Microglia-Mediated Neurodegeneration in Perinatal Brain Injuries

 ,
,  ,
,
Abstract
1. Perinatal Brain Disorders
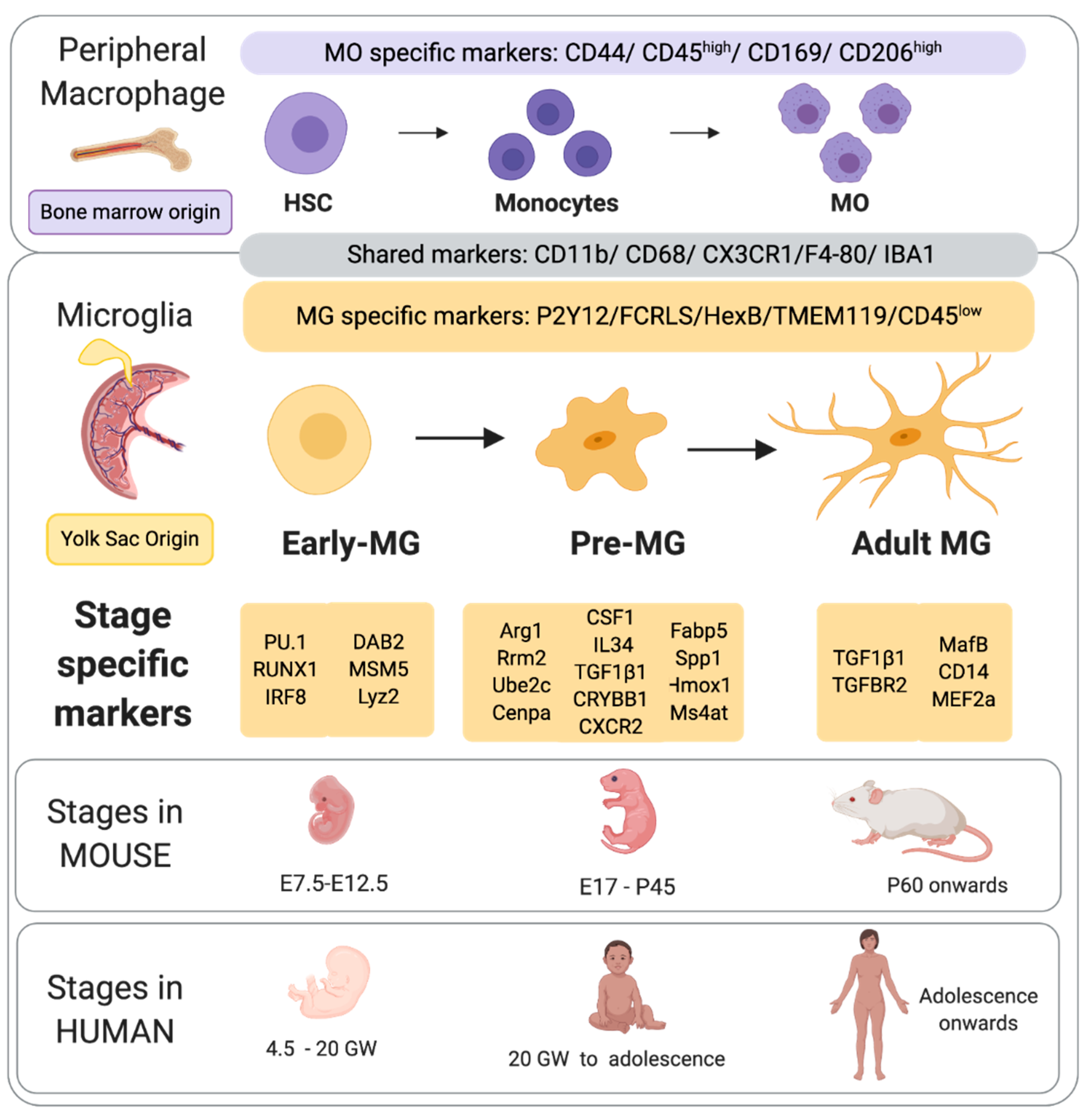
2. Origins, Development and Heterogeneity of Microglia
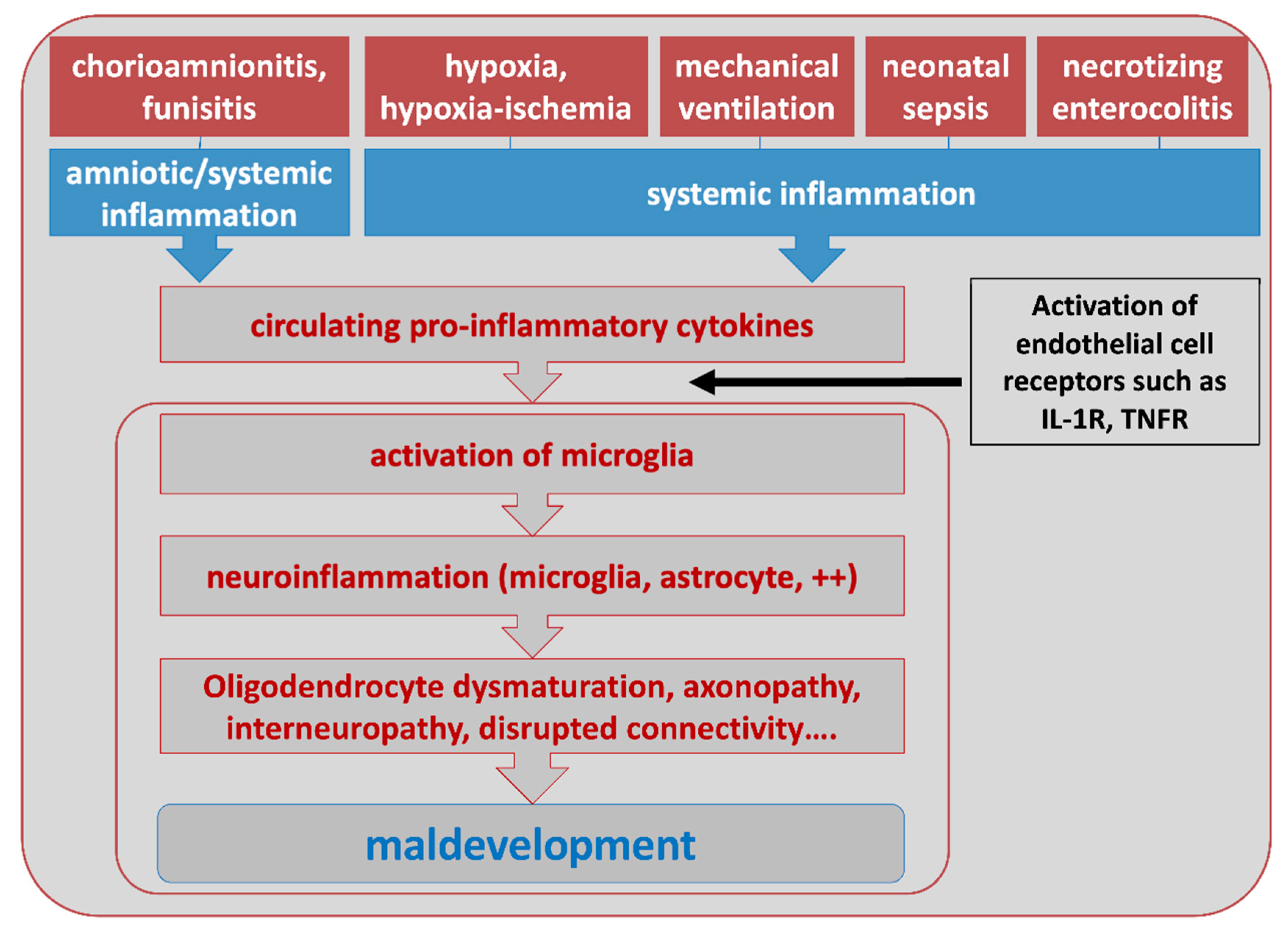
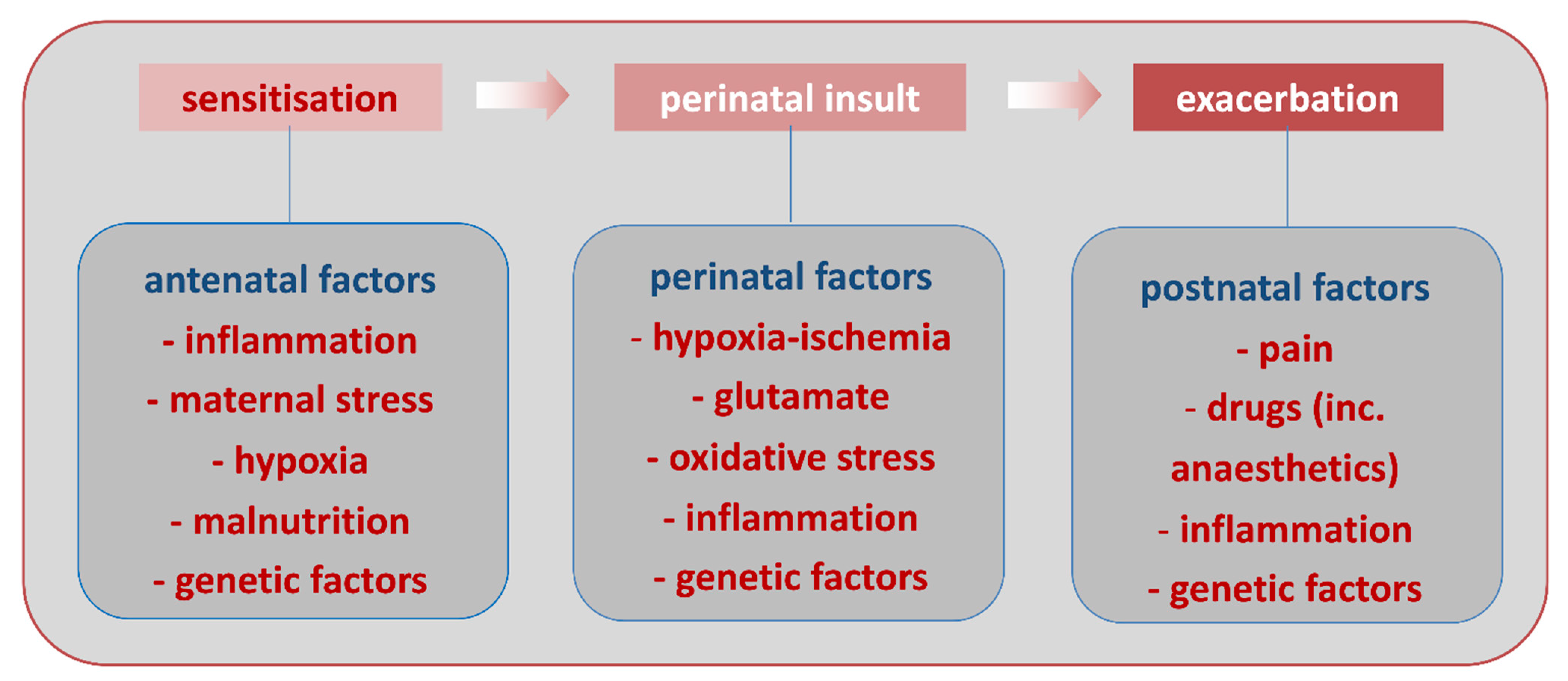
3. Role of Systemic Inflammation in PBIs
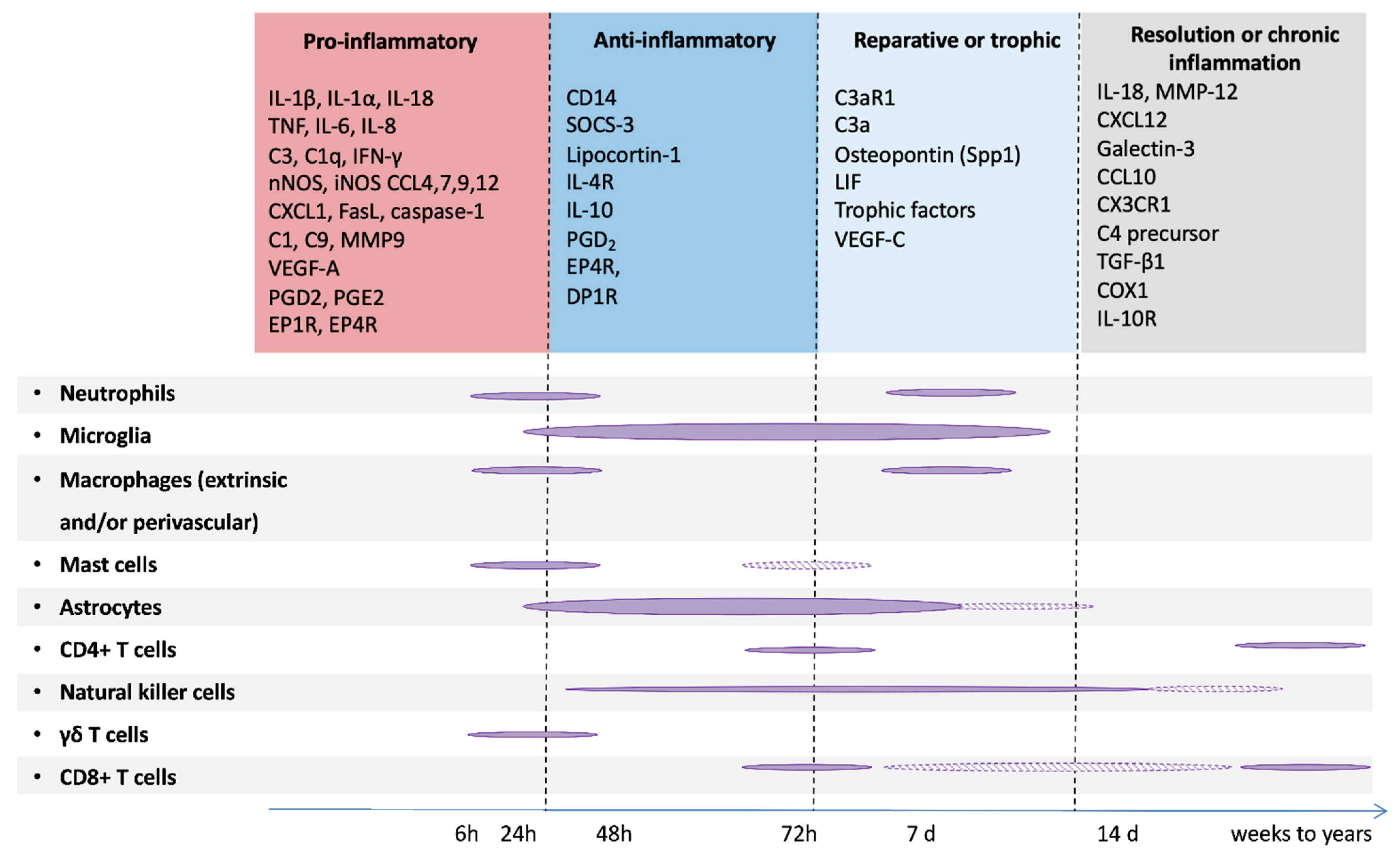
4. Role of Microglia in PBIs
5. Limitations of Microglial Studies in the Immature Animal
6. Microglia and Other Cellular Brain Partners
6.1. Microglia Effects on Oligodendrocyte Progenitor Cell Maturation and Myelin Deficit in PBIs
6.2. Microglia Interaction with Astrocytes in PBIs
6.3. Microglia Effects on Neuronal Cell Death in PBIs
6.4. Microglia Effects on Interneurons in PBIs
6.5. Microglia Effects on Neuronal Connectivity in PBIs
7. Microglia, a Target for Neuroprotection in PBIs?
8. Perspectives and Potential Relevance for Other Brain Disorders
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PBI | perinatal brain injury; |
| EoFGR | encephalopathy of fetal growth restriction; |
| EoP | encephalopathy of prematurity; |
| NE | neonatal encephalopathy; |
| NS | neonatal stroke; |
| ASD | autism spectrum disorders; |
| MS | multiple sclerosis; |
| PD | Parkinson’s disease; |
| PSD95 | post synaptic density 95; |
| AD | Alzheimer’s Disease; |
| TBI | traumatic brain injury |
References
- Burke, C.; Gobe, G. Pontosubicular apoptosis (“necrosis”) in human neonates with intrauterine growth retardation and placental infarction. Virchows Arch. 2005, 446, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Schiering, I.A.; de Haan, T.R.; Niermeijer, J.M.; Koelman, J.H.; Majoie, C.B.; Reneman, L.; Aronica, E. Correlation between clinical and histologic findings in the human neonatal hippocampus after perinatal asphyxia. J. Neuropathol. Exp. Neurol. 2014, 73, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Titomanlio, L.; Fernandez-Lopez, D.; Manganozzi, L.; Moretti, R.; Vexler, Z.S.; Gressens, P. Pathophysiology and neuroprotection of global and focal perinatal brain injury: Lessons from animal models. Pediatr. Neurol. 2015, 52, 566–584. [Google Scholar] [CrossRef] [PubMed]
- Ball, G.; Pazderova, L.; Chew, A.; Tusor, N.; Merchant, N.; Arichi, T.; Allsop, J.M.; Cowan, F.M.; Edwards, A.D.; Counsell, S.J. Thalamocortical Connectivity Predicts Cognition in Children Born Preterm. Cereb. Cortex 2015, 25, 4310–4318. [Google Scholar] [CrossRef] [PubMed]
- Pandit, A.S.; Robinson, E.; Aljabar, P.; Ball, G.; Gousias, I.S.; Wang, Z.; Hajnal, J.V.; Rueckert, D.; Counsell, S.J.; Montana, G.; et al. Whole-brain mapping of structural connectivity in infants reveals altered connection strength associated with growth and preterm birth. Cereb. Cortex 2014, 24, 2324–2333. [Google Scholar] [CrossRef] [PubMed]
- Stolp, H.B.; Fleiss, B.; Arai, Y.; Supramaniam, V.; Vontell, R.; Birtles, S.; Yates, A.G.; Baburamani, A.A.; Thornton, C.; Rutherford, M.; et al. Interneuron Development Is Disrupted in Preterm Brains With Diffuse White Matter Injury: Observations in Mouse and Human. Front. Physiol. 2019, 10, 955. [Google Scholar] [CrossRef]
- Heinonen, K.; Eriksson, J.G.; Lahti, J.; Kajantie, E.; Pesonen, A.K.; Tuovinen, S.; Osmond, C.; Raikkonen, K. Late preterm birth and neurocognitive performance in late adulthood: A birth cohort study. Pediatrics 2015, 135, e818–825. [Google Scholar] [CrossRef]
- Cheong, J.L.; Bainbridge, A.; Anderson, P.J.; Lee, K.J.; Burnett, A.C.; Thompson, D.K.; Roberts, G.; Wood, S.J.; Doyle, L.W.; Robertson, N.J. Altered posterior cingulate brain metabolites and cognitive dysfunction in preterm adolescents. Pediatr. Res. 2016, 79, 716–722. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, J.E.; Begbie, F.D.; Trojanowski, J.Q.; Smith, D.H.; Stewart, W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013, 136, 28–42. [Google Scholar] [CrossRef]
- Lin, C.Y.; Chang, Y.C.; Wang, S.T.; Lee, T.Y.; Lin, C.F.; Huang, C.C. Altered inflammatory responses in preterm children with cerebral palsy. Ann. Neurol. 2010, 68, 204–212. [Google Scholar] [CrossRef]
- Robertson, N.J.; Cox, I.J.; Cowan, F.M.; Counsell, S.J.; Azzopardi, D.; Edwards, A.D. Cerebral intracellular lactic alkalosis persisting months after neonatal encephalopathy measured by magnetic resonance spectroscopy. Pediatr. Res. 1999, 46, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Ziebell, J.M.; Rowe, R.K.; Muccigrosso, M.M.; Reddaway, J.T.; Adelson, P.D.; Godbout, J.P.; Lifshitz, J. Aging with a traumatic brain injury: Could behavioral morbidities and endocrine symptoms be influenced by microglial priming? Brain Behav. Immun. 2017, 59, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.C.; Anderson, P.J.; Cheong, J.; Doyle, L.W.; Davey, C.G.; Wood, S.J. Prevalence of psychiatric diagnoses in preterm and full-term children, adolescents and young adults: A meta-analysis. Psychol. Med. 2011, 41, 2463–2474. [Google Scholar] [CrossRef] [PubMed]
- Ewing-Cobbs, L.; Prasad, M.R.; Kramer, L.; Cox, C.S., Jr.; Baumgartner, J.; Fletcher, S.; Mendez, D.; Barnes, M.; Zhang, X.; Swank, P. Late intellectual and academic outcomes following traumatic brain injury sustained during early childhood. J. Neurosurg. 2006, 105, 287–296. [Google Scholar] [CrossRef]
- Lahti, M.; Eriksson, J.G.; Heinonen, K.; Kajantie, E.; Lahti, J.; Wahlbeck, K.; Tuovinen, S.; Pesonen, A.K.; Mikkonen, M.; Osmond, C.; et al. Late preterm birth, post-term birth, and abnormal fetal growth as risk factors for severe mental disorders from early to late adulthood. Psychol. Med. 2015, 45, 985–999. [Google Scholar] [CrossRef]
- Wu, C.W.; Huang, S.W.; Lin, J.W.; Liou, T.H.; Chou, L.C.; Lin, H.W. Risk of stroke among patients with cerebral palsy: A population-based cohort study. Dev. Med. Child. Neurol. 2017, 59, 52–56. [Google Scholar] [CrossRef]
- Fleiss, B.; Gressens, P. Tertiary mechanisms of brain damage: A new hope for treatment of cerebral palsy? Lancet Neurol. 2012, 11, 556–566. [Google Scholar] [CrossRef]
- Aden, U. Neonatal stroke is not a harmless condition. Stroke 2009, 40, 1948–1949. [Google Scholar] [CrossRef][Green Version]
