Redox Signaling from Mitochondria: Signal Propagation and Its Targets



Abstract
1. Preface
2. Elevations of Mitochondrial Superoxide Formation
2.1. Mechanisms of Mitochondrial Superoxide Generation
2.2. The Interplay between ROS, Mitochondrial Anion Channels, and Mitochondrial Permeability Transition
2.3. The Interplay between ROS and Mitochondrial Ca2+ Uniporter and Mitochondrial Ca2+ Antiporters
2.4. ROS and Mitochondrial K+ Channels
2.5. Mitochondrial ROS and Voltage-Dependent Anion-Selective Channels
3. Redox Signal Spreading Out of Mitochondria
3.1. Hypothetical Redox Signaling by Superoxide Diffusion?
3.2. H2O2 Routes up to OMM
4. Redox Signal Spreading within the Cytosol
4.1. Diffusion of H2O2
4.2. Reactions of Thiol-Containing Proteins
4.3. Peroxiredoxin Family
4.4. Floodgate Model
4.5. Signaling via Redox Relay
4.6. Mitochondrial Peroxiredoxins
4.7. Glutathione Peroxidases
4.8. Amplification of Cytosolic ROS Production (Signaling) by Stimulation of NADPH Oxidases by Mitochondrial ROS
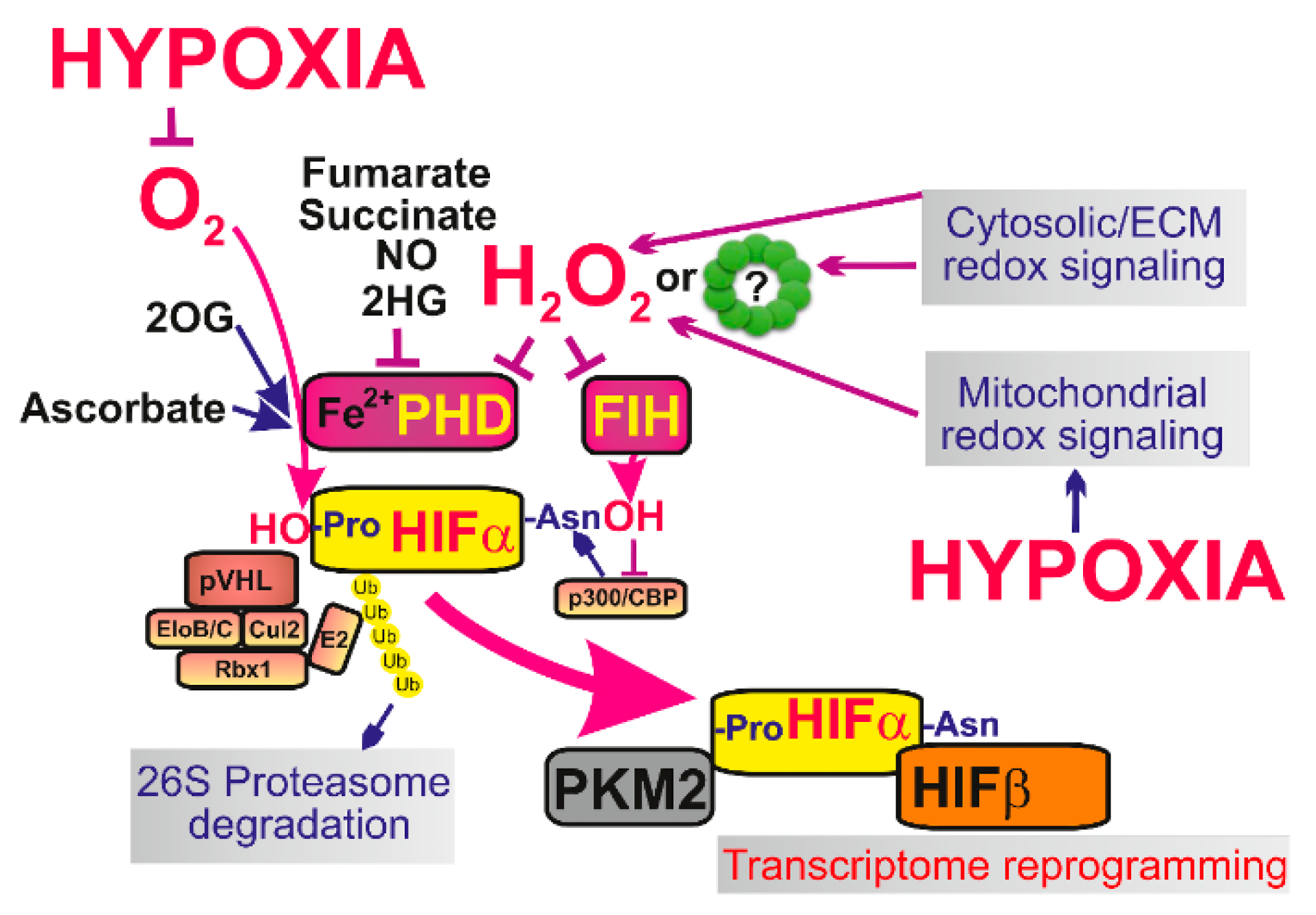
5. Mitochondrial Redox Signaling at Hypoxia
5.1. Hypoxia-Inducible Factor
