivity to apoptotic induction and reduction of atherosclerosis in mice. J. Clin. Investig. 2008, 118, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Osto, E.; Alexey, K.; Pavani, M.; Alexander, A.; Christian, B.; Lucia, R.; Arnold, V.E.; Iliceto, S.; Volpe, M.; Lüscher, T.F.; et al. Inhibition of protein kinase Cbeta prevents foam cell formation by reducing scavenger receptor A expression in human macrophages. Circulation 2008, 118, 2174–2182. [Google Scholar] [CrossRef] [Green Version]
- Harja, E.; Chang, J.S.; Lu, Y.; Leitges, M.; Zou, Y.S.; Schmidt, A.M.; Yan, S. Mice deficient in PKCbeta and apolipoprotein E display decreased atherosclerosis. Faseb. J. 2009, 23, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Tse, K.; Tse, H.; Sidney, J.; Sette, A.; Ley, K. T cells in atherosclerosis. Int. Immunol. 2013, 25, 615–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sage, A.P.; Tsiantoulas, D.; Binder, C.J.; Mallat, Z. The role of B cells in atherosclerosis. Nat. Rev. Cardiol. 2018, 16, 180–196. [Google Scholar] [CrossRef]
- McDermott, M.I.; Wang, Y.; Wakelam, M.; Bankaitis, V. Mammalian phospholipase D: Function, and therapeutics. Prog. Lipid Res. 2020, 78, 101018. [Google Scholar] [CrossRef] [PubMed]
- Divecha, N.; Mieke, R.; Jonathan, R.H.; Sabine, D.; Mar, F.-B.; Lauran, O.; Kahlid, M.S.; Michael, W.; Clive, D. Interaction of the type Ialpha PIPkinase with phospholipase D: A role for the local generation of phosphatidylinositol 4, 5-bisphosphate in the regulation of PLD2 activity. EMBO J. 2000, 19, 5440–5449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruntz, R.C.; Lindsley, C.W.; Brown, H.A. Phospholipase D Signaling Pathways and Phosphatidic Acid as Therapeutic Targets in Cancer. Pharmacol. Rev. 2014, 66, 1033–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Kim, Y.; Lee, S.D.; Lopez, I.; Arnold, R.S.; Lambeth, J.; Suh, P.-G.; Ryu, S.H. Selective activation of phospholipase D2 by unsaturated fatty acid. FEBS Lett. 1999, 454, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Cambronero, J. New Concepts in Phospholipase D Signaling in Inflammation and Cancer. Sci. World J. 2010, 10, 1356–1369. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, K.C.; Ross, A.H.; Qiu, R.G.; Symons, M.; Exton, J.H. Activation of rat liver phospholipase D by the small GTP-binding protein RhoA. J. Biol. Chem. 1994, 269, 25951–25954. [Google Scholar]
- Hammond, S.M.; Jenco, J.M.; Nakashima, S.; Cadwallader, K.; Gu, Q.; Cook, S.; Nozawa, Y.; Prestwich, G.D.; Frohman, M.A.; Morris, A.J. Characterization of two alternately spliced forms of phospholipase D1. Activation of the purified enzymes by phosphatidylinositol 4,5-bisphosphate, ADP-ribosylation factor, and Rho family monomeric GTP-binding proteins and protein kinase C-alpha. J. Biol. Chem. 1997, 272, 3860–3868. [Google Scholar] [CrossRef] [Green Version]
- Du, G.; Huang, P.; Liang, B.T.; Frohman, M.A. Phospholipase D2 Localizes to the Plasma Membrane and Regulates Angiotensin II Receptor Endocytosis. Mol. Biol. Cell 2004, 15, 1024–1030. [Google Scholar] [CrossRef] [Green Version]
- Manifava, M.; Sugars, J.; Ktistakis, N.T. Modification of Catalytically Active Phospholipase D1 with Fatty Acidin Vivo. J. Biol. Chem. 1999, 274, 1072–1077. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.D.; Pineda, C.; Chiu, W.S.; Bowen, R.; Deeg, M.A. Glycosylphosphatidylinositol-Specific Phospholipase D Is Expressed by Macrophages in Human Atherosclerosis and Colocalizes With Oxidation Epitopes. Circulation 1999, 99, 2876–2882. [Google Scholar] [CrossRef] [Green Version]