- Colella, M.; Frerot, A.; Novais, A.R.B.; Baud, O. Neonatal and Long-Term Consequences of Fetal Growth Restriction. Curr. Pediatr. Rev. 2018, 14, 212–218. [Google Scholar] [CrossRef]
- De Vries, L.S.; Glass, H.C. Preface. Handb. Clin. Neurol. 2019, 162, ix. [Google Scholar] [CrossRef]
- Fleiss, B.; Tann, C.J.; Degos, V.; Sigaut, S.; Van Steenwinckel, J.; Schang, A.L.; Kichev, A.; Robertson, N.J.; Mallard, C.; Hagberg, H.; et al. Inflammation-induced sensitization of the brain in term infants. Dev. Med. Child Neurol. 2015, 57 (Suppl. 3), 17–28. [Google Scholar] [CrossRef]
- Fleiss, B.; Wong, F.; Brownfoot, F.; Shearer, I.K.; Baud, O.; Walker, D.W.; Gressens, P.; Tolcos, M. Knowledge Gaps and Emerging Research Areas in Intrauterine Growth Restriction-Associated Brain Injury. Front. Endocrinol. 2019, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Bokobza, C.; Van Steenwinckel, J.; Mani, S.; Mezger, V.; Fleiss, B.; Gressens, P. Neuroinflammation in preterm babies and autism spectrum disorders. Pediatr. Res. 2019, 85, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Linsell, L.; Malouf, R.; Johnson, S.; Morris, J.; Kurinczuk, J.J.; Marlow, N. Prognostic Factors for Behavioral Problems and Psychiatric Disorders in Children Born Very Preterm or Very Low Birth Weight: A Systematic Review. J. Dev. Behav. Pediatr. 2016, 37, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Montagna, A.; Nosarti, C. Socio-Emotional Development Following Very Preterm Birth: Pathways to Psychopathology. Front. Psychol. 2016, 7, 80. [Google Scholar] [CrossRef] [PubMed]
- Lawson, L.J.; Perry, V.H.; Gordon, S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience 1992, 48, 405–415. [Google Scholar] [CrossRef]
- Tay, T.L.; Savage, J.C.; Hui, C.W.; Bisht, K.; Tremblay, M.E. Microglia across the lifespan: From origin to function in brain development, plasticity and cognition. J. Physiol. 2017, 595, 1929–1945. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Hoeffel, G.; Wang, Y.; Greter, M.; See, P.; Teo, P.; Malleret, B.; Leboeuf, M.; Low, D.; Oller, G.; Almeida, F.; et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J. Exp. Med. 2012, 209, 1167–1181. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Rosenbauer, F.; Tenen, D.G. Transcription factors in myeloid development: Balancing differentiation with transformation. Nat. Rev. Immunol. 2007, 7, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Holscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zvi, A.; Lacoste, B.; Kur, E.; Andreone, B.J.; Mayshar, Y.; Yan, H.; Gu, C. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 2014, 509, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Vargas Aguilar, S.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; Zelada Gonzalez, F.; Perrin, P.; et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, aad8670. [Google Scholar] [CrossRef]
- Spittau, B.; Dokalis, N.; Prinz, M. The Role of TGFbeta Signaling in Microglia Maturation and Activation. Trends Immunol. 2020, 41, 836–848. [Google Scholar] [CrossRef]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Masuda, T.; Amann, L.; Sankowski, R.; Staszewski, O.; Lenz, M.; d´Errico, P.; Snaidero, N.; Costa Jordao, M.J.; Bottcher, C.; Kierdorf, K.; et al. Novel Hexb-based tools for studying microglia in the CNS. Nat. Immunol. 2020, 21, 802–815. [Google Scholar] [CrossRef]
- Otis, E.M.; Brent, R. Equivalent ages in mouse and human embryos. Anat. Rec. 1954, 120, 33–63. [Google Scholar] [CrossRef]
- Andersen, S.L. Trajectories of brain development: Point of vulnerability or window of opportunity? Neurosci. Biobehav. Rev. 2003, 27, 3–18. [Google Scholar] [CrossRef]
- Swinnen, N.; Smolders, S.; Avila, A.; Notelaers, K.; Paesen, R.; Ameloot, M.; Brone, B.; Legendre, P.; Rigo, J.M. Complex invasion pattern of the cerebral cortex bymicroglial cells during development of the mouse embryo. Glia 2013, 61, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Arnold, T.; Betsholtz, C. Correction: The importance of microglia in the development of the vasculature in the central nervous system. Vasc. Cell 2013, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Verney, C.; Monier, A.; Fallet-Bianco, C.; Gressens, P. Early microglial colonization of the human forebrain and possible involvement in periventricular white-matter injury of preterm infants. J. Anat. 2010, 217, 436–448. [Google Scholar] [CrossRef]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Alliot, F.; Godin, I.; Pessac, B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res. Dev. Brain Res. 1999, 117, 145–152. [Google Scholar] [CrossRef]
- Bardehle, S.; Kruger, M.; Buggenthin, F.; Schwausch, J.; Ninkovic, J.; Clevers, H.; Snippert, H.J.; Theis, F.J.; Meyer-Luehmann, M.; Bechmann, I.; et al. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat. Neurosci. 2013, 16, 580–586. [Google Scholar] [CrossRef]
- De, S.; Van Deren, D.; Peden, E.; Hockin, M.; Boulet, A.; Titen, S.; Capecchi, M.R. Two distinct ontogenies confer heterogeneity to mouse brain microglia. Development 2018, 145. [Google Scholar] [CrossRef]
- Antonson, A.M.; Lawson, M.A.; Caputo, M.P.; Matt, S.M.; Leyshon, B.J.; Johnson, R.W. Maternal viral infection causes global alterations in porcine fetal microglia. Proc. Natl. Acad. Sci. USA 2019, 116, 20190–20200. [Google Scholar] [CrossRef]
- Cunningham, C.L.; Martinez-Cerdeno, V.; Noctor, S.C. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J. Neurosci. 2013, 33, 4216–4233. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Yeh, H.; Ikezu, T. Transcriptional and Epigenetic Regulation of Microglia in Health and Disease. Trends Mol. Med. 2019, 25, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.D.; Sautkulis, L.N.; Danielson, P.E.; Cooper, J.; Hasel, K.W.; Hilbush, B.S.; Sutcliffe, J.G.; Carson, M.J. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 2002, 83, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- De Haas, A.H.; Boddeke, H.W.; Biber, K. Region-specific expression of immunoregulatory proteins on microglia in the healthy CNS. Glia 2008, 56, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.M.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P.; et al. An environment-dependent transcriptional network specifies human microglia identity. Science 2017, 356. [Google Scholar] [CrossRef] [PubMed]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Olah, M.; Biber, K.; Vinet, J.; Boddeke, H.W. Microglia phenotype diversity. CNS Neurol. Disord. Drug Targets 2011, 10, 108–118. [Google Scholar] [CrossRef]