5.2. Role of Mitochondrial Redox Signaling in Hypoxic Adaptation
5.3. Mechanism of Complex III Initiated Mitochondrial Redox Signaling in Hypoxic Adaptation
5.4. Mechanism of Complex I Initiated Mitochondrial Redox Signaling in Hypoxic Adaptation
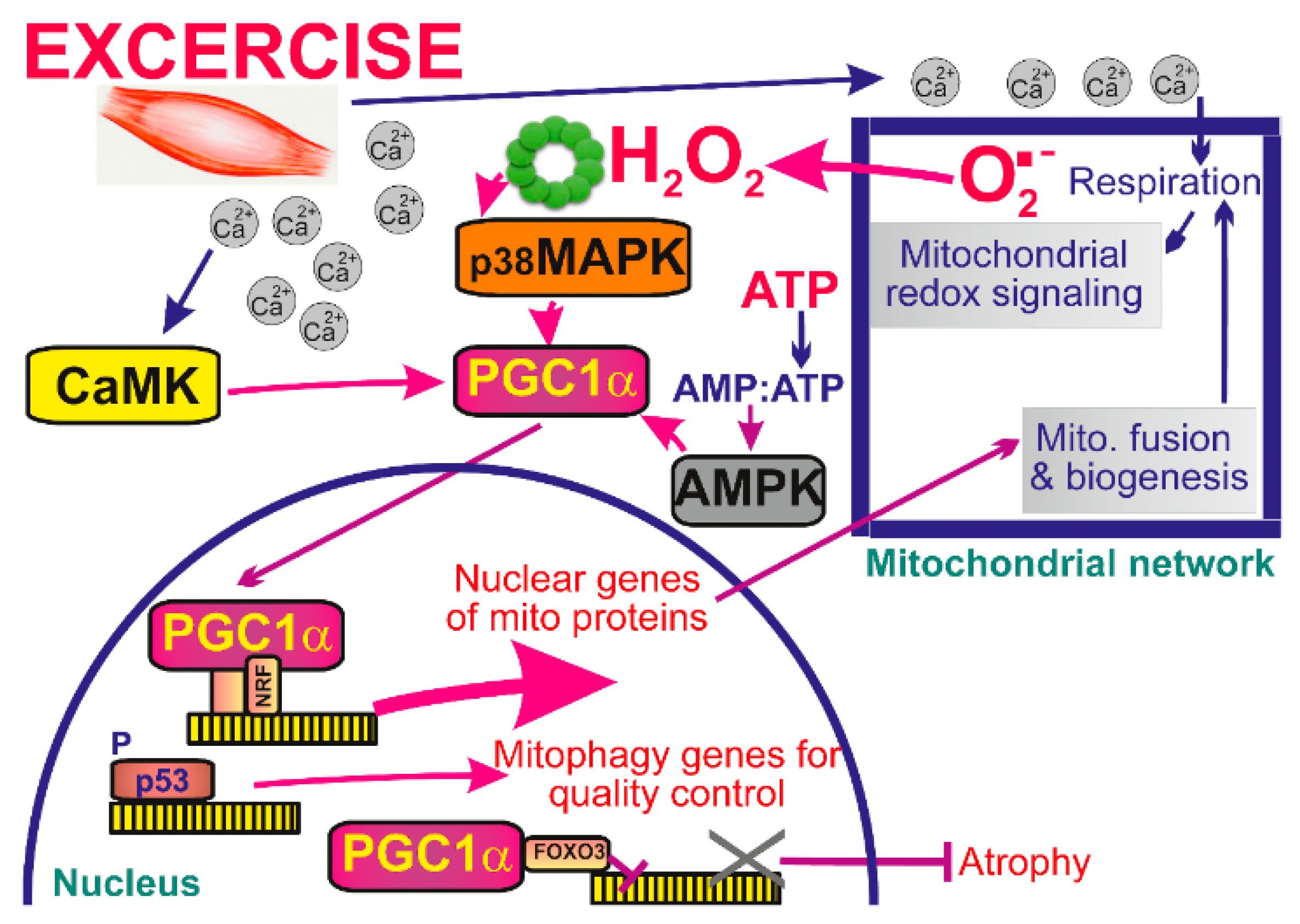
6. Mitochondrial Redox Signaling in Skeletal Muscle
6.1. Exercise Evoked Signaling in Skeletal Muscle
6.2. Exercise Evoked Mitochondrial Signaling Targets PGC1α in Skeletal Muscle
6.3. Mitochondrial Network in Skeletal Muscle
6.4. Mechanisms of Superoxide Elevation for Mitochondrial Redox Signaling in Skeletal Muscle
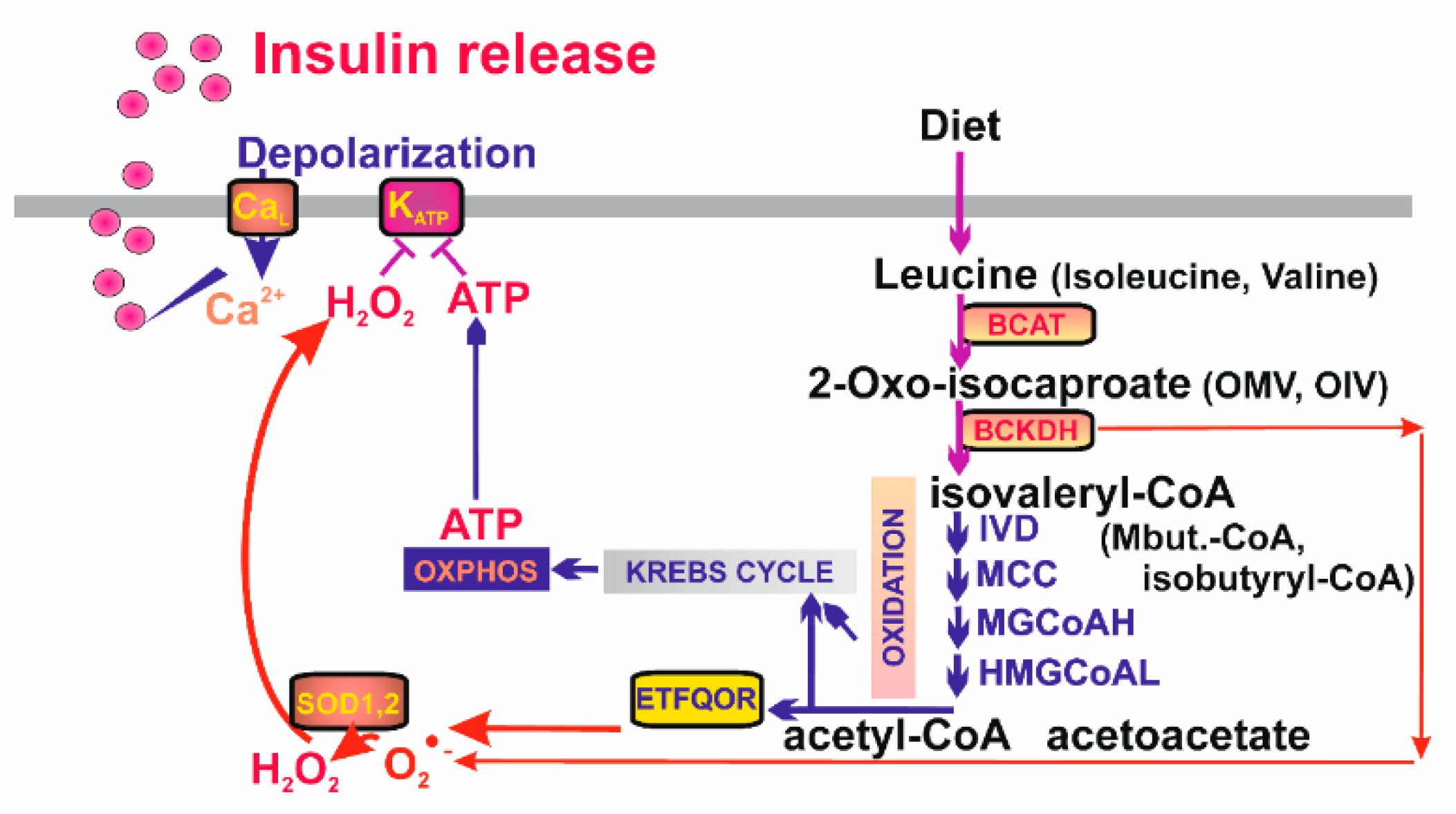
7. Mitochondrial Signaling in Pancreatic β-Cells
7.1. Mitochondrial Signaling during Fatty Acid Stimulated Insulin Secretion in Pancreatic β-Cells
7.2. Intramitochondrial Signaling to iPLA2γ Amplifies GPR40 Response
7.3. Mitochondrial Signaling Insulin Secretion Stimulated by Branched Chain Keto-Acids in Pancreatic β-Cells
8. Mitochondrial Signaling in Immune Cells
8.1. Mitochondrial Role and Signaling in Innate Immune Cells