- Corrotte, M.; Chasserot-Golaz, S.; Huang, P.; Du, G.; Ktistakis, N.T.; Frohman, M.A.; Vitale, N.; Bader, M.-F.; Grant, N.J. Dynamics and Function of Phospholipase D and Phosphatidic Acid During Phagocytosis. Traffic 2006, 7, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, R.; Henkels, K.M.; Wrenshall, L.E.; Kanaho, Y.; Di Paolo, G.; Frohman, M.A.; Gomez-Cambronero, J. Oxidized LDL phagocytosis during foam cell formation in atherosclerotic plaques relies on a PLD2-CD36 functional interdependence. J. Leukoc. Biol. 2018, 103, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Melendez, A.J.; Bruetschy, L.; Floto, R.A.; Harnett, M.M.; Allen, J.M. Functional coupling of FcγRI to nicotinamide adenine dinucleotide phosphate (reduced form) oxidative burst and immune complex trafficking requires the activation of phospholipase D1. Blood 2001, 98, 3421–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, F.; Gordon, M.L.; Robinson, J.J.; Galvani, D.W.; Edwards, S.W. Phospholipase D-dependent and-independent activation of the neutrophil NADPH oxidase. Biosci. Rep. 1994, 14, 91–102. [Google Scholar] [CrossRef]
- Bréchard, S.; Plançon, S.; Tschirhart, E.J. New Insights into the Regulation of Neutrophil NADPH Oxidase Activity in the Phagosome: A Focus on the Role of Lipid and Ca2+Signaling. Antioxid. Redox Signal. 2013, 18, 661–676. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Wakita, S.; Suganami, A.; Tamura, Y.; Hanada, K.; Murayama, T. Modulation of the activity of cytosolic phospholipase A2α (cPLA2α) by cellular sphingolipids and inhibition of cPLA2α by sphingomyelin[S]. J. Lipid Res. 2009, 51, 720–728. [Google Scholar] [CrossRef] [Green Version]
- Kramer, R.M.; Sharp, J.D. Structure, function and regulation of Ca2+-sensitive cytosolic phospholipase A2 (cPLA2). FEBS Lett. 1997, 410, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Kudo, I.; Murakami, M. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat. 2002, 68, 3–58. [Google Scholar] [CrossRef]
- Choukroun, G.J.; Marshansky, V.; Gustafson, C.E.; McKee, M.; Hajjar, R.J.; Rosenzweig, A.; Brown, D.; Bonventre, J.V. Cytosolic phospholipase A2 regulates Golgi structure and modulates intracellular trafficking of membrane proteins. J. Clin. Investig. 2000, 106, 983–993. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.-L.; Wartmann, M.; Lin, A.Y.; Knopf, J.L.; Seth, A.; Davis, R.J. cPLA2 is phosphorylated and activated by MAP kinase. Cell 1993, 72, 269–278. [Google Scholar] [CrossRef]
- Trelle, S.; Reichenbach, S.; Wandel, S.; Hildebrand, P.; Tschannen, B.; Villiger, P.M.; Egger, M.; Jüni, P. Cardiovascular safety of non-steroidal anti-inflammatory drugs: Network meta-analysis. BMJ 2011, 342, c7086. [Google Scholar] [CrossRef] [Green Version]
- Schanstra, J.P.; Luong, T.T.; Makridakis, M.; Van Linthout, S.; Lygirou, V.; Latosisnska, A.; Alesutan, I.; Boehme, B.; Schelski, N.; Von Lewinski, D.; et al. Systems biology identifies cytosolic PLA2 as a target in vascular calcification treatment. JCI Insight 2019, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Oestvang, J.; Bonnefont-Rousselot, D.; Ninio, E.; Hakala, J.K.; Johansen, B.; Anthonsen, M.W. Modification of LDL with human secretory phospholipase A2or sphingomyelinase promotes its arachidonic acid-releasing propensity. J. Lipid Res. 2004, 45, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Csaki, L.S.; Reue, K. Lipins: Multifunctional Lipid Metabolism Proteins. Annu. Rev. Nutr. 2010, 30, 257–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donkor, J.; Sariahmetoglu, M.; Dewald, J.; Brindley, D.N.; Reue, K. Three Mammalian Lipins Act as Phosphatidate Phosphatases with Distinct Tissue Expression Patterns. J. Biol. Chem. 2006, 282, 3450–3457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, G.