- Stowell, R.D.; Wong, E.L.; Batchelor, H.N.; Mendes, M.S.; Lamantia, C.E.; Whitelaw, B.S.; Majewska, A.K. Cerebellar microglia are dynamically unique and survey Purkinje neurons in vivo. Dev. Neurobiol. 2018, 78, 627–644. [Google Scholar] [CrossRef]
- Savchenko, V.L.; McKanna, J.A.; Nikonenko, I.R.; Skibo, G.G. Microglia and astrocytes in the adult rat brain: Comparative immunocytochemical analysis demonstrates the efficacy of lipocortin 1 immunoreactivity. Neuroscience 2000, 96, 195–203. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Sholar, P.W.; Bilbo, S.D. Sex differences in microglial colonization of the developing rat brain. J. Neurochem. 2012, 120, 948–963. [Google Scholar] [CrossRef]
- Bordt, E.A.; Ceasrine, A.M.; Bilbo, S.D. Microglia and sexual differentiation of the developing brain: A focus on ontogeny and intrinsic factors. Glia 2020, 68, 1085–1099. [Google Scholar] [CrossRef]
- Bodhankar, S.; Lapato, A.; Chen, Y.; Vandenbark, A.A.; Saugstad, J.A.; Offner, H. Role for microglia in sex differences after ischemic stroke: Importance of M2. Metab. Brain Dis. 2015, 30, 1515–1529. [Google Scholar] [CrossRef] [PubMed]
- Kodama, L.; Gan, L. Do Microglial Sex Differences Contribute to Sex Differences in Neurodegenerative Diseases? Trends Mol. Med. 2019, 25, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Favrais, G.; van de Looij, Y.; Fleiss, B.; Ramanantsoa, N.; Bonnin, P.; Stoltenburg-Didinger, G.; Lacaud, A.; Saliba, E.; Dammann, O.; Gallego, J.; et al. Systemic inflammation disrupts the developmental program of white matter. Ann. Neurol. 2011, 70, 550–565. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Gelosa, P.; Castiglioni, L.; Cimino, M.; Rizzi, N.; Pepe, G.; Lolli, F.; Marcello, E.; Sironi, L.; Vegeto, E.; et al. Sex-Specific Features of Microglia from Adult Mice. Cell Rep. 2018, 23, 3501–3511. [Google Scholar] [CrossRef] [PubMed]
- Hanamsagar, R.; Alter, M.D.; Block, C.S.; Sullivan, H.; Bolton, J.L.; Bilbo, S.D. Generation of a microglial developmental index in mice and in humans reveals a sex difference in maturation and immune reactivity. Glia 2018, 66, 460. [Google Scholar] [CrossRef]
- Geirsdottir, L.; David, E.; Keren-Shaul, H.; Weiner, A.; Bohlen, S.C.; Neuber, J.; Balic, A.; Giladi, A.; Sheban, F.; Dutertre, C.A.; et al. Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell 2020, 181, 746. [Google Scholar] [CrossRef]
- Bendix, I.; Miller, S.L.; Winterhager, E. Editorial: Causes and Consequences of Intrauterine Growth Restriction. Front. Endocrinol. 2020, 11, 205. [Google Scholar] [CrossRef]
- Lai, J.C.Y.; Rocha-Ferreira, E.; Ek, C.J.; Wang, X.; Hagberg, H.; Mallard, C. Immune responses in perinatal brain injury. Brain Behav. Immun. 2017, 63, 210–223. [Google Scholar] [CrossRef]
- O’Shea, T.M.; Allred, E.N.; Dammann, O.; Hirtz, D.; Kuban, K.C.; Paneth, N.; Leviton, A.; Investigators, E.s. The ELGAN study of the brain and related disorders in extremely low gestational age newborns. Early Hum. Dev. 2009, 85, 719–725. [Google Scholar] [CrossRef]
- Dammann, O.; Leviton, A. Intermittent or sustained systemic inflammation and the preterm brain. Pediatr. Res. 2014, 75, 376–380. [Google Scholar] [CrossRef]
- Wixey, J.A.; Chand, K.K.; Colditz, P.B.; Bjorkman, S.T. Review: Neuroinflammation in intrauterine growth restriction. Placenta 2017, 54, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, H.; Mallard, C.; Ferriero, D.M.; Vannucci, S.J.; Levison, S.W.; Vexler, Z.S.; Gressens, P. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 2015, 11, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Abu Jawdeh, E.G.; Huang, H.; Westgate, P.M.; Patwardhan, A.; Bada, H.; Bauer, J.A.; Giannone, P. Intermittent Hypoxemia in Preterm Infants: A Potential Proinflammatory Process. Am. J. Perinatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Martinello, K.A.; Meehan, C.; Avdic-Belltheus, A.; Lingam, I.; Ragab, S.; Hristova, M.; Tann, C.J.; Peebles, D.; Hagberg, H.; Wolfs, T.G.A.M.; et al. Acute LPS sensitization and continuous infusion exacerbates hypoxic brain injury in a piglet model of neonatal encephalopathy. Sci. Rep. 2019, 9, 10184–10117. [Google Scholar] [CrossRef]
- Adén, U.; Favrais, G.; Plaisant, F.; Winerdal, M.; Felderhoff-Mueser, U.; Lampa, J.; Lelièvre, V.; Gressens, P. Systemic inflammation sensitizes the neonatal brain to excitotoxicity through a pro-/anti-inflammatory imbalance: Key role of TNFalpha pathway and protection by etanercept. Brain Behav. Immun. 2010, 24, 747. [Google Scholar] [CrossRef] [PubMed]
- Favrais, G.; Schwendimann, L.; Gressens, P.; Lelièvre, V. Cyclooxygenase-2 mediates the sensitizing effects of systemic IL-1-beta on excitotoxic brain lesions in newborn mice. Neurobiol. Dis. 2007, 25, 496–505. [Google Scholar] [CrossRef]
- Degos, V.; Peineau, S.; Nijboer, C.; Kaindl, A.M.; Sigaut, S.; Favrais, G.; Plaisant, F.; Teissier, N.; Gouadon, E.; Lombet, A.; et al. G protein-coupled receptor kinase 2 and group I metabotropic glutamate receptors mediate inflammation-induced sensitization to excitotoxic neurodegeneration. Ann. Neurol. 2013, 73, 667–678. [Google Scholar] [CrossRef]
- Azzopardi, D.; Strohm, B.; Edwards, A.D.; Halliday, H.; Juszczak, E.; Levene, M.; Thoresen, M.; Whitelaw, A.; Brocklehurst, P.; Steering, G.; et al. Treatment of asphyxiated newborns with moderate hypothermia in routine clinical practice: How cooling is managed in the UK outside a clinical trial. Arch. Dis. Child. Fetal Neonatal. Ed. 2009, 94, F260–F264. [Google Scholar] [CrossRef]
- Falck, M.; Osredkar, D.; Wood, T.R.; Maes, E.; Flatebo, T.; Sabir, H.; Thoresen, M. Neonatal Systemic Inflammation Induces Inflammatory Reactions and Brain Apoptosis in a Pathogen-Specific Manner. Neonatology 2018, 113, 212–220. [Google Scholar] [CrossRef]
- Du, X.; Fleiss, B.; Li, H.; D’Angelo, B.; Sun, Y.; Zhu, C.; Hagberg, H.; Levy, O.; Mallard, C.; Wang, X. Systemic stimulation of TLR2 impairs neonatal mouse brain development. PLoS ONE 2011, 6, e19583. [Google Scholar] [CrossRef]
- Korzeniewski, S.J.; Romero, R.; Cortez, J.; Pappas, A.; Schwartz, A.G.; Kim, C.J.; Kim, J.S.; Kim, Y.M.; Yoon, B.H.; Chaiworapongsa, T.; et al. A “multi-hit” model of neonatal white matter injury: Cumulative contributions of chronic placental inflammation, acute fetal inflammation and postnatal inflammatory events. J. Perinat. Med. 2014, 42, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.R.; Liddelow, S.A.; Dziegielewska, K.M. Barrier mechanisms in the developing brain. Front. Pharmacol. 2012, 3, 46. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lopez, D.; Faustino, J.; Daneman, R.; Zhou, L.; Lee, S.Y.; Derugin, N.; Wendland, M.F.; Vexler, Z.S. Blood-brain barrier permeability is increased after acute adult stroke but not neonatal stroke in the rat. J. Neurosci. 2012, 32, 9588–9600. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lopez, D.; Faustino, J.; Klibanov, A.L.; Derugin, N.; Blanchard, E.; Simon, F.; Leib, S.L.; Vexler, Z.S. Microglial Cells Prevent Hemorrhage in Neonatal Focal Arterial Stroke. J. Neurosci. 2016, 36, 2881–2893. [Google Scholar] [CrossRef] [PubMed]
- Hill-Yardin, E.L.; McKeown, S.J.; Novarino, G.; Grabrucker, A.M. Extracerebral Dysfunction in Animal Models of Autism Spectrum Disorder. Adv. Anat. Embryol. Cell Biol. 2017, 224, 159–187. [Google Scholar] [CrossRef] [PubMed]
- Prince, A.L.; Ma, J.; Kannan, P.S.; Alvarez, M.; Gisslen, T.; Harris, R.A.; Sweeney, E.L.; Knox, C.L.; Lambers, D.S.; Jobe, A.H.; et al. The placental membrane microbiome is altered among subjects with spontaneous preterm birth with and without chorioamnionitis. Am. J. Obstet. Gynecol. 2016, 214, 627.e1–627.e16. [Google Scholar] [CrossRef] [PubMed]
- Underwood, M.A.; Sohn, K. The Microbiota of the Extremely Preterm Infant. Clin. Perinatol. 2017, 44, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Hosie, S.; Ellis, M.; Swaminathan, M.; Ramalhosa, F.; Seger, G.O.; Balasuriya, G.K.; Gillberg, C.; Rastam, M.; Churilov, L.; McKeown, S.J.; et al. Gastrointestinal dysfunction in patients and mice expressing the autism-associated R451C mutation in neuroligin-3. Autism Res. 2019, 12, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Mallard, C.; Tremblay, M.E.; Vexler, Z.S. Microglia and Neonatal Brain Injury. Neuroscience 2019, 405, 68–76. [Google Scholar] [CrossRef]
- Van Steenwinckel, J.; Schang, A.L.; Krishnan, M.L.; Degos, V.; Delahaye-Duriez, A.; Bokobza, C.; Csaba, Z.; Verdonk, F.; Montane, A.; Sigaut, S.; et al. Decreased microglial Wnt/beta-catenin signalling drives microglial pro-inflammatory activation in the developing brain. Brain 2019, 142, 3806–3833. [Google Scholar] [CrossRef] [PubMed]
- Verney, C.; Pogledic, I.; Biran, V.; Adle-Biassette, H.; Fallet-Bianco, C.; Gressens, P. Microglial reaction in axonal crossroads is a hallmark of noncystic periventricular white matter injury in very preterm infants. J. Neuropathol. Exp. Neurol. 2012, 71, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Zinni, M.; Colella, M.; Batista Novais, A.R.; Baud, O.; Mairesse, J. Modulating the Oxytocin System During the Perinatal Period: A New Strategy for Neuroprotection of the Immature Brain? Front. Neurol. 2018, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, M.L.; Van Steenwinckel, J.; Schang, A.L.; Yan, J.; Arnadottir, J.; Le Charpentier, T.; Csaba, Z.; Dournaud, P.; Cipriani, S.; Auvynet, C.; et al. Integrative genomics of microglia implicates DLG4 (PSD95) in the white matter development of preterm infants. Nat. Commun. 2017, 8, 428. [Google Scholar] [CrossRef] [PubMed]
- Denker, S.P.; Ji, S.; Dingman, A.; Lee, S.Y.; Derugin, N.; Wendland, M.F.; Vexler, Z.S. Macrophages are comprised of resident brain microglia not infiltrating peripheral monocytes acutely after neonatal stroke. J. Neurochem. 2007, 100, 893–904. [Google Scholar] [CrossRef]
- Bona, E.; Andersson, A.L.; Blomgren, K.; Gilland, E.; Puka-Sundvall, M.; Gustafson, K.; Hagberg, H. Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatr. Res. 1999, 45, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Albertsson, A.M.; Zhang, X.; Vontell, R.; Bi, D.; Bronson, R.T.; Supramaniam, V.; Baburamani, A.A.; Hua, S.; Nazmi, A.; Cardell, S.; et al. gammadelta T Cells Contribute to Injury in the Developing Brain. Am. J. Pathol. 2018, 188, 757–767. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–1746. [Google Scholar] [CrossRef]
- Faraco, G.; Park, L.; Anrather, J.; Iadecola, C. Brain perivascular macrophages: Characterization and functional roles in health and disease. J. Mol. Med. (Berl.) 2017, 95, 1143–1152. [Google Scholar] [CrossRef]
- Hawkes, C.A.; Gentleman, S.M.; Nicoll, J.A.; Carare, R.O. Prenatal high-fat diet alters the cerebrovasculature and clearance of beta-amyloid in adult offspring. J. Pathol. 2015, 235, 619–631. [Google Scholar] [CrossRef] [PubMed]
- McNamara, N.B.; Miron, V.E. Microglia in developing white matter and perinatal brain injury. Neurosci. Lett. 2020, 714, 134539. [Google Scholar] [CrossRef] [PubMed]
- Faustino, J.V.; Wang, X.; Johnson, C.E.; Klibanov, A.; Derugin, N.; Wendland, M.F.; Vexler, Z.S. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J. Neurosci. 2011, 31, 12992–13001. [Google Scholar] [CrossRef] [PubMed]
- Chhor, V.; Moretti, R.; Le Charpentier, T.; Sigaut, S.; Lebon, S.; Schwendimann, L.; Ore, M.V.; Zuiani, C.; Milan, V.; Josserand, J.; et al. Role of microglia in a mouse model of paediatric traumatic brain injury. Brain. Behav. Immun. 2017, 63, 197–209. [Google Scholar] [CrossRef]
- Tsuji, M.; Wilson, M.A.; Lange, M.S.; Johnston, M.V. Minocycline worsens hypoxic-ischemic brain injury in a neonatal mouse model. Exp. Neurol. 2004, 189, 58–65. [Google Scholar] [CrossRef]