8.2. Redox Signaling and Other ROS Effects in the Establishment of Inflammasome
8.3. ROS Signaling and Mitochondrial ROS Related to T Cell Activation
8.4. ROS Signaling and Mitochondrial ROS Related to B Cell Activation
9. Mitochondria and Kinase Signaling
10. Future Perspectives
Conflicts of Interest
References
- Parvez, S.; Long, M.J.C.; Poganik, J.R.; Aye, Y. Redox Signaling by Reactive Electrophiles and Oxidants. Chem. Rev. 2018, 118, 8798–8888. [Google Scholar] [CrossRef] [PubMed]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Skoko, J.J.; Attaran, S.; Neumann, C.A. Signals Getting Crossed in the Entanglement of Redox and Phosphorylation Pathways: Phosphorylation of Peroxiredoxin Proteins Sparks Cell Signaling. Antioxid. Basel 2019, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Plecita-Hlavata, L.; Tauber, J.; Li, M.; Zhang, H.; Flockton, A.R.; Pullamsetti, S.S.; Chelladurai, P.; D’Alessandro, A.; El Kasmi, K.C.; Jezek, P.; et al. Constitutive Reprogramming of Fibroblast Mitochondrial Metabolism in Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2016, 55, 47–57. [Google Scholar] [CrossRef]