-S.; Wu, W.-I.; Carman, G.M. TheSaccharomyces cerevisiaeLipin Homolog Is a Mg2+-dependent Phosphatidate Phosphatase Enzyme. J. Biol. Chem. 2006, 281, 9210–9218. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Rui, B.-B.; Tang, L.-Y.; Hu, C.-M. Lipin Family Proteins—Key Regulators in Lipid Metabolism. Ann. Nutr. Metab. 2014, 66, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Gropler, M.C.; Chen, Z.; Leone, T.C.; Croce, M.A.; Harris, T.E.; Lawrence, J.C., Jr.; Kelly, D.P. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006, 4, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Harris, T.E.; Huffman, T.A.; Chi, A.; Shabanowitz, J.; Hunt, N.F.; Kumar, A.; Lawrence, J.C. Insulin Controls Subcellular Localization and Multisite Phosphorylation of the Phosphatidic Acid Phosphatase, Lipin 1. J. Biol. Chem. 2006, 282, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Donkor, J.; Zhang, P.; Wong, S.; O’Loughlin, L.; Dewald, J.; Kok, B.P.; Brindley, D.N.; Reue, K. A Conserved Serine Residue Is Required for the Phosphatidate Phosphatase Activity but Not the Transcriptional Coactivator Functions of Lipin-1 and Lipin-2. J. Biol. Chem. 2009, 284, 29968–29978. [Google Scholar] [CrossRef] [Green Version]
- Péterfy, M.; Phan, J.; Xu, P.; Reue, K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat. Genet. 2001, 27, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Han, G.-S.; Carman, G.M. Characterization of the HumanLPIN1-encoded Phosphatidate Phosphatase Isoforms. J. Biol. Chem. 2010, 285, 14628–14638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartz, R.; Li, W.-H.; Venables, B.; Zehmer, J.K.; Roth, M.R.; Welti, R.; Anderson, R.G.W.; Liu, P.; Chapman, K.D. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J. Lipid Res. 2007, 48, 837–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navratil, A.R.; Vozenilek, A.; Cardelli, J.A.; Green, J.M.; Thomas, M.J.; Sorci-Thomas, M.; Orr, A.W.; Woolard, M.D. Lipin-1 contributes to modified low-density lipoprotein-elicited macrophage pro-inflammatory responses. Atherosclerosis 2015, 242, 424–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, R.; Coleman, R.A.; Lee, D.P. Enzymes of triacylglycerol synthesis and their regulation. Prog. Lipid Res. 2004, 43, 134–176. [Google Scholar] [CrossRef]
- Zhang, P.; Reue, K. Lipin proteins and glycerolipid metabolism: Roles at the ER membrane and beyond. Biochim. Biophys. Acta (BBA)-Biomembr. 2017, 1859, 1583–1595. [Google Scholar] [CrossRef] [PubMed]
- Kwiatek, J.M.; Carman, G.M. Yeast phosphatidic acid phosphatase Pah1 hops and scoots along the membrane phospholipid bilayer. J. Lipid Res. 2020, 61, 1232–1243. [Google Scholar] [CrossRef]
- Murphy, D.J. The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog. Lipid Res. 2001, 40, 325–438. [Google Scholar] [CrossRef]
- Meana, C.; Pena, L.; Lordén, G.; Esquinas, E.; Guijas, C.; Valdearcos, M.; Balsinde, J.; Balboa, M.A. Lipin-1 Integrates Lipid Synthesis with Proinflammatory Responses during TLR Activation in Macrophages. J. Immunol. 2014, 193, 4614–4622. [Google Scholar] [CrossRef] [Green Version]
- Valdearcos, M.; Esquinas, E.; Meana, C.; Gil-De-Gómez, L.; Guijas, C.; Balsinde, J.; Balboa, M.A. Subcellular Localization and Role of Lipin-1 in Human Macrophages. J. Immunol. 2011, 186, 6004–6013. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Kwon, S.; Ham, S.; Lee, D.; Park, H.H.; Yamaoka, Y.; Jeong, D.; Artan, M.; Altintas, O.; Park, S.; et al. Caenorhabditis elegans Lipin 1 moderates the lifespan-shortening effects of dietary glucose by maintaining ω-6 polyunsaturated fatty acids. Aging Cell 2020, 19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Verity, M.A.; Reue, K. Lipin-1 Regulates Autophagy Clearance and Intersects with Statin Drug Effects in Skeletal Muscle. Cell Metab. 2014, 20, 267–279. [Google Scholar] [CrossRef] [Green Version]
- Alshudukhi, A.A.; Zhu, J.; Huang, D.; Jama, A.; Smith, J.D.; Wang, Q.J.; Esser, K.A.; Ren, H. Lipin-1 regulates Bnip3–mediated mitophagy in glycolytic muscle. FASEB J. 2018, 32, 6796–6807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR Complex 1 Regulates Lipin 1 Localization to Control the SREBP Pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, C.; Lieggi, N.T.; Barry, D.; Mooney, D.; De Gaetano, M.; James, W.G.; McClelland, S.; Barry, M.C.; Escoubet-Lozach, L.; Li, A.C.; et al. Macrophage PPAR gamma Co-activator-1 alpha participates in repressing foam cell formation and atherosclerosis in response to conjugated linoleic acid. EMBO Mol. Med. 2013, 5, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.E.; Bae, E.; Jeong, D.-Y.; Kim, M.-J.; Jin, W.-J.; Park, S.-W.; Han, G.-S.; Carman, G.M.; Koh, E.; Kim, K.-S. Lipin1 regulates PPARγ transcriptional activity. Biochem. J. 2013, 453, 49–60. [Google Scholar] [CrossRef]
- Soskic, S.; Dobutović, B.D.; Sudar, E.; Obradovic, M.; Nikolic, D.; Zaric, B.; Stojanović, S.Đ.; Stokić, E.J.; Mikhailidis, D.P.; Isenovic, E.R. Peroxisome Proliferator-Activated Receptors and Atherosclerosis. Angiology 2011, 62, 523–534. [Google Scholar] [CrossRef]
- Chen, H.; Shi, R.; Luo, B.; Yang, X.; Qiu, L.; Xiong, J.; Jiang, M.; Liu, Y.; Zhang, Z.-R.; Wu, Y. Macrophage peroxisome proliferator-activated receptor γ deficiency delays skin wound healing through impairing apoptotic cell clearance in mice. Cell Death Dis. 2015, 6, e1597. [Google Scholar] [CrossRef] [Green Version]
- Chandran, S.; Schilke, R.M.; Blackburn, C.M.R.; Yurochko, A.; Mirza, R.; Scott, R.S.; Finck, B.N.; Woolard, M.D. Lipin-1 Contributes to IL-4 Mediated Macrophage Polarization. Front. Immunol. 2020, 11, 787. [Google Scholar] [CrossRef]
- Kim, H.B.; Kumar, A.; Wang, L.; Liu, G.-H.; Keller, S.R.; Lawrence, J.C.; Finck, B.N.; Harris, T.E. Lipin 1 Represses NFATc4 Transcriptional Activity in Adipocytes to Inhibit Secretion of Inflammatory Factors. Mol. Cell. Biol. 2010, 30, 3126–3139. [Google Scholar] [CrossRef] [Green Version]
- Grkovich, A.; Johnson, C.A.; Buczynski, M.W.; Dennis, E.A. Lipopolysaccharide-induced Cyclooxygenase-2 Expression in Human U937 Macrophages Is Phosphatidic Acid Phosphohydrolase-1-dependent. J. Biol. Chem. 2006, 281, 32978–32987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grkovich, A.; Armando, A.; Quehenberger, O.; Dennis, E.A. TLR-4 mediated group IVA phospholipase A2 activation is phosphatidic acid phosphohydrolase 1 and protein kinase C dependent. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2009, 1791, 975–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vozenilek, A.E.; Navratil, A.R.; Green, J.M.; Coleman, D.T.; Blackburn, C.M.; Finney, A.C.; Pearson, B.H.; Chrast, R.; Finck, B.N.; Klein, R.L.; et al. Macrophage-Associated Lipin-1 Enzymatic Activity Contributes to Modified Low-Density Lipoprotein-Induced Proinflammatory Signaling and Atherosclerosis. Arter. Thromb. Vasc. Biol. 2017, 38, 324–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meana, C.; García-Rostán, G.; Peña, L.; Lordén, G.; Cubero, Á.; Orduña, A.; Győrffy, B.; Balsinde, J.; Balboa, M.A. The phosphatidic acid phosphatase lipin-1 facilitates inflammation-driven colon carcinogenesis. JCI Insight 2018, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, G.G.; Collier, S.L.; Chen, Z.; Eaton, J.M.; Connolly, A.M.; Bucelli, R.C.; Pestronk, A.; Harris, T.E.; Finck, B.N.; Zschocke, J. Rhabdomyolysis-Associated Mutations in Human LPIN1 Lead to Loss of Phosphatidic Acid Phosphohydrolase Activity. JIMD Rep. 2015, 23, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Schweitzer, G.G.; Collier, S.L.; Chen, Z.; McCommis, K.S.; Pittman, S.K.; Yoshino, J.; Matkovich, S.; Hsu, F.-F.; Chrast, R.; Eaton, J.M.; et al. Loss of lipin 1-mediated phosphatidic acid phosphohydrolase activity in muscle leads to skeletal myopathy in mice. FASEB J. 2018, 33, 652–667. [Google Scholar] [CrossRef]
- Reue, K.; Wang, H. Mammalian lipin phosphatidic acid phosphatases in lipid synthesis and beyond: Metabolic and inflammatory disorders. J. Lipid Res. 2019, 60, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Reue, K.; Doolittle, M.H. Naturally occurring mutations in mice affecting lipid transport and metabolism. J. Lipid Res. 1996, 37, 1387–1405. [Google Scholar]
- Li, Y.Y.; Zhou, J.Y. Role of lipin-1 in the pathogenesis of alcoholic fatty liver disease. Zhonghua gan zang bing za zhi = Zhonghua ganzangbing zazhi = Chin. J. Hepatol. 2016, 24, 237–240. [Google Scholar]
- Barrett, T.J. Macrophages in Atherosclerosis Regression. Arter. Thromb. Vasc. Biol. 2020, 40, 20–33. [Google Scholar] [CrossRef]
- Reiss, A.B.; E Vagell, M. PPARgamma activity in the vessel wall: Anti-atherogenic properties. Curr. Med. Chem. 2006, 13, 3227–3238. [Google Scholar] [CrossRef] [PubMed]
- Staels, B. PPARgamma and atherosclerosis. Curr. Med. Res. Opin. 2005, 21, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Taketa, K.; Matsumura, T.; Yano, M.; Ishii, N.; Senokuchi, T.; Motoshima, H.; Murata, Y.; Kim-Mitsuyama, S.; Kawada, T.; Itabe, H.; et al. Oxidized Low Density Lipoprotein Activates Peroxisome Proliferator-activated Receptor-α (PPARα) and PPARγ through MAPK-dependent COX-2 Expression in Macrophages. J. Biol. Chem. 2008, 283, 9852–9862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornmueller, K.; Vidakovic, I.; Prassl, R. Artificial High Density Lipoprotein Nanoparticles in Cardiovascular Research. Molecules 2019, 24, 2829. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schilke, R.M.; Blackburn, C.M.R.; Bamgbose, T.T.; Woolard, M.D. Interface of Phospholipase Activity, Immune Cell Function, and Atherosclerosis. Biomolecules 2020, 10, 1449. https://doi.org/10.3390/biom10101449
Schilke RM, Blackburn CMR, Bamgbose TT, Woolard MD. Interface of Phospholipase Activity, Immune Cell Function, and Atherosclerosis. Biomolecules. 2020; 10(10):1449. https://doi.org/10.3390/biom10101449
Chicago/Turabian StyleSchilke, Robert M., Cassidy M. R. Blackburn, Temitayo T. Bamgbose, and Matthew D. Woolard. 2020. "Interface of Phospholipase Activity, Immune Cell Function, and Atherosclerosis" Biomolecules 10, no. 10: 1449. https://doi.org/10.3390/biom10101449
APA StyleSchilke, R. M., Blackburn, C. M. R., Bamgbose, T. T., & Woolard, M. D. (2020). Interface of Phospholipase Activity, Immune Cell Function, and Atherosclerosis. Biomolecules, 10(10), 1449. https://doi.org/10.3390/biom10101449