- Tsuji, S.; Di Martino, E.; Mukai, T.; Tsuji, S.; Murakami, T.; Harris, R.A.; Blomgren, K.; Aden, U. Aggravated brain injury after neonatal hypoxic ischemia in microglia-depleted mice. J. Neuroinflamm. 2020, 17, 111. [Google Scholar] [CrossRef]
- Gordon, P.H.; Moore, D.H.; Miller, R.G.; Florence, J.M.; Verheijde, J.L.; Doorish, C.; Hilton, J.F.; Spitalny, G.M.; MacArthur, R.B.; Mitsumoto, H.; et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: A phase III randomised trial. Lancet Neurol. 2007, 6, 1045–1053. [Google Scholar] [CrossRef]
- Hanlon, L.A.; Huh, J.W.; Raghupathi, R. Minocycline Transiently Reduces Microglia/Macrophage Activation but Exacerbates Cognitive Deficits Following Repetitive Traumatic Brain Injury in the Neonatal Rat. J. Neuropathol. Exp. Neurol. 2016, 75, 214–226. [Google Scholar] [CrossRef]
- Szalay, G.; Martinecz, B.; Lenart, N.; Kornyei, Z.; Orsolits, B.; Judak, L.; Csaszar, E.; Fekete, R.; West, B.L.; Katona, G.; et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun. 2016, 7, 11499. [Google Scholar] [CrossRef]
- Fumagalli, M.; Lombardi, M.; Gressens, P.; Verderio, C. How to reprogram microglia toward beneficial functions. Glia 2018, 66, 2531–2549. [Google Scholar] [CrossRef]
- Boardman, J.P.; Walley, A.; Ball, G.; Takousis, P.; Krishnan, M.L.; Hughes-Carre, L.; Aljabar, P.; Serag, A.; King, C.; Merchant, N.; et al. Common genetic variants and risk of brain injury after preterm birth. Pediatrics 2014, 133, e1655–e1663. [Google Scholar] [CrossRef]
- Krishnan, M.L.; Wang, Z.; Aljabar, P.; Ball, G.; Mirza, G.; Saxena, A.; Counsell, S.J.; Hajnal, J.V.; Montana, G.; Edwards, A.D. Machine learning shows association between genetic variability in PPARG and cerebral connectivity in preterm infants. Proc. Natl. Acad. Sci. USA 2017, 114, 13744–13749. [Google Scholar] [CrossRef] [PubMed]
- Mairesse, J.; Zinni, M.; Pansiot, J.; Hassan-Abdi, R.; Demene, C.; Colella, M.; Charriaut-Marlangue, C.; Rideau Batista Novais, A.; Tanter, M.; Maccari, S.; et al. Oxytocin receptor agonist reduces perinatal brain damage by targeting microglia. Glia 2019, 67, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Miller, S.P. Brain injury in premature neonates: A primary cerebral dysmaturation disorder? Ann. Neurol. 2014, 75, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Kaindl, A.M.; Favrais, G.; Gressens, P. Molecular mechanisms involved in injury to the preterm brain. J. Child Neurol. 2009, 24, 1112–1118. [Google Scholar] [CrossRef]
- Frost, P.S.; Barros-Aragao, F.; da Silva, R.T.; Venancio, A.; Matias, I.; Lyra, E.S.N.M.; Kincheski, G.C.; Pimentel-Coelho, P.M.; De Felice, F.G.; Gomes, F.C.A.; et al. Neonatal infection leads to increased susceptibility to Abeta oligomer-induced brain inflammation, synapse loss and cognitive impairment in mice. Cell Death Dis. 2019, 10, 323. [Google Scholar] [CrossRef] [PubMed]
- Altamentova, S.; Rumajogee, P.; Hong, J.; Beldick, S.R.; Park, S.J.; Yee, A.; Fehlings, M.G. Methylprednisolone Reduces Persistent Post-ischemic Inflammation in a Rat Hypoxia-Ischemia Model of Perinatal Stroke. Transl. Stroke Res. 2020, 11, 1117–1136. [Google Scholar] [CrossRef]
- Cai, Z.; Fan, L.W.; Kaizaki, A.; Tien, L.T.; Ma, T.; Pang, Y.; Lin, S.; Lin, R.C.; Simpson, K.L. Neonatal systemic exposure to lipopolysaccharide enhances susceptibility of nigrostriatal dopaminergic neurons to rotenone neurotoxicity in later life. Dev. Neurosci. 2013, 35, 155–171. [Google Scholar] [CrossRef]
- Wang, X.; Hagberg, H.; Nie, C.; Zhu, C.; Ikeda, T.; Mallard, C. Dual role of intrauterine immune challenge on neonatal and adult brain vulnerability to hypoxia-ischemia. J. Neuropathol. Exp. Neurol. 2007, 66, 552–561. [Google Scholar] [CrossRef]
- Mattei, D.; Ivanov, A.; Ferrai, C.; Jordan, P.; Guneykaya, D.; Buonfiglioli, A.; Schaafsma, W.; Przanowski, P.; Deuther-Conrad, W.; Brust, P.; et al. Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl. Psychiatry 2017, 7, e1120. [Google Scholar] [CrossRef]
- Xie, C.; Zhou, K.; Wang, X.; Blomgren, K.; Zhu, C. Therapeutic benefits of delayed lithium administration in the neonatal rat after cerebral hypoxia-ischemia. PLoS ONE 2014, 9, e107192. [Google Scholar] [CrossRef]
- Ramlackhansingh, A.F.; Brooks, D.J.; Greenwood, R.J.; Bose, S.K.; Turkheimer, F.E.; Kinnunen, K.M.; Gentleman, S.; Heckemann, R.A.; Gunanayagam, K.; Gelosa, G.; et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann. Neurol. 2011, 70, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Dammann, O. Persistent neuro-inflammation in cerebral palsy: A therapeutic window of opportunity? Acta Paediatr. 2007, 96, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Dammann, O. Paediatric neurology: The many faces of development. Lancet Neurol. 2007, 6, 12–14. [Google Scholar] [CrossRef]
- Desplats, P.; Gutierrez, A.M.; Antonelli, M.C.; Frasch, M.G. Microglial memory of early life stress and inflammation: Susceptibility to neurodegeneration in adulthood. Neurosci. Biobehav. Rev. 2020, 117, 232–242. [Google Scholar] [CrossRef]
- Squarzoni, P.; Oller, G.; Hoeffel, G.; Pont-Lezica, L.; Rostaing, P.; Low, D.; Bessis, A.; Ginhoux, F.; Garel, S. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014, 8, 1271–1279. [Google Scholar] [CrossRef]
- Lizen, B.; Claus, M.; Jeannotte, L.; Rijli, F.M.; Gofflot, F. Perinatal induction of Cre recombination with tamoxifen. Transgenic Res. 2015, 24, 1065–1077. [Google Scholar] [CrossRef]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef]
- Van Tilborg, E.; Heijnen, C.J.; Benders, M.J.; van Bel, F.; Fleiss, B.; Gressens, P.; Nijboer, C.H. Impaired oligodendrocyte maturation in preterm infants: Potential therapeutic targets. Prog. Neurobiol. 2016, 136, 28–49. [Google Scholar] [CrossRef]
- Tahraoui, S.L.; Marret, S.; Bodenant, C.; Leroux, P.; Dommergues, M.A.; Evrard, P.; Gressens, P. Central role of microglia in neonatal excitotoxic lesions of the murine periventricular white matter. Brain Pathol. 2001, 11, 56–71. [Google Scholar] [CrossRef]
- Wlodarczyk, A.; Holtman, I.R.; Krueger, M.; Yogev, N.; Bruttger, J.; Khorooshi, R.; Benmamar-Badel, A.; de Boer-Bergsma, J.J.; Martin, N.A.; Karram, K.; et al. A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. 2017, 36, 3292–3308. [Google Scholar] [CrossRef] [PubMed]
- Jiao, M.; Li, X.; Chen, L.; Wang, X.; Yuan, B.; Liu, T.; Dong, Q.; Mei, H.; Yin, H. Neuroprotective effect of astrocyte-derived IL-33 in neonatal hypoxic-ischemic brain injury. J. Neuroinflamm. 2020, 17, 251. [Google Scholar] [CrossRef] [PubMed]
- Shiow, L.R.; Favrais, G.; Schirmer, L.; Schang, A.L.; Cipriani, S.; Andres, C.; Wright, J.N.; Nobuta, H.; Fleiss, B.; Gressens, P.; et al. Reactive astrocyte COX2-PGE2 production inhibits oligodendrocyte maturation in neonatal white matter injury. Glia 2017, 65, 2024–2037. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Truttmann, A.C.; Ginet, V.; Puyal, J. Current Evidence on Cell Death in Preterm Brain Injury in Human and Preclinical Models. Front. Cell Dev. Biol. 2020, 8, 27. [Google Scholar] [CrossRef]
- Renolleau, S.; Fau, S.; Goyenvalle, C.; Joly, L.M.; Chauvier, D.; Jacotot, E.; Mariani, J.; Charriaut-Marlangue, C. Specific caspase inhibitor Q-VD-OPh prevents neonatal stroke in P7 rat: A role for gender. J. Neurochem. 2007, 100, 1062–1071. [Google Scholar] [CrossRef]
- Askalan, R.; Gabarin, N.; Armstrong, E.A.; Fang Liu, Y.; Couchman, D.; Yager, J.Y. Mechanisms of neurodegeneration after severe hypoxic-ischemic injury in the neonatal rat brain. Brain Res. 2015, 1629, 94–103. [Google Scholar] [CrossRef]
- Thornton, C.; Leaw, B.; Mallard, C.; Nair, S.; Jinnai, M.; Hagberg, H. Cell Death in the Developing Brain after Hypoxia-Ischemia. Front. Cell. Neurosci. 2017, 11, 248. [Google Scholar] [CrossRef]
- Wu, Y.; Song, J.; Wang, Y.; Wang, X.; Culmsee, C.; Zhu, C. The Potential Role of Ferroptosis in Neonatal Brain Injury. Front. Neurosci. 2019, 13, 115. [Google Scholar] [CrossRef]
- Alberi, L.; Chi, Z.; Kadam, S.D.; Mulholland, J.D.; Dawson, V.L.; Gaiano, N.; Comi, A.M. Neonatal stroke in mice causes long-term changes in neuronal Notch-2 expression that may contribute to prolonged injury. Stroke 2010, 41, S64–S71. [Google Scholar] [CrossRef]
- Leviton, A.; Gressens, P. Neuronal damage accompanies perinatal white-matter damage. Trends Neurosci. 2007, 30, 473–478. [Google Scholar] [CrossRef]
- Dommergues, M.A.; Plaisant, F.; Verney, C.; Gressens, P. Early microglial activation following neonatal excitotoxic brain damage in mice: A potential target for neuroprotection. Neuroscience 2003, 121, 619–628. [Google Scholar] [CrossRef]
- Frade, J.M.; Barde, Y.A. Microglia-derived nerve growth factor causes cell death in the developing retina. Neuron 1998, 20, 35–41. [Google Scholar] [CrossRef]
- Lepiemme, F.; Silva, G.C.; Nguyen, L. Chapter 16—Mechanisms of tangential migration of interneurons in the developing forebrain. In Cellular Migration and Formation of Axons and Dendrites, 2nd ed.; Rubenstein, J., Rakic, P., Chen, B., Kwan, K.Y., Kolodkin, A., Anton, E., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 345–363. [Google Scholar] [CrossRef]
- Panda, S.; Dohare, P.; Jain, S.; Parikh, N.; Singla, P.; Mehdizadeh, R.; Klebe, D.W.; Kleinman, G.M.; Cheng, B.; Ballabh, P. Estrogen Treatment Reverses Prematurity-Induced Disruption in Cortical Interneuron Population. J. Neurosci. 2018, 38, 7378–7391. [Google Scholar] [CrossRef] [PubMed]
- Ardalan, M.; Svedin, P.; Baburamani, A.A.; Supramaniam, V.G.; Ek, J.; Hagberg, H.; Mallard, C. Dysmaturation of Somatostatin Interneurons Following Umbilical Cord Occlusion in Preterm Fetal Sheep. Front. Physiol. 2019, 10, 563. [Google Scholar] [CrossRef]
- Vaes, J.E.G.; Kosmeijer, C.M.; Kaal, M.; van Vliet, R.; Brandt, M.J.V.; Benders, M.; Nijboer, C.H. Regenerative Therapies to Restore Interneuron Disturbances in Experimental Models of Encephalopathy of Prematurity. Int. J. Mol. Sci. 2020, 22. [Google Scholar] [CrossRef]
- Thion, M.S.; Garel, S. On place and time: Microglia in embryonic and perinatal brain development. Curr. Opin. Neurobiol. 2017, 47, 121–130. [Google Scholar] [CrossRef]
- Park, G.H.; Noh, H.; Shao, Z.; Ni, P.; Qin, Y.; Liu, D.; Beaudreault, C.P.; Park, J.S.; Abani, C.P.; Park, J.M.; et al. Activated microglia cause metabolic disruptions in developmental cortical interneurons that persist in interneurons from individuals with schizophrenia. Nat. Neurosci. 2020, 23, 1352–1364. [Google Scholar] [CrossRef]
- Ball, G.; Aljabar, P.; Arichi, T.; Tusor, N.; Cox, D.; Merchant, N.; Nongena, P.; Hajnal, J.V.; Edwards, A.D.; Counsell, S.J. Machine-learning to characterise neonatal functional connectivity in the preterm brain. Neuroimage 2016, 124, 267–275. [Google Scholar] [CrossRef]
- Smyser, C.D.; Wheelock, M.D.; Limbrick, D.D., Jr.; Neil, J.J. Neonatal brain injury and aberrant connectivity. Neuroimage 2019, 185, 609–623. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Rosenzweig, J.M.; Mishra, M.K.; Alshehri, W.; Brancusi, F.; McLane, M.; Almalki, A.; Bahabry, R.; Arif, H.; Rozzah, R.; et al. Maternal dendrimer-based therapy for inflammation-induced preterm birth and perinatal brain injury. Sci. Rep. 2017, 7, 6106. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Dai, H.; Navath, R.S.; Balakrishnan, B.; Jyoti, A.; Janisse, J.; Romero, R.; Kannan, R.M. Dendrimer-based postnatal therapy for neuroinflammation and cerebral palsy in a rabbit model. Sci. Transl. Med. 2012, 4, 130ra146. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Ferrari, R.; De Paola, M.; Rossi, F.; Mariani, A.; Caron, I.; Sammali, E.; Peviani, M.; Dell’Oro, V.; Colombo, C.; et al. Polymeric nanoparticle system to target activated microglia/macrophages in spinal cord injury. J. Control. Release 2014, 174, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Rossi, F.; Ferrari, R.; Mariani, A.; De Paola, M.; Caron, I.; Fiordaliso, F.; Bisighini, C.; Sammali, E.; Colombo, C.; et al. Selective nanovector mediated treatment of activated proinflammatory microglia/macrophages in spinal cord injury. ACS Nano 2013, 7, 9881–9895. [Google Scholar] [CrossRef]
- Das, A.; Chai, J.C.; Kim, S.H.; Lee, Y.S.; Park, K.S.; Jung, K.H.; Chai, Y.G. Transcriptome sequencing of microglial cells stimulated with TLR3 and TLR4 ligands. BMC Genom. 2015, 16, 517. [Google Scholar] [CrossRef]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Ore, M.V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Savman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef]
- Van der Poel, M.; Ulas, T.; Mizee, M.R.; Hsiao, C.C.; Miedema, S.S.M.; Adelia; Schuurman, K.G.; Helder, B.; Tas, S.W.; Schultze, J.L.; et al. Transcriptional profiling of human microglia reveals grey-white matter heterogeneity and multiple sclerosis-associated changes. Nat. Commun. 2019, 10, 1139. [Google Scholar] [CrossRef]
- Hart, A.D.; Wyttenbach, A.; Perry, V.H.; Teeling, J.L. Age related changes in microglial phenotype vary between CNS regions: Grey versus white matter differences. Brain. Behav. Immun. 2012, 26, 754–765. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271 e256. [Google Scholar] [CrossRef]
- Nair, S.; Sobotka, K.S.; Joshi, P.; Gressens, P.; Fleiss, B.; Thornton, C.; Mallard, C.; Hagberg, H. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia 2019. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; McBean, G.; Cindric, M.; Egea, J.; López, M.G.; Rada, P.; Zarkovic, N.; Cuadrado, A. Redox Control of Microglial Function: Molecular Mechanisms and Functional Significance. Antioxid. Redox Signal. 2014, 21, 1766–1801. [Google Scholar] [CrossRef] [PubMed]
- Cho, S. The Class B Scavenger Receptor CD36 Mediates Free Radical Production and Tissue Injury in Cerebral Ischemia. J. Neurosci. 2005, 25, 2504–2512. [Google Scholar] [CrossRef]
- Li, F.; Faustino, J.; Woo, M.S.; Derugin, N.; Vexler, Z.S. Lack of the scavenger receptor CD36 alters microglial phenotypes after neonatal stroke. J. Neurochem. 2015, 135, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Fink, A.; Doblhammer, G. Effect of pioglitazone medication on the incidence of dementia. Ann. Neurol. 2015, 78, 284–294. [Google Scholar] [CrossRef]
- Yamanaka, M.; Ishikawa, T.; Griep, A.; Axt, D.; Kummer, M.P.; Heneka, M.T. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J. Neurosci. 2012, 32, 17321–17331. [Google Scholar] [CrossRef]
- Hardie, D.G. Sensing of energy and nutrients by AMP-activated protein kinase. Am. J. Clin. Nutr. 2011, 93, 891S–896S. [Google Scholar] [CrossRef]
- Song, X.M.; Yu, Q.; Dong, X.; Yang, H.O.; Zeng, K.W.; Li, J.; Tu, P.F. Aldose reductase inhibitors attenuate beta-amyloid-induced TNF-alpha production in microlgia via ROS-PKC-mediated NF-kappaB and MAPK pathways. Int. Immunopharmacol. 2017, 50, 30–37. [Google Scholar] [CrossRef]
- Zhang, Q.; Bian, G.; Chen, P.; Liu, L.; Yu, C.; Liu, F.; Xue, Q.; Chung, S.K.; Song, B.; Ju, G.; et al. Aldose Reductase Regulates Microglia/Macrophages Polarization Through the cAMP Response Element-Binding Protein After Spinal Cord Injury in Mice. Mol. Neurobiol. 2016, 53, 662–676. [Google Scholar] [CrossRef]
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenet. 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Madore, C.; Leyrolle, Q.; Morel, L.; Rossitto, M.; Greenhalgh, A.D.; Delpech, J.C.; Martinat, M.; Bosch-Bouju, C.; Bourel, J.; Rani, B.; et al. Essential omega-3 fatty acids tune microglial phagocytosis of synaptic elements in the mouse developing brain. Nat. Commun. 2020, 11, 6133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hu, X.; Yang, W.; Gao, Y.; Chen, J. Omega-3 polyunsaturated fatty acid supplementation confers long-term neuroprotection against neonatal hypoxic-ischemic brain injury through anti-inflammatory actions. Stroke 2010, 41, 2341–2347. [Google Scholar] [CrossRef] [PubMed]
- Tskitishvili, E.; Nisolle, M.; Munaut, C.; Pequeux, C.; Gerard, C.; Noel, A.; Foidart, J.M. Estetrol attenuates neonatal hypoxic-ischemic brain injury. Exp. Neurol. 2014, 261, 298–307. [Google Scholar] [CrossRef]
- Carty, M.L.; Wixey, J.A.; Reinebrant, H.E.; Gobe, G.; Colditz, P.B.; Buller, K.M. Ibuprofen inhibits neuroinflammation and attenuates white matter damage following hypoxia-ischemia in the immature rodent brain. Brain Res. 2011, 1402, 9–19. [Google Scholar] [CrossRef]
- Camara-Lemarroy, C.R.; Metz, L.; Meddings, J.B.; Sharkey, K.A.; Wee Yong, V. The intestinal barrier in multiple sclerosis: Implications for pathophysiology and therapeutics. Brain 2018, 141, 1900–1916. [Google Scholar] [CrossRef]
- Carlessi, A.S.; Borba, L.A.; Zugno, A.I.; Quevedo, J.; Reus, G.Z. Gut microbiota-brain axis in depression: The role of neuroinflammation. Eur. J. Neurosci. 2019. [Google Scholar] [CrossRef]
- Donega, V.; Nijboer, C.H.; Braccioli, L.; Slaper-Cortenbach, I.; Kavelaars, A.; van Bel, F.; Heijnen, C.J. Intranasal administration of human MSC for ischemic brain injury in the mouse: In vitro and in vivo neuroregenerative functions. PLoS ONE 2014, 9, e112339. [Google Scholar] [CrossRef]
- Shariati, A.; Nemati, R.; Sadeghipour, Y.; Yaghoubi, Y.; Baghbani, R.; Javidi, K.; Zamani, M.; Hassanzadeh, A. Mesenchymal stromal cells (MSCs) for neurodegenerative disease: A promising frontier. Eur. J. Cell Biol. 2020, 99, 151097. [Google Scholar] [CrossRef]
- Nair, S.; Rocha-Ferreira, E.; Fleiss, B.; Nijboer, C.H.; Gressens, P.; Mallard, C.; Hagberg, H. Neuroprotection offered by mesenchymal stem cells in perinatal brain injury: Role of mitochondria, inflammation and reactive oxygen species. J. Neurochem. 2020. [Google Scholar] [CrossRef]
- Gao, J.; Su, G.; Liu, J.; Zhang, J.; Zhou, J.; Liu, X.; Tian, Y.; Zhang, Z. Mechanisms of Inhibition of Excessive Microglial Activation by Melatonin. J. Mol. Neurosci. 2020, 70, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Liddell, J.R. Are Astrocytes the Predominant Cell Type for Activation of Nrf2 in Aging and Neurodegeneration? Antioxidants 2017, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Correa, F.; Mallard, C.; Nilsson, M.; Sandberg, M. Activated microglia decrease histone acetylation and Nrf2-inducible anti-oxidant defence in astrocytes: Restoring effects of inhibitors of HDACs, p38 MAPK and GSK3beta. Neurobiol. Dis. 2011, 44, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Guardia Clausi, M.; Levison, S.W. Delayed ALK5 inhibition improves functional recovery in neonatal brain injury. J. Cereb. Blood Flow Metab. 2017, 37, 787–800. [Google Scholar] [CrossRef]
- Kim, B.H.; Guardia Clausi, M.; Frondelli, M.; Nnah, I.C.; Saqcena, C.; Dobrowolski, R.; Levison, S.W. Age-Dependent Effects of ALK5 Inhibition and Mechanism of Neuroprotection in Neonatal Hypoxic-Ischemic Brain Injury. Dev. Neurosci. 2017, 39, 338–351. [Google Scholar] [CrossRef]
- Bennet, L.; Van Den Heuij, L.; Dean, J.M.; Drury, P.; Wassink, G.; Gunn, A.J. Neural plasticity and the Kennard Principle—Does it work for the preterm brain? Clin. Exp. Pharmacol. Physiol. 2013. [Google Scholar] [CrossRef]
- Anderson, V.; Spencer-Smith, M.; Wood, A. Do children really recover better? Neurobehavioural plasticity after early brain insult. Brain 2011, 134, 2197–2221. [Google Scholar] [CrossRef]
- Ginhoux, F.; Garel, S. The mysterious origins of microglia. Nat. Neurosci. 2018, 21, 897–899. [Google Scholar] [CrossRef]
- Colella, M.; Zinni, M.; Pansiot, J.; Cassanello, M.; Mairesse, J.; Ramenghi, L.; Baud, O. Modulation of Microglial Activation by Adenosine A2a Receptor in Animal Models of Perinatal Brain Injury. Front. Neurol. 2018, 9, 605. [Google Scholar] [CrossRef]
- Dean, J.M.; Wang, X.; Kaindl, A.M.; Gressens, P.; Fleiss, B.; Hagberg, H.; Mallard, C. Microglial MyD88 signaling regulates acute neuronal toxicity of LPS-stimulated microglia in vitro. Brain. Behav. Immun. 2010, 24, 776–783. [Google Scholar] [CrossRef]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef]
- Hellstrom Erkenstam, N.; Smith, P.L.; Fleiss, B.; Nair, S.; Svedin, P.; Wang, W.; Bostrom, M.; Gressens, P.; Hagberg, H.; Brown, K.L.; et al. Temporal Characterization of Microglia/Macrophage Phenotypes in a Mouse Model of Neonatal Hypoxic-Ischemic Brain Injury. Front. Cell. Neurosci. 2016, 10, 286. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.; Leger, P.L.; Besson, V.C.; Csaba, Z.; Pansiot, J.; Di Criscio, L.; Gentili, A.; Titomanlio, L.; Bonnin, P.; Baud, O.; et al. Sildenafil, a cyclic GMP phosphodiesterase inhibitor, induces microglial modulation after focal ischemia in the neonatal mouse brain. J. Neuroinflamm. 2016, 13, 95. [Google Scholar] [CrossRef] [PubMed]
- Supramaniam, V.; Vontell, R.; Srinivasan, L.; Wyatt-Ashmead, J.; Hagberg, H.; Rutherford, M. Microglia activation in the extremely preterm human brain. Pediatr. Res. 2013, 73, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Vontell, R.; Supramaniam, V.; Thornton, C.; Wyatt-Ashmead, J.; Mallard, C.; Gressens, P.; Rutherford, M.; Hagberg, H. Toll-like receptor 3 expression in glia and neurons alters in response to white matter injury in preterm infants. Dev. Neurosci. 2013, 35, 130–139. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PBIs | Key Features (by Severity) |
|---|---|
| EoFGR [19] | Reduced cortical grey matter volume Altered cortical gyrification Impaired myelination Reduced connectivity |
| EoP [22,23] | Diffuse white matter injury (including cerebellum) Punctate white matter cystic lesions Reduced grey matter microstructure Reduced connectivity (in particular thalamocortical connectivity) Intra-ventricular hemorrhages |
| NE [22] | Destructive lesions of cortical areas Destructive lesions of basal ganglia Impaired myelination of the posterior limb of the internal capsule (PLIC) |
| NS [20] | Destructive lesions of arterial or venous territories |
| Molecular Targets | Candidate Interventions |
|---|---|
| PPAR receptor | Pioglitazone, Rosiglitazone, Metformin [167,168] |
| AMP-related kinase (AMPK) | Metformin [169] |
| Aldose reductase | Sorbinil, Zopolrestat, Fidarestat, Tolrestat [170,171] |
| Sirtuin 1 | Resveratrol [172] |
| Wnt pathway | Wnt pathway agonists (via 3DNA nanoparticle delivery) [91] |
| MicroRNAs (miRs) | miR mimics and antago-miRs (via 3DNA nanoparticle delivery) [110] |
| Omega3-omega6 fatty acid balance | Enriched omega3 fatty acid nutrition [173,174] |
| Steroid receptors | Estetrol (E4) [175] |
| Cyclo-oxygenase-2 (COX)-2 | Nimesulide, Indomethacin [133,176] |
| Leukotriens | Montelukast |
| Microbiota and gut | Prebiotics, probiotics, faecal transplant [177,178] |
| Multiple targets | Mesenchymal stem cells [179,180,181], melatonin [182] |
| Redox balance, Nrf2 | GSH: GSSG, N-acetyl-cysteine, sulforaphane [183], p38 [184] |
| Transforming Growth Factor Beta | ALK5 [185,186] |
| Brain Disorders | Potential Similarities with PBIs |
|---|---|
| ASDs | Pro-inflammatory MG activation Myelin defects Impaired connectivity Impaired synaptogenesis Impaired microbiota Sex bias |
| MS | Pro-inflammatory MG activation Reactive astrocytes OPC maturation arrest Myelin defects |
| PD and AD | Pro-inflammatory MG activation Reactive astrocytes Protracted neuronal cell death Sensitization, systemic inflammation |
| TBI | Pro-inflammatory MG activation Impaired astrocyte functions Myelin defect Tertiary phase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fleiss, B.; Van Steenwinckel, J.; Bokobza, C.; K. Shearer, I.; Ross-Munro, E.; Gressens, P. Microglia-Mediated Neurodegeneration in Perinatal Brain Injuries. Biomolecules 2021, 11, 99. https://doi.org/10.3390/biom11010099
Fleiss B, Van Steenwinckel J, Bokobza C, K. Shearer I, Ross-Munro E, Gressens P. Microglia-Mediated Neurodegeneration in Perinatal Brain Injuries. Biomolecules. 2021; 11(1):99. https://doi.org/10.3390/biom11010099
Chicago/Turabian StyleFleiss, Bobbi, Juliette Van Steenwinckel, Cindy Bokobza, Isabelle K. Shearer, Emily Ross-Munro, and Pierre Gressens. 2021. "Microglia-Mediated Neurodegeneration in Perinatal Brain Injuries" Biomolecules 11, no. 1: 99. https://doi.org/10.3390/biom11010099
APA StyleFleiss, B., Van Steenwinckel, J., Bokobza, C., K. Shearer, I., Ross-Munro, E., & Gressens, P. (2021). Microglia-Mediated Neurodegeneration in Perinatal Brain Injuries. Biomolecules, 11(1), 99. https://doi.org/10.3390/biom11010099