- D’Alessandro, A.; El Kasmi, K.C.; Plecitá-Hlavatá, L.; Ježek, P.; Li, M.; Zhang, H.; Gupte, S.A.; Stenmark, K.R. Hallmarks of Pulmonary Hypertension: Mesenchymal and Inflammatory Cell Metabolic Reprogramming. Antioxid. Redox Signal. 2018, 28, 230–250. [Google Scholar] [CrossRef]
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cocheme, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012, 125, 801–806. [Google Scholar] [CrossRef]
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 2016, 100, 86–93. [Google Scholar] [CrossRef]
- Indo, H.P.; Hawkins, C.L.; Nakanishi, I.; Matsumoto, K.I.; Matsui, H.; Suenaga, S.; Davies, M.J.; St Clair, D.K.; Ozawa, T.; Majima, H.J. Role of Mitochondrial Reactive Oxygen Species in the Activation of Cellular Signals, Molecules, and Function. In Pharmacology of Mitochondria; Singh, H., Sheu, S.S., Eds.; Springer: Basel, Switzerland, 2017; Volume 240, pp. 439–456. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef]
- Ježek, P.; Hlavatá, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol. 2005, 37, 2478–2503. [Google Scholar] [CrossRef] [PubMed]
- Plecitá-Hlavatá, L.; Ježek