Microglia in Alzheimer’s Disease in the Context of Tau Pathology



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Tau Protein
2.1. Expression, Isoforms, and Localization
2.2. Structure and Post-Translational Modifications of Tau
2.3. Tau Aggregation
2.4. Main Functions of Tau
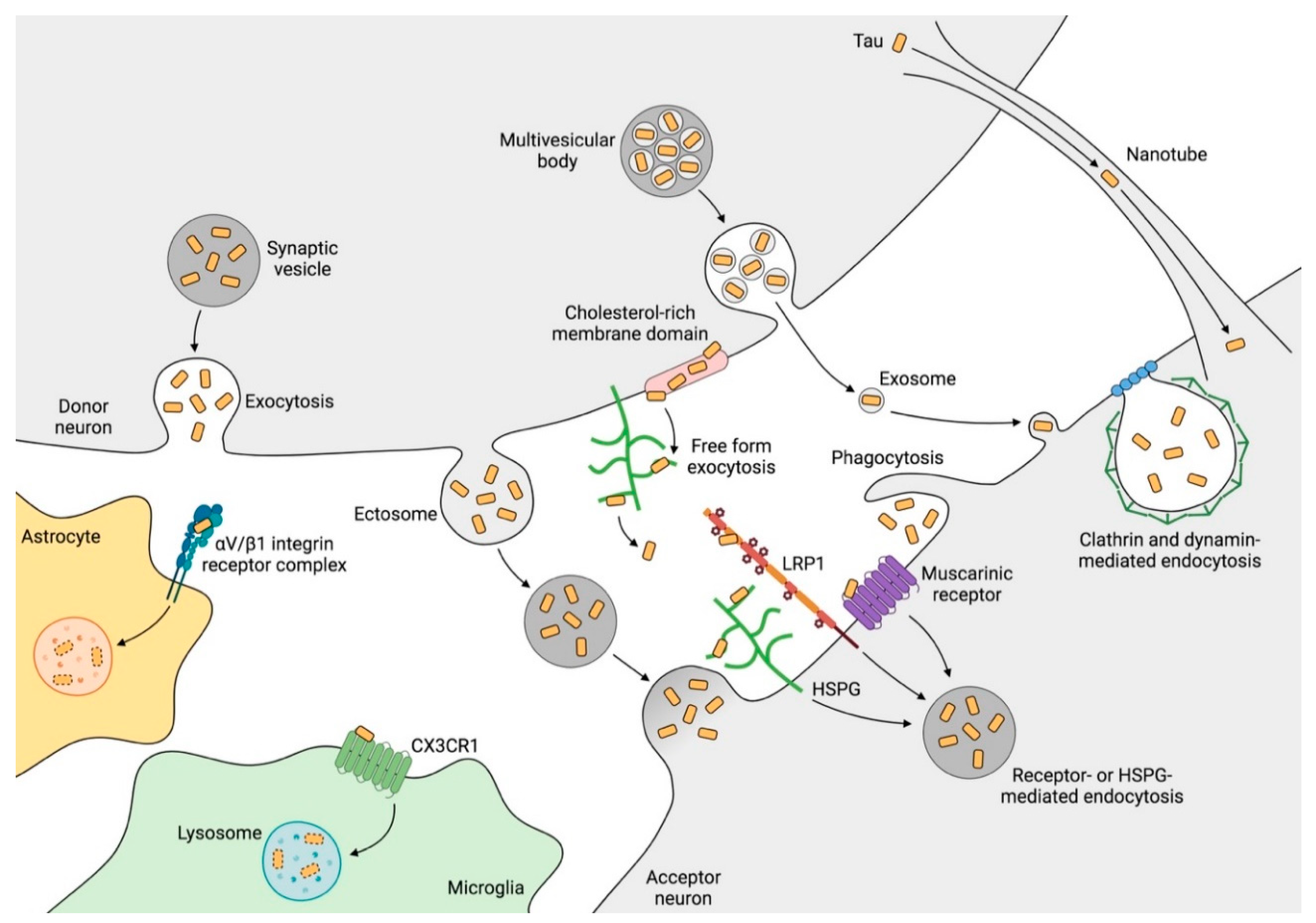
2.5. Mechanisms of Tau Propagation
3. Microglia
3.1. History and Origin of Microglia
3.2. Physiological Functions of Microglia
3.3. Contribution of Microglia to Tau Pathology
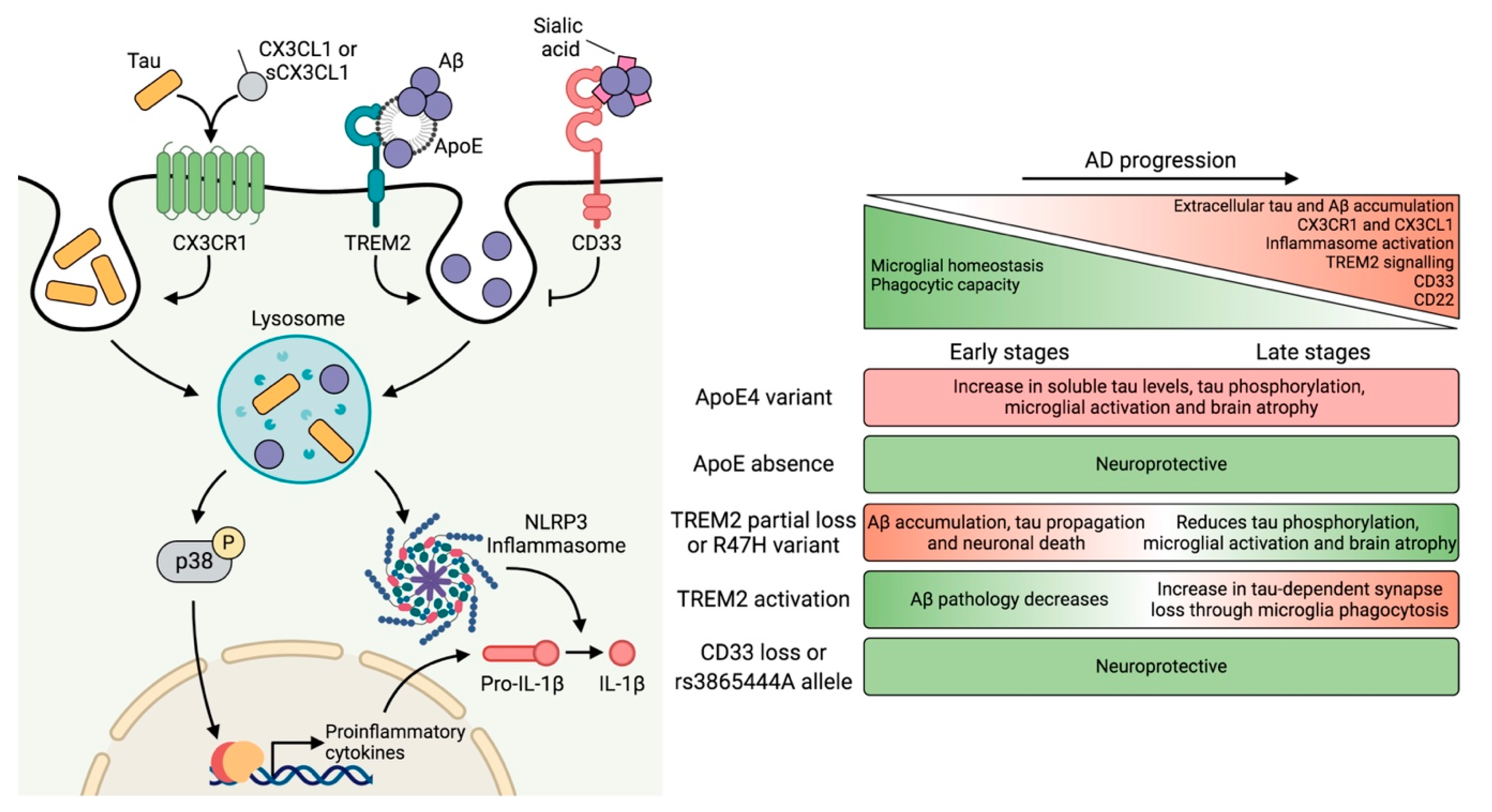
4. Genetic Risk Factors and Their Impact on Microglial Function
4.1. APOE
4.2. TREM2
4.3. CD33
5. Neuron–Microglia Crosstalk: Implications of the CX3CL1–CX3CR1 Axis and Downstream Signaling
6. Extracellular Soluble Tau as the Main Driving Force of Toxicity
7. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Katzman, R. The prevalence and malignancy of Alzheimer disease. A major killer. Arch. Neurol. 1976, 33, 217–218. [Google Scholar] [CrossRef]
- Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, A.M.; Winblad, B.; Jönsson, L.; Liu, Z.; Prince, M. The worldwide costs of dementia 2015 and comparisons with 2010. Alzheimer’s Dement. 2017, 13, 1–7. [Google Scholar] [CrossRef]
- Nichols, E.; Szoeke, C.E.I.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef]
- Alzheimer, A. Über einen eigenartigen schweren Erkrankungsprozeß der Hirnrinde. Neurol. Zentralblatt 1906, 23, 1129–1136. [Google Scholar]
- Alzheimer, A. Über eine eigenartige Erkrankung der Hirnrinde. Allg. Zeitschrift fur Psychiatr. und Psych. Medizin 1907, 64, 146–148. [Google Scholar]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T. The C-terminus of the beta protein is critical in amyloidogenesis. Ann. N. Y. Acad. Sci. 1993, 695, 144–148. [Google Scholar] [CrossRef]
- Maggio, J.E.; Stimson, E.R.; Ghilardi, J.R.; Allen, C.J.; Dahl, C.E.; Whitcomb, D.C.; Vigna, S.R.; Vinters, H.V.; Labenski, M.E.; Mantyh, P.W. Reversible in vitro growth of Alzheimer disease beta-amyloid plaques by deposition of labeled amyloid peptide. Proc. Natl. Acad. Sci. USA 1992, 89, 5462–5466. [Google Scholar] [CrossRef]
- Kidd, M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature 1963, 197, 192–193. [Google Scholar] [CrossRef]
- Crowther, R.A.; Wischik, C.M. Image reconstruction of the Alzheimer paired helical filament. EMBO J. 1985, 4, 3661–3665. [Google Scholar] [CrossRef]
- von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.M.; Mandelkow, E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Tcw, J.; Goate, A.M. Genetics of β-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2017, 7, a024539. [Google Scholar] [CrossRef]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef]
- Bierer, L.M.; Hof, P.R.; Purohit, D.P.; Carlin, L.; Schmeidler, J.; Davis, K.L.; Perl, D.P. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch. Neurol. 1995, 52, 81–88. [Google Scholar] [CrossRef]
- Giannakopoulos, P.; Herrmann, F.R.; Bussière, T.; Bouras, C.; Kövari, E.; Perl, D.P.; Morrison, J.H.; Gold, G.; Hof, P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef]
- La Joie, R.; Visani, A.V.; Baker, S.L.; Brown, J.A.; Bourakova, V.; Cha, J.; Chaudhary, K.; Edwards, L.; Iaccarino, L.; Janabi, M.; et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci. Transl. Med. 2020, 12, eaau5732. [Google Scholar] [CrossRef]
- Rapoport, M.; Dawson, H.N.; Binder, L.I.; Vitek, M.P.; Ferreira, A. Tau is essential to β-amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 6364–6369. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.-Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef]
- Mintun, M.A.; Larossa, G.N.; Sheline, Y.I.; Dence, C.S.; Lee, S.Y.; Mach, R.H.; Klunk, W.E.; Mathis, C.A.; DeKosky, S.T.; Morris, J.C. [11C]PIB in a nondemented population: Potential antecedent marker of Alzheimer disease. Neurology 2006, 67, 446–452. [Google Scholar] [CrossRef]
- Aizenstein, H.J.; Nebes, R.D.; Saxton, J.A.; Price, J.C.; Mathis, C.A.; Tsopelas, N.D.; Ziolko, S.K.; James, J.A.; Snitz, B.E.; Houck, P.R.; et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch. Neurol. 2008, 65, 1509–1517. [Google Scholar] [CrossRef]
- Meyer, P.-F.; Savard, M.; Poirier, J.; Labonté, A.; Rosa-Neto, P.; Weitz, T.M.; Town, T.; Breitner, J. Alzheimer’s Disease Neuroimaging Initiative; PREVENT-AD Research Group Bi-directional Association of Cerebrospinal Fluid Immune Markers with Stage of Alzheimer’s Disease Pathogenesis. J. Alzheimers. Dis. 2018, 63, 577–590. [Google Scholar] [CrossRef]
- Suárez-Calvet, M.; Morenas-Rodríguez, E.; Kleinberger, G.; Schlepckow, K.; Araque Caballero, M.Á.; Franzmeier, N.; Capell, A.; Fellerer, K.; Nuscher, B.; Eren, E.; et al. Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol. Neurodegener. 2019, 14, 1. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef]
- LoPresti, P.; Szuchet, S.; Papasozomenos, S.C.; Zinkowski, R.P.; Binder, L.I. Functional implications for the microtubule-associated protein tau: Localization in oligodendrocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 10369–10373. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef]
- Neve, R.L.; Harris, P.; Kosik, K.S.; Kurnit, D.M.; Donlon, T.A. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 1986, 387, 271–280. [Google Scholar] [CrossRef]
- Andreadis, A. Tau gene alternative splicing: Expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim. Biophys. Acta 2005, 1739, 91–103. [Google Scholar] [CrossRef]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef]
- Bruch, J.; Xu, H.; De Andrade, A.; Höglinger, G. Mitochondrial complex 1 inhibition increases 4-repeat isoform tau by SRSF2 upregulation. PLoS ONE 2014, 9, e113070. [Google Scholar] [CrossRef]
- Smith, P.Y.; Delay, C.; Girard, J.; Papon, M.-A.; Planel, E.; Sergeant, N.; Buée, L.; Hébert, S.S. MicroRNA-132 loss is associated with tau exon 10 inclusion in progressive supranuclear palsy. Hum. Mol. Genet. 2011, 20, 4016–4024. [Google Scholar] [CrossRef]
- Orozco, D.; Tahirovic, S.; Rentzsch, K.; Schwenk, B.M.; Haass, C.; Edbauer, D. Loss of fused in sarcoma (FUS) promotes pathological Tau splicing. EMBO Rep. 2012, 13, 759–764. [Google Scholar] [CrossRef]
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 1985, 101, 1371–1378. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef]
- Loomis, P.A.; Howard, T.H.; Castleberry, R.P.; Binder, L.I. Identification of nuclear tau isoforms in human neuroblastoma cells. Proc. Natl. Acad. Sci. USA 1990, 87, 8422–8426. [Google Scholar] [CrossRef]
- Rady, R.M.; Zinkowski, R.P.; Binder, L.I. Presence of tau in isolated nuclei from human brain. Neurobiol. Aging 1995, 16, 479–486. [Google Scholar] [CrossRef]
- Brandt, R.; Léger, J.; Lee, G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol. 1995, 131, 1327–1340. [Google Scholar] [CrossRef]
- Ait-Bouziad, N.; Lv, G.; Mahul-Mellier, A.-L.; Xiao, S.; Zorludemir, G.; Eliezer, D.; Walz, T.; Lashuel, H.A. Discovery and characterization of stable and toxic Tau/phospholipid oligomeric complexes. Nat. Commun. 2017, 8, 1678. [Google Scholar] [CrossRef]
- Liu, C.; Götz, J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS ONE 2013, 8, e84849. [Google Scholar] [CrossRef]
- Chen, J.; Kanai, Y.; Cowan, N.J.; Hirokawa, N. Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 1992, 360, 674–677. [Google Scholar] [CrossRef]
- Frappier, T.F.; Georgieff, I.S.; Brown, K.; Shelanski, M.L. Tau regulation of microtubule-microtubule spacing and bundling. J. Neurochem. 1994, 63, 2288–2294. [Google Scholar] [CrossRef]
- Lee, G.; Neve, R.L.; Kosik, K.S. The microtubule binding domain of tau protein. Neuron 1989, 2, 1615–1624. [Google Scholar] [CrossRef]
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 1998, 111, 3167–3177. [Google Scholar]
- Tapia-Rojas, C.; Cabezas-Opazo, F.; Deaton, C.A.; Vergara, E.H.; Johnson, G.V.W.; Quintanilla, R.A. It’s all about tau. Prog. Neurobiol. 2019, 175, 54–76. [Google Scholar] [CrossRef]
- Ercan, E.; Eid, S.; Weber, C.; Kowalski, A.; Bichmann, M.; Behrendt, A.; Matthes, F.; Krauss, S.; Reinhardt, P.; Fulle, S.; et al. A validated antibody panel for the characterization of tau post-translational modifications. Mol. Neurodegener. 2017, 12, 87. [Google Scholar] [CrossRef]
- Kanemaru, K.; Takio, K.; Miura, R.; Titani, K.; Ihara, Y. Fetal-type phosphorylation of the tau in paired helical filaments. J. Neurochem. 1992, 58, 1667–1675. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef]
- Biernat, J.; Gustke, N.; Drewes, G.; Mandelkow, E.M.; Mandelkow, E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: Distinction between PHF-like immunoreactivity and microtubule binding. Neuron 1993, 11, 153–163. [Google Scholar] [CrossRef]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.-L.; et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef]
- Dickey, C.A.; Kamal, A.; Lundgren, K.; Klosak, N.; Bailey, R.M.; Dunmore, J.; Ash, P.; Shoraka, S.; Zlatkovic, J.; Eckman, C.B.; et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Investig. 2007, 117, 648–658. [Google Scholar] [CrossRef]
- Bhaskar, K.; Yen, S.-H.; Lee, G. Disease-related modifications in tau affect the interaction between Fyn and Tau. J. Biol. Chem. 2005, 280, 35119–35125. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Garwood, C.J.; Wray, S.; Price, C.; Kellie, S.; Perera, T.; Zvelebil, M.; Yang, A.; Sheppard, P.W.; Varndell, I.M.; et al. Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J. Biol. Chem. 2008, 283, 18177–18186. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Götz, J. Phosphorylated Tau interacts with c-Jun N-terminal kinase-interacting protein 1 (JIP1) in Alzheimer disease. J. Biol. Chem. 2009, 284, 20909–20916. [Google Scholar] [CrossRef]
- Hanger, D.P. Tau Phosphorylation Sites. Available online: https://docs.google.com/spreadsheets/d/1hGYs1ZcupmTnbB7n6qs1r_WVTXHt1O7NBLyKBN7EOUQ/edit#gid=0. (accessed on 15 July 2020).
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Buée-Scherrer, V.; Goedert, M. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases in intact cells. FEBS Lett. 2002, 515, 151–154. [Google Scholar] [CrossRef]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.-X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 2005, 22, 1942–1950. [Google Scholar] [CrossRef]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef]
- Lee, V.M.-Y.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Dickson, D.W.; Kouri, N.; Murray, M.E.; Josephs, K.A. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J. Mol. Neurosci. 2011, 45, 384–389. [Google Scholar] [CrossRef]
- Alonso, A.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef]
- Haase, C.; Stieler, J.T.; Arendt, T.; Holzer, M. Pseudophosphorylation of tau protein alters its ability for self-aggregation. J. Neurochem. 2004, 88, 1509–1520. [Google Scholar] [CrossRef]
- Xia, Y.; Prokop, S.; Gorion, K.-M.M.; Kim, J.D.; Sorrentino, Z.A.; Bell, B.M.; Manaois, A.N.; Chakrabarty, P.; Davies, P.; Giasson, B.I. Tau Ser208 phosphorylation promotes aggregation and reveals neuropathologic diversity in Alzheimer’s disease and other tauopathies. Acta Neuropathol. Commun. 2020, 8, 88. [Google Scholar] [CrossRef]
- Schneider, A.; Biernat, J.; von Bergen, M.; Mandelkow, E.; Mandelkow, E.-M. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 1999, 38, 3549–3558. [Google Scholar] [CrossRef]
- Wang, Y.; Garg, S.; Mandelkow, E.-M.; Mandelkow, E. Proteolytic processing of tau. Biochem. Soc. Trans. 2010, 38, 955–961. [Google Scholar] [CrossRef]
- Montejo de Garcini, E.; Avila, J. In vitro conditions for the self-polymerization of the microtubule-associated protein, tau factor. J. Biochem. 1987, 102, 1415–1421. [Google Scholar] [CrossRef]
- Hong, M.; Zhukareva, V.; Vogelsberg-Ragaglia, V.; Wszolek, Z.; Reed, L.; Miller, B.I.; Geschwind, D.H.; Bird, T.D.; McKeel, D.; Goate, A.; et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 1998, 282, 1914–1917. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar] [CrossRef]
- Kampers, T.; Friedhoff, P.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996, 399, 344–349. [Google Scholar] [CrossRef]
- Cowan, C.M.; Mudher, A. Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 2013, 4, 114. [Google Scholar] [CrossRef]
- Mirbaha, H.; Chen, D.; Morazova, O.A.; Ruff, K.M.; Sharma, A.M.; Liu, X.; Goodarzi, M.; Pappu, R.V.; Colby, D.W.; Mirzaei, H.; et al. Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife 2018, 7, e36584. [Google Scholar] [CrossRef]
- Schweers, O.; Schönbrunn-Hanebeck, E.; Marx, A.; Mandelkow, E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J. Biol. Chem. 1994, 269, 24290–24297. [Google Scholar]
- Wille, H.; Drewes, G.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J. Cell Biol. 1992, 118, 573–584. [Google Scholar] [CrossRef]
- Sahara, N.; Maeda, S.; Murayama, M.; Suzuki, T.; Dohmae, N.; Yen, S.-H.; Takashima, A. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur. J. Neurosci. 2007, 25, 3020–3029. [Google Scholar] [CrossRef]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, M.; Yoshiike, Y.; Kim, H.; Miyasaka, T.; Murayama, S.; Ikai, A.; Takashima, A. Granular tau oligomers as intermediates of tau filaments. Biochemistry 2007, 46, 3856–3861. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef]
- Zhang, W.; Tarutani, A.; Newell, K.L.; Murzin, A.G.; Matsubara, T.; Falcon, B.; Vidal, R.; Garringer, H.J.; Shi, Y.; Ikeuchi, T.; et al. Novel tau filament fold in corticobasal degeneration. Nature 2020, 580, 283–287. [Google Scholar] [CrossRef]
- Arakhamia, T.; Lee, C.E.; Carlomagno, Y.; Duong, D.M.; Kundinger, S.R.; Wang, K.; Williams, D.; DeTure, M.; Dickson, D.W.; Cook, C.N.; et al. Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell 2020, 180, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Sun, X.; Austin, T.O.; Muralidharan, H.; Jean, D.C.; Liu, M.; Yu, W.; Baas, P.W. Tau Does Not Stabilize Axonal Microtubules but Rather Enables Them to Have Long Labile Domains. Curr. Biol. 2018, 28, 2181–2189. [Google Scholar] [CrossRef] [PubMed]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L.F. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef]
- Caceres, A.; Kosik, K.S. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature 1990, 343, 461–463. [Google Scholar] [CrossRef]
- Kimura, T.; Whitcomb, D.J.; Jo, J.; Regan, P.; Piers, T.; Heo, S.; Brown, C.; Hashikawa, T.; Murayama, M.; Seok, H.; et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2014, 369, 20130144. [Google Scholar] [CrossRef]
- Ahmed, T.; Van der Jeugd, A.; Blum, D.; Galas, M.-C.; D’Hooge, R.; Buee, L.; Balschun, D. Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol. Aging 2014, 35, 2474–2478. [Google Scholar] [CrossRef]
- Pallas-Bazarra, N.; Jurado-Arjona, J.; Navarrete, M.; Esteban, J.A.; Hernández, F.; Ávila, J.; Llorens-Martín, M. Novel function of Tau in regulating the effects of external stimuli on adult hippocampal neurogenesis. EMBO J. 2016, 35, 1417–1436. [Google Scholar] [CrossRef]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef]
- Harada, A.; Oguchi, K.; Okabe, S.; Kuno, J.; Terada, S.; Ohshima, T.; Sato-Yoshitake, R.; Takei, Y.; Noda, T.; Hirokawa, N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 1994, 369, 488–491. [Google Scholar] [CrossRef]
- van Hummel, A.; Bi, M.; Ippati, S.; van der Hoven, J.; Volkerling, A.; Lee, W.S.; Tan, D.C.S.; Bongers, A.; Ittner, A.; Ke, Y.D.; et al. No Overt Deficits in Aged Tau-Deficient C57Bl/6.Mapttm1(EGFP)Kit GFP Knockin Mice. PLoS ONE 2016, 11, e0163236. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Spoerri, L.; Ciccotosto, G.D.; Wright, D.K.; Wong, B.X.W.; Adlard, P.A.; Cherny, R.A.; Lam, L.Q.; et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012, 18, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Ayton, S.; Moon, S.; Zhang, Q.; Volitakis, I.; Finkelstein, D.I.; Bush, A.I. Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol. Neurodegener. 2014, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- de Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef]
- DeVos, S.L.; Corjuc, B.T.; Oakley, D.H.; Nobuhara, C.K.; Bannon, R.N.; Chase, A.; Commins, C.; Gonzalez, J.A.; Dooley, P.M.; Frosch, M.P.; et al. Synaptic Tau Seeding Precedes Tau Pathology in Human Alzheimer’s Disease Brain. Front. Neurosci. 2018, 12, 267. [Google Scholar] [CrossRef]
- Wegmann, S.; Bennett, R.E.; Delorme, L.; Robbins, A.B.; Hu, M.; McKenzie, D.; Kirk, M.J.; Schiantarelli, J.; Tunio, N.; Amaral, A.C.; et al. Experimental evidence for the age dependence of tau protein spread in the brain. Sci. Adv. 2019, 5, eaaw6404. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Clavaguera, F.; Potier, M.-C. The prion-like propagation hypothesis in Alzheimer’s and Parkinson’s disease. Curr. Opin. Neurol. 2019, 32, 266–271. [Google Scholar] [CrossRef]
- Blennow, K.; Wallin, A.; Agren, H.; Spenger, C.; Siegfried, J.; Vanmechelen, E. Tau protein in cerebrospinal fluid: A biochemical marker for axonal degeneration in Alzheimer disease? Mol. Chem. Neuropathol. 1995, 26, 231–245. [Google Scholar] [CrossRef]
- Gómez-Ramos, A.; Díaz-Hernández, M.; Cuadros, R.; Hernández, F.; Avila, J. Extracellular tau is toxic to neuronal cells. FEBS Lett. 2006, 580, 4842–4850. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.W.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Sokolow, S.; Henkins, K.M.; Bilousova, T.; Gonzalez, B.; Vinters, H.V.; Miller, C.A.; Cornwell, L.; Poon, W.W.; Gylys, K.H. Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer’s disease. J. Neurochem. 2015, 133, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Simón, D.; García-García, E.; Royo, F.; Falcón-Pérez, J.M.; Avila, J. Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett. 2012, 586, 47–54. [Google Scholar] [CrossRef]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.Y.; et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef]
- Dujardin, S.; Bégard, S.; Caillierez, R.; Lachaud, C.; Delattre, L.; Carrier, S.; Loyens, A.; Galas, M.-C.; Bousset, L.; Melki, R.; et al. Ectosomes: A new mechanism for non-exosomal secretion of tau protein. PLoS ONE 2014, 9, e100760. [Google Scholar] [CrossRef]
- Chai, X.; Dage, J.L.; Citron, M. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol. Dis. 2012, 48, 356–366. [Google Scholar] [CrossRef]
- Katsinelos, T.; Zeitler, M.; Dimou, E.; Karakatsani, A.; Müller, H.-M.; Nachman, E.; Steringer, J.P.; Ruiz de Almodovar, C.; Nickel, W.; Jahn, T.R. Unconventional Secretion Mediates the Trans-cellular Spreading of Tau. Cell Rep. 2018, 23, 2039–2055. [Google Scholar] [CrossRef]
- Merezhko, M.; Brunello, C.A.; Yan, X.; Vihinen, H.; Jokitalo, E.; Uronen, R.-L.; Huttunen, H.J. Secretion of Tau via an Unconventional Non-vesicular Mechanism. Cell Rep. 2018, 25, 2027–2035. [Google Scholar] [CrossRef]
- Wu, J.W.; Herman, M.; Liu, L.; Simoes, S.; Acker, C.M.; Figueroa, H.; Steinberg, J.I.; Margittai, M.; Kayed, R.; Zurzolo, C.; et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 2013, 288, 1856–1870. [Google Scholar] [CrossRef]
- Gómez-Ramos, A.; Díaz-Hernández, M.; Rubio, A.; Miras-Portugal, M.T.; Avila, J. Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell. Neurosci. 2008, 37, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Morozova, V.; Cohen, L.S.; Makki, A.E.-H.; Shur, A.; Pilar, G.; El Idrissi, A.; Alonso, A.D. Normal and Pathological Tau Uptake Mediated by M1/M3 Muscarinic Receptors Promotes Opposite Neuronal Changes. Front. Cell. Neurosci. 2019, 13, 403. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138-47. [Google Scholar] [CrossRef]
- Calafate, S.; Flavin, W.; Verstreken, P.; Moechars, D. Loss of Bin1 Promotes the Propagation of Tau Pathology. Cell Rep. 2016, 17, 931–940. [Google Scholar] [CrossRef]
- Tardivel, M.; Bégard, S.; Bousset, L.; Dujardin, S.; Coens, A.; Melki, R.; Buée, L.; Colin, M. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 2016, 4, 117. [Google Scholar] [CrossRef]
- Brunello, C.A.; Merezhko, M.; Uronen, R.-L.; Huttunen, H.J. Mechanisms of secretion and spreading of pathological tau protein. Cell. Mol. Life Sci. 2019. [Google Scholar] [CrossRef]
- Flavin, W.P.; Bousset, L.; Green, Z.C.; Chu, Y.; Skarpathiotis, S.; Chaney, M.J.; Kordower, J.H.; Melki, R.; Campbell, E.M. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 2017, 134, 629–653. [Google Scholar] [CrossRef]
- Luo, W.; Liu, W.; Hu, X.; Hanna, M.; Caravaca, A.; Paul, S.M. Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci. Rep. 2015, 5, 11161. [Google Scholar] [CrossRef]
- Bolós, M.; Llorens-Martín, M.; Jurado-Arjona, J.; Hernández, F.; Rábano, A.; Avila, J. Direct Evidence of Internalization of Tau by Microglia In Vitro and In Vivo. J. Alzheimers. Dis. 2015, 50, 77–87. [Google Scholar] [CrossRef]
- Piacentini, R.; Li Puma, D.D.; Mainardi, M.; Lazzarino, G.; Tavazzi, B.; Arancio, O.; Grassi, C. Reduced gliotransmitter release from astrocytes mediates tau-induced synaptic dysfunction in cultured hippocampal neurons. Glia 2017, 65, 1302–1316. [Google Scholar] [CrossRef] [PubMed]
- Perea, J.R.; López, E.; Díez-Ballesteros, J.C.; Ávila, J.; Hernández, F.; Bolós, M. Extracellular Monomeric Tau Is Internalized by Astrocytes. Front. Neurosci. 2019, 13, 442. [Google Scholar] [CrossRef] [PubMed]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef] [PubMed]
- Hopp, S.C.; Lin, Y.; Oakley, D.; Roe, A.D.; DeVos, S.L.; Hanlon, D.; Hyman, B.T. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflammation 2018, 15, 269. [Google Scholar] [CrossRef] [PubMed]
- Martini-Stoica, H.; Cole, A.L.; Swartzlander, D.B.; Chen, F.; Wan, Y.-W.; Bajaj, L.; Bader, D.A.; Lee, V.M.Y.; Trojanowski, J.Q.; Liu, Z.; et al. TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J. Exp. Med. 2018, 215, 2355–2377. [Google Scholar] [CrossRef]
- Narasimhan, S.; Changolkar, L.; Riddle, D.M.; Kats, A.; Stieber, A.; Weitzman, S.A.; Zhang, B.; Li, Z.; Roberson, E.D.; Trojanowski, J.Q.; et al. Human tau pathology transmits glial tau aggregates in the absence of neuronal tau. J. Exp. Med. 2020, 217, e20190783. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, A.; Bichmann, M.; Ercan-Herbst, E.; Haberkant, P.; Schöndorf, D.C.; Wolf, M.; Fahim, S.A.; Murolo, E.; Ehrnhoefer, D.E. Asparagine endopeptidase cleaves tau at N167 after uptake into microglia. Neurobiol. Dis. 2019, 130, 104518. [Google Scholar] [CrossRef]
- Metschnikoff, E. Lecture on Phagocytosis and Immunity. Br. Med. J. 1891, 1, 213–217. [Google Scholar] [CrossRef]
- Achúcarro, N. Cellules allongées et Stäbchenzellen: Cellules neurogliques et cellules granulo-adipeuses à la corne dámmon du lapin. Trab. Lab. Investig. Biológica la Univ. Complut. Madrid 1909, 4, 2–15. [Google Scholar]
- Ramón y Cajal, S. Contribución al conocimiento de la neuroglía del cerebro humano. Trab. Lab. Investig. Biológica Univ. Complut. Madrid 1913, XI, 215–315. [Google Scholar]
- Río-Hortega, P. El “tercer elemento” de los centros nerviosos. I. La microglía en estado normal. Boletín Soc. Española Biol. 1919, VIII, 67–82. [Google Scholar]
- Río-Hortega, P. El “tercer elemento” de los centros nerviosos. II. Intervención de la microglía en los procesos patológicos (células en bastoncito y cuerpos gránulo-adiposos). Boletín Soc. Española Biol. 1919, VIII, 91–103. [Google Scholar]
- Río-Hortega, P. El “tercer elemento” de los centros nerviosos. III. Naturaleza probable de la microglía. Boletín Soc. Española Biol. 1919, VIII, 108–121. [Google Scholar]
- Río-Hortega, P. El “tercer elemento” de los centros nerviosos. IV. Poder fagocitario y movilidad de la microglía. Boletín Soc. Española Biol. 1919, VIII, 154–171. [Google Scholar]
- Tremblay, M.-È.; Lecours, C.; Samson, L.; Sánchez-Zafra, V.; Sierra, A. From the Cajal alumni Achúcarro and Río-Hortega to the rediscovery of never-resting microglia. Front. Neuroanat. 2015, 9, 1–10. [Google Scholar] [CrossRef]
- Sierra, A.; Paolicelli, R.C.; Kettenmann, H. Cien Años de Microglía: Milestones in a Century of Microglial Research. Trends Neurosci. 2019, 42, 778–792. [Google Scholar] [CrossRef]
- Alliot, F.; Godin, I.; Pessac, B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res. Dev. Brain Res. 1999, 117, 145–152. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. V Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef]
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep. 2017, 18, 391–405. [Google Scholar] [CrossRef]
- Réu, P.; Khosravi, A.; Bernard, S.; Mold, J.E.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; et al. The Lifespan and Turnover of Microglia in the Human Brain. Cell Rep. 2017, 20, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; Coenen, V.A.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Böttcher, C.; Schlickeiser, S.; Sneeboer, M.A.M.; Kunkel, D.; Knop, A.; Paza, E.; Fidzinski, P.; Kraus, L.; Snijders, G.J.L.; Kahn, R.S.; et al. Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat. Neurosci. 2019, 22, 78–90. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef]
- Colonna, M.; Brioschi, S. Neuroinflammation and neurodegeneration in human brain at single-cell resolution. Nat. Rev. Immunol. 2020, 20, 81–82. [Google Scholar] [CrossRef]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271. [Google Scholar] [CrossRef]
- Villa, A.; Gelosa, P.; Castiglioni, L.; Cimino, M.; Rizzi, N.; Pepe, G.; Lolli, F.; Marcello, E.; Sironi, L.; Vegeto, E.; et al. Sex-Specific Features of Microglia from Adult Mice. Cell Rep. 2018, 23, 3501–3511. [Google Scholar] [CrossRef]
- Yanguas-Casás, N. Physiological sex differences in microglia and their relevance in neurological disorders. Neuroimmunol. Neuroinflamm. 2020. [Google Scholar] [CrossRef]
- Kodama, L.; Guzman, E.; Etchegaray, J.I.; Li, Y.; Sayed, F.A.; Zhou, L.; Zhou, Y.; Zhan, L.; Le, D.; Udeochu, J.C.; et al. Microglial microRNAs mediate sex-specific responses to tau pathology. Nat. Neurosci. 2020, 23, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Peri, F.; Nüsslein-Volhard, C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell 2008, 133, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.P.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Aparicio, I.; Paris, I.; Sierra-Torre, V.; Plaza-Zabala, A.; Rodríguez-Iglesias, N.; Márquez-Ropero, M.; Beccari, S.; Huguet, P.; Abiega, O.; Alberdi, E.; et al. Microglia Actively Remodel Adult Hippocampal Neurogenesis through the Phagocytosis Secretome. J. Neurosci. 2020, 40, 1453–1482. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef]
- Miyamoto, A.; Wake, H.; Ishikawa, A.W.; Eto, K.; Shibata, K.; Murakoshi, H.; Koizumi, S.; Moorhouse, A.J.; Yoshimura, Y.; Nabekura, J. Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 2016, 7, 12540. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.T.; Dorman, L.C.; Pan, S.; Vainchtein, I.D.; Han, R.T.; Nakao-Inoue, H.; Taloma, S.E.; Barron, J.J.; Molofsky, A.B.; Kheirbek, M.A.; et al. Microglial Remodeling of the Extracellular Matrix Promotes Synapse Plasticity. Cell 2020, 182, 388–403. [Google Scholar] [CrossRef]
- Cserép, C.; Pósfai, B.; Lénárt, N.; Fekete, R.; László, Z.I.; Lele, Z.; Orsolits, B.; Molnár, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef]
- Wlodarczyk, A.; Holtman, I.R.; Krueger, M.; Yogev, N.; Bruttger, J.; Khorooshi, R.; Benmamar-Badel, A.; de Boer-Bergsma, J.J.; Martin, N.A.; Karram, K.; et al. A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. 2017, 36, 3292–3308. [Google Scholar] [CrossRef]
- Fantin, A.; Vieira, J.M.; Gestri, G.; Denti, L.; Schwarz, Q.; Prykhozhij, S.; Peri, F.; Wilson, S.W.; Ruhrberg, C. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 2010, 116, 829–840. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.-B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- Madry, C.; Kyrargyri, V.; Arancibia-Cárcamo, I.L.; Jolivet, R.; Kohsaka, S.; Bryan, R.M.; Attwell, D. Microglial Ramification, Surveillance, and Interleukin-1β Release Are Regulated by the Two-Pore Domain K+ Channel THIK-1. Neuron 2018, 97, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Nakanishi, H.; Nabekura, J.; Akaike, N. AMPA-kainate subtypes of glutamate receptor in rat cerebral microglia. J. Neurosci. 2000, 20, 251–258. [Google Scholar] [CrossRef]
- Alzheimer, A. Beiträge zur Kenntnis der pathologischen Neuroglia und ihrer Beziehungen zu den Abbauvorgängen im Nervengewebe. In Histologische und histopathologische Arbeiten über Die Grosshirnrinde mit Besonderer Berücksichtigung der pathologischen Anatomie der Geisteskrankheiten; Gustav Fischer: Jena, Germany, 1910; pp. 401–562. [Google Scholar]
- Cras, P.; Kawai, M.; Siedlak, S.; Perry, G. Microglia are associated with the extracellular neurofibrillary tangles of Alzheimer disease. Brain Res. 1991, 558, 312–314. [Google Scholar] [CrossRef]
- El Khoury, J.; Hickman, S.E.; Thomas, C.A.; Cao, L.; Silverstein, S.C.; Loike, J.D. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature 1996, 382, 716–719. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.-Y. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Holmes, B.B.; Furman, J.L.; Mahan, T.E.; Yamasaki, T.R.; Mirbaha, H.; Eades, W.C.; Belaygorod, L.; Cairns, N.J.; Holtzman, D.M.; Diamond, M.I. Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E4376–E4385. [Google Scholar] [CrossRef]
- van Olst, L.; Verhaege, D.; Franssen, M.; Kamermans, A.; Roucourt, B.; Carmans, S.; Ytebrouck, E.; van der Pol, S.M.A.; Wever, D.; Popovic, M.; et al. Microglial activation arises after aggregation of phosphorylated-tau in a neuron-specific P301S tauopathy mouse model. Neurobiol. Aging 2020, 89, 89–98. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet. Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef]
- Luo, X.-G.; Ding, J.-Q.; Chen, S.-D. Microglia in the aging brain: Relevance to neurodegeneration. Mol. Neurodegener. 2010, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Gottfried-Blackmore, A.C.; McEwen, B.S.; Bulloch, K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 2007, 55, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Matyszak, M.K.; Fearn, S. Altered antigen expression of microglia in the aged rodent CNS. Glia 1993, 7, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef]
- Han, J.; Zhu, K.; Zhang, X.-M.; Harris, R.A. Enforced microglial depletion and repopulation as a promising strategy for the treatment of neurological disorders. Glia 2019, 67, 217–231. [Google Scholar] [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.; Fryatt, G.; Cleal, M.; Obst, J.; Pipi, E.; Monzón-Sandoval, J.; Ribe, E.; Winchester, L.; Webber, C.; Nevado, A.; et al. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain 2019, 142, 3243–3264. [Google Scholar] [CrossRef] [PubMed]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Sosna, J.; Philipp, S.; Albay, R.; Reyes-Ruiz, J.M.; Baglietto-Vargas, D.; LaFerla, F.M.; Glabe, C.G. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef]
- Gale, S.C.; Gao, L.; Mikacenic, C.; Coyle, S.M.; Rafaels, N.; Murray Dudenkov, T.; Madenspacher, J.H.; Draper, D.W.; Ge, W.; Aloor, J.J.; et al. APOε4 is associated with enhanced in vivo innate immune responses in human subjects. J. Allergy Clin. Immunol. 2014, 134, 127–134. [Google Scholar] [CrossRef]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef]
- Bales, K.R.; Verina, T.; Dodel, R.C.; Du, Y.; Altstiel, L.; Bender, M.; Hyslop, P.; Johnstone, E.M.; Little, S.P.; Cummins, D.J.; et al. Lack of apolipoprotein E dramatically reduces amyloid β-peptide deposition. Nat. Genet. 1997, 17, 263–264. [Google Scholar] [CrossRef]
- Jiang, Q.; Lee, C.Y.D.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. ApoE Promotes the Proteolytic Degradation of Aβ. Neuron 2008, 58, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, J.D.; Ulland, T.K.; Mahan, T.E.; Nyström, S.; Nilsson, K.P.; Song, W.M.; Zhou, Y.; Reinartz, M.; Choi, S.; Jiang, H.; et al. ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med. 2018, 215, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Goedert, M.; Weisgraber, K.H.; Dong, L.M.; Jakes, R.; Huang, D.Y.; Pericak-Vance, M.; Schmechel, D.; Roses, A.D. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: Implications for Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 11183–11186. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Manis, M.; Long, J.; Wang, K.; Sullivan, P.M.; Remolina Serrano, J.; Hoyle, R.; Holtzman, D.M. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med. 2019, 216, 2546–2561. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Merlini, M.; Rafalski, V.A.; Rios Coronado, P.E.; Gill, T.M.; Ellisman, M.; Muthukumar, G.; Subramanian, K.S.; Ryu, J.K.; Syme, C.A.; Davalos, D.; et al. Fibrinogen Induces Microglia-Mediated Spine Elimination and Cognitive Impairment in an Alzheimer’s Disease Model. Neuron 2019, 101, 1099–1108. [Google Scholar] [CrossRef]
- Atagi, Y.; Liu, C.-C.; Painter, M.M.; Chen, X.-F.; Verbeeck, C.; Zheng, H.; Li, X.; Rademakers, R.; Kang, S.S.; Xu, H.; et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J. Biol. Chem. 2015, 290, 26043–26050. [Google Scholar] [CrossRef]
- Bailey, C.C.; DeVaux, L.B.; Farzan, M. The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. J. Biol. Chem. 2015, 290, 26033–26042. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Ulland, T.K.; Colonna, M. TREM2—A key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Bemiller, S.M.; McCray, T.J.; Allan, K.; Formica, S.V.; Xu, G.; Wilson, G.; Kokiko-Cochran, O.N.; Crish, S.D.; Lasagna-Reeves, C.A.; Ransohoff, R.M.; et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegener. 2017, 12, 74. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Remolina Serrano, J.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Gratuze, M.; Narasimhan, S.; Jain, N.; Koscal, L.J.; Jiang, H.; Manis, M.; Colonna, M.; Lee, V.M.Y.; Ulrich, J.D.; et al. TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. 2019, 22, 1217–1222. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.G.; Sauerbeck, A.D.; St-Pierre, M.-K.; Xiong, M.; Kim, N.; Serrano, J.R.; Tremblay, M.-È.; Kummer, T.T.; Colonna, M.; et al. Impact of TREM2R47H variant on tau pathology–induced gliosis and neurodegeneration. J. Clin. Investig. 2020, 130, 4954–4968. [Google Scholar] [CrossRef] [PubMed]
- Paloneva, J.; Manninen, T.; Christman, G.; Hovanes, K.; Mandelin, J.; Adolfsson, R.; Bianchin, M.; Bird, T.; Miranda, R.; Salmaggi, A.; et al. Mutations in Two Genes Encoding Different Subunits of a Receptor Signaling Complex Result in an Identical Disease Phenotype. Am. J. Hum. Genet. 2002, 71, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Brosseron, F.; Kolbe, C.; Santarelli, F.; Carvalho, S.; Antonell, A.; Castro-Gomez, S.; Tacik, P.; Namasivayam, A.A.; Mangone, G.; Schneider, R.; et al. Multicenter Alzheimer’s and Parkinson’s disease immune biomarker verification study. Alzheimers. Dement. 2020, 16, 292–304. [Google Scholar] [CrossRef]
- Ewers, M.; Franzmeier, N.; Suárez-Calvet, M.; Morenas-Rodriguez, E.; Caballero, M.A.A.; Kleinberger, G.; Piccio, L.; Cruchaga, C.; Deming, Y.; Dichgans, M.; et al. Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci. Transl. Med. 2019, 11, eaav6221. [Google Scholar] [CrossRef] [PubMed]
- Ewers, M.; Biechele, G.; Suárez-Calvet, M.; Sacher, C.; Blume, T.; Morenas-Rodriguez, E.; Deming, Y.; Piccio, L.; Cruchaga, C.; Kleinberger, G.; et al. Higher CSF sTREM2 and microglia activation are associated with slower rates of beta-amyloid accumulation. EMBO Mol. Med. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Lange, C.; Mullin, K.; Parkinson, M.; Hsiao, M.; Hogan, M.F.; Schjeide, B.M.M.; Hooli, B.; DiVito, J.; Ionita, I.; et al. Genome-wide Association Analysis Reveals Putative Alzheimer’s Disease Susceptibility Loci in Addition to APOE. Am. J. Hum. Genet. 2008, 83, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.-C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.-S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef]
- Freeman, S.; Kelm, S.; Barber, E.; Crocker, P. Characterization of CD33 as a new member of the sialoadhesin family of cellular interaction molecules. Blood 1995, 85, 2005–2012. [Google Scholar] [CrossRef]
- Pillai, S.; Netravali, I.A.; Cariappa, A.; Mattoo, H. Siglecs and Immune Regulation. Annu. Rev. Immunol. 2012, 30, 357–392. [Google Scholar] [CrossRef]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s Disease Risk Gene CD33 Inhibits Microglial Uptake of Amyloid Beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Patel, S.; Federico, A.N.; Choi, S.H.; Innes, B.J.; Oram, M.K.; Cereghetti, G.; McGinty, D.; Anselmo, A.; Sadreyev, R.I.; et al. TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer’s Disease. Neuron 2019, 103, 820–835. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zaidi, T.; Iqbal, K.; Grundke-Iqbal, I.; Merkle, R.K.; Gong, C.-X. Role of glycosylation in hyperphosphorylation of tau in Alzheimer’s disease. FEBS Lett. 2002, 512, 101–106. [Google Scholar] [CrossRef]
- Nagamine, S.; Yamazaki, T.; Makioka, K.; Fujita, Y.; Ikeda, M.; Takatama, M.; Okamoto, K.; Yokoo, H.; Ikeda, Y. Hypersialylation is a common feature of neurofibrillary tangles and granulovacuolar degenerations in Alzheimer’s disease and tauopathy brains. Neuropathology 2016, 36, 333–345. [Google Scholar] [CrossRef]
- Zlotnik, A.; Yoshie, O. The chemokine superfamily revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef]
- Hatori, K.; Nagai, A.; Heisel, R.; Ryu, J.K.; Kim, S.U. Fractalkine and fractalkine receptors in human neurons and glial cells. J. Neurosci. Res. 2002, 69, 418–426. [Google Scholar] [CrossRef]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef]
- Bazan, J.F.; Bacon, K.B.; Hardiman, G.; Wang, W.; Soo, K.; Rossi, D.; Greaves, D.R.; Zlotnik, A.; Schall, T.J. A new class of membrane-bound chemokine with a CX3C motif. Nature 1997, 385, 640–644. [Google Scholar] [CrossRef]
- Al-Aoukaty, A.; Rolstad, B.; Giaid, A.; Maghazachi, A.A. MIP-3alpha, MIP-3beta and fractalkine induce the locomotion and the mobilization of intracellular calcium, and activate the heterotrimeric G proteins in human natural killer cells. Immunology 1998, 95, 618–624. [Google Scholar] [CrossRef]
- Chandrasekar, B.; Mummidi, S.; Perla, R.P.; Bysani, S.; Dulin, N.O.; Liu, F.; Melby, P.C. Fractalkine (CX3CL1) stimulated by nuclear factor kappaB (NF-kappaB)-dependent inflammatory signals induces aortic smooth muscle cell proliferation through an autocrine pathway. Biochem. J. 2003, 373, 547–558. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bisht, K.; Tremblay, M.-È. Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front. Cell. Neurosci. 2014, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Zujovic, V.; Benavides, J.; Vigé, X.; Carter, C.; Taupin, V. Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia 2000, 29, 305–315. [Google Scholar] [CrossRef]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Innamorato, N.G.; Jaworski, T.; Rábano, A.; Kügler, S.; Van Leuven, F.; Cuadrado, A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 2014, 137, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Bolós, M.; Llorens-Martín, M.; Perea, J.; Jurado-Arjona, J.; Rábano, A.; Hernández, F.; Avila, J. Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol. Neurodegener. 2017, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Fuster-Matanzo, A.; Jurado-Arjona, J.; Benvegnù, S.; García, E.; Martín-Maestro, P.; Gómez-Sintes, R.; Hernández, F.; Ávila, J. Glycogen synthase kinase-3β regulates fractalkine production by altering its trafficking from Golgi to plasma membrane: Implications for Alzheimer’s disease. Cell. Mol. Life Sci. 2017, 74, 1153–1163. [Google Scholar] [CrossRef]
- Lee, S.; Varvel, N.H.; Konerth, M.E.; Xu, G.; Cardona, A.E.; Ransohoff, R.M.; Lamb, B.T. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol. 2010, 177, 2549–2562. [Google Scholar] [CrossRef]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of tau pathology by the microglial fractalkine receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S.T. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [CrossRef]
- Castro-Sánchez, S.; García-Yagüe, Á.J.; Kügler, S.; Lastres-Becker, I. CX3CR1-deficient microglia shows impaired signalling of the transcription factor NRF2: Implications in tauopathies. Redox Biol. 2019, 22, 101118. [Google Scholar] [CrossRef]
- Gyoneva, S.; Hosur, R.; Gosselin, D.; Zhang, B.; Ouyang, Z.; Cotleur, A.C.; Peterson, M.; Allaire, N.; Challa, R.; Cullen, P.; et al. Cx3cr1-deficient microglia exhibit a premature aging transcriptome. Life Sci. Alliance 2019, 2, e201900453. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; Coleman, U.; Kingery-Gallagher, N.D.; El Khoury, J. Heterozygous CX3CR1 Deficiency in Microglia Restores Neuronal β-Amyloid Clearance Pathways and Slows Progression of Alzheimer’s Like-Disease in PS1-APP Mice. Front. Immunol. 2019, 10, 2780. [Google Scholar] [CrossRef] [PubMed]
- Nash, K.R.; Lee, D.C.; Hunt, J.B.; Morganti, J.M.; Selenica, M.-L.; Moran, P.; Reid, P.; Brownlow, M.; Guang-Yu Yang, C.; Savalia, M.; et al. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol. Aging 2013, 34, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Bemiller, S.M.; Maphis, N.M.; Formica, S.V.; Wilson, G.N.; Miller, C.M.; Xu, G.; Kokiko-Cochran, O.N.; Kim, K.-W.; Jung, S.; Cannon, J.L.; et al. Genetically enhancing the expression of chemokine domain of CX3CL1 fails to prevent tau pathology in mouse models of tauopathy. J. Neuroinflamm. 2018, 15, 278. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; He, W.; Gayen, M.; Benoit, M.R.; Luo, X.; Hu, X.; Yan, R. Activated CX3CL1/Smad2 Signals Prevent Neuronal Loss and Alzheimer’s Tau Pathology-Mediated Cognitive Dysfunction. J. Neurosci. 2020, 40, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.N.; Subbarayan, M.S.; Grimmig, B.; Weesner, J.A.; Moss, L.; Peters, M.; Weeber, E.; Nash, K.; Bickford, P.C. Two forms of CX3CL1 display differential activity and rescue cognitive deficits in CX3CL1 knockout mice. J. Neuroinflamm. 2020, 17, 157. [Google Scholar] [CrossRef]
- Bondareff, W.; Mountjoy, C.Q.; Roth, M.; Hauser, D.L. Neurofibrillary degeneration and neuronal loss in Alzheimer’s disease. Neurobiol. Aging 1989, 10, 709–715. [Google Scholar] [CrossRef]
- Morsch, R.; Simon, W.; Coleman, P.D. Neurons may live for decades with neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 1999, 58, 188–197. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Wegmann, S.; Kopeikina, K.J.; Hawkes, J.; Rudinskiy, N.; Andermann, M.L.; Spires-Jones, T.L.; Bacskai, B.J.; Hyman, B.T. Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 510–514. [Google Scholar] [CrossRef]
- Andorfer, C.; Acker, C.M.; Kress, Y.; Hof, P.R.; Duff, K.; Davies, P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 2005, 25, 5446–5454. [Google Scholar] [CrossRef]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef]
- Sydow, A.; Van der Jeugd, A.; Zheng, F.; Ahmed, T.; Balschun, D.; Petrova, O.; Drexler, D.; Zhou, L.; Rune, G.; Mandelkow, E.; et al. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J. Neurosci. 2011, 31, 2511–2525. [Google Scholar] [CrossRef] [PubMed]
- Van der Jeugd, A.; Hochgräfe, K.; Ahmed, T.; Decker, J.M.; Sydow, A.; Hofmann, A.; Wu, D.; Messing, L.; Balschun, D.; D’Hooge, R.; et al. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol. 2012, 123, 787–805. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Wegmann, S.; Dujardin, S.; Commins, C.; Schiantarelli, J.; Klickstein, N.; Kamath, T.V.; Carlson, G.A.; Nelken, I.; Hyman, B.T. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci. 2019, 22, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Pickett, E.K.; Herrmann, A.G.; McQueen, J.; Abt, K.; Dando, O.; Tulloch, J.; Jain, P.; Dunnett, S.; Sohrabi, S.; Fjeldstad, M.P.; et al. Amyloid Beta and Tau Cooperate to Cause Reversible Behavioral and Transcriptional Deficits in a Model of Alzheimer’s Disease. Cell Rep. 2019, 29, 3592–3604. [Google Scholar] [CrossRef]
- Dujardin, S.; Commins, C.; Lathuiliere, A.; Beerepoot, P.; Fernandes, A.R.; Kamath, T.V.; De Los Santos, M.B.; Klickstein, N.; Corjuc, D.L.; Corjuc, B.T.; et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat. Med. 2020, 26, 1256–1263. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Hernández, M.; Gómez-Ramos, A.; Rubio, A.; Gómez-Villafuertes, R.; Naranjo, J.R.; Miras-Portugal, M.T.; Avila, J. Tissue-nonspecific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J. Biol. Chem. 2010, 285, 32539–32548. [Google Scholar] [CrossRef]
- Vardy, E.R.L.C.; Kellett, K.A.B.; Cocklin, S.L.; Hooper, N.M. Alkaline phosphatase is increased in both brain and plasma in Alzheimer’s disease. Neurodegener. Dis. 2012, 9, 31–37. [Google Scholar] [CrossRef]
- Curtis, D.; Bakaya, K.; Sharma, L.; Bandyopadhyay, S. Weighted burden analysis of exome-sequenced late-onset Alzheimer’s cases and controls provides further evidence for a role for PSEN1 and suggests involvement of the PI3K/Akt/GSK-3β and WNT signalling pathways. Ann. Hum. Genet. 2020, 1–12. [Google Scholar] [CrossRef]
- Doens, D.; Fernández, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Gunawardana, C.G.; Mehrabian, M.; Wang, X.; Mueller, I.; Lubambo, I.B.; Jonkman, J.E.N.; Wang, H.; Schmitt-Ulms, G. The Human Tau Interactome: Binding to the Ribonucleoproteome, and Impaired Binding of the Proline-to-Leucine Mutant at Position 301 (P301L) to Chaperones and the Proteasome. Mol. Cell. Proteomics 2015, 14, 3000–3014. [Google Scholar] [CrossRef] [PubMed]
- Stefanoska, K.; Volkerling, A.; Bertz, J.; Poljak, A.; Ke, Y.D.; Ittner, L.M.; Ittner, A. An N-terminal motif unique to primate tau enables differential protein-protein interactions. J. Biol. Chem. 2018, 293, 3710–3719. [Google Scholar] [CrossRef] [PubMed]
- Luck, K.; Kim, D.-K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020. [Google Scholar] [CrossRef]
- Wang, P.; Ye, Y. An integrin receptor complex mediates filamentous Tau-induced activation of primary astrocytes. bioRxiv 2020, .95. [Google Scholar] [CrossRef]
- Pooler, A.M.; Usardi, A.; Evans, C.J.; Philpott, K.L.; Noble, W.; Hanger, D.P. Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol. Aging 2012, 33, 431–e27. [Google Scholar] [CrossRef]
- Arrasate, M.; Pérez, M.; Avila, J. Tau dephosphorylation at tau-1 site correlates with its association to cell membrane. Neurochem. Res. 2000, 25, 43–50. [Google Scholar] [CrossRef]
- Ait-Bouziad, N.; Chiki, A.; Limorenko, G.; Xiao, S.; Eliezer, D.; Lashuel, H.A. Phosphorylation of the overlooked tyrosine 310 regulates the structure, aggregation, and microtubule- and lipid-binding properties of Tau. J. Biol. Chem. 2020, 295. [Google Scholar] [CrossRef]
- Momtazmanesh, S.; Perry, G.; Rezaei, N. Toll-like receptors in Alzheimer’s disease. J. Neuroimmunol. 2020, 348, 577362. [Google Scholar] [CrossRef]
- Perea, J.R.; Ávila, J.; Bolós, M. Dephosphorylated rather than hyperphosphorylated Tau triggers a pro-inflammatory profile in microglia through the p38 MAPK pathway. Exp. Neurol. 2018, 310, 14–21. [Google Scholar] [CrossRef]
- Gjoneska, E.; Pfenning, A.R.; Mathys, H.; Quon, G.; Kundaje, A.; Tsai, L.-H.; Kellis, M. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 2015, 518, 365–369. [Google Scholar] [CrossRef]
- Adsera, C.B.; Park, Y.P.; Meuleman, W.; Kellis, M. Integrative analysis of 10,000 epigenomic maps across 800 samples for regulatory genomics and disease dissection. bioRxiv 2019, 810291. [Google Scholar] [CrossRef]
- Wendeln, A.-C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Häsler, L.M.; Wild, K.; Skodras, A.; et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef]
- Unger, M.S.; Li, E.; Scharnagl, L.; Poupardin, R.; Altendorfer, B.; Mrowetz, H.; Hutter-Paier, B.; Weiger, T.M.; Heneka, M.T.; Attems, J.; et al. CD8+ T-cells infiltrate Alzheimer’s disease brains and regulate neuronal- and synapse-related gene expression in APP-PS1 transgenic mice. Brain. Behav. Immun. 2020. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef]
- Minter, M.R.; Hinterleitner, R.; Meisel, M.; Zhang, C.; Leone, V.; Zhang, X.; Oyler-Castrillo, P.; Zhang, X.; Musch, M.W.; Shen, X.; et al. Antibiotic-induced perturbations in microbial diversity during post-natal development alters amyloid pathology in an aged APPSWE/PS1ΔE9 murine model of Alzheimer’s disease. Sci. Rep. 2017, 7, 10411. [Google Scholar] [CrossRef]
- Dodiya, H.B.; Kuntz, T.; Shaik, S.M.; Baufeld, C.; Leibowitz, J.; Zhang, X.; Gottel, N.; Zhang, X.; Butovsky, O.; Gilbert, J.A.; et al. Sex-specific effects of microbiome perturbations on cerebral Aβ amyloidosis and microglia phenotypes. J. Exp. Med. 2019, 216, 1542–1560. [Google Scholar] [CrossRef]
- Elmore, M.R.P.; Hohsfield, L.A.; Kramár, E.A.; Soreq, L.; Lee, R.J.; Pham, S.T.; Najafi, A.R.; Spangenberg, E.E.; Wood, M.A.; West, B.L.; et al. Replacement of microglia in the aged brain reverses cognitive, synaptic, and neuronal deficits in mice. Aging Cell 2018, 17, e12832. [Google Scholar] [CrossRef]
- Pluvinage, J.V.; Haney, M.S.; Smith, B.A.H.; Sun, J.; Iram, T.; Bonanno, L.; Li, L.; Lee, D.P.; Morgens, D.W.; Yang, A.C.; et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 2019, 568, 187–192. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perea, J.R.; Bolós, M.; Avila, J. Microglia in Alzheimer’s Disease in the Context of Tau Pathology. Biomolecules 2020, 10, 1439. https://doi.org/10.3390/biom10101439
Perea JR, Bolós M, Avila J. Microglia in Alzheimer’s Disease in the Context of Tau Pathology. Biomolecules. 2020; 10(10):1439. https://doi.org/10.3390/biom10101439
Chicago/Turabian StylePerea, Juan Ramón, Marta Bolós, and Jesús Avila. 2020. "Microglia in Alzheimer’s Disease in the Context of Tau Pathology" Biomolecules 10, no. 10: 1439. https://doi.org/10.3390/biom10101439
APA StylePerea, J. R., Bolós, M., & Avila, J. (2020). Microglia in Alzheimer’s Disease in the Context of Tau Pathology. Biomolecules, 10(10), 1439. https://doi.org/10.3390/biom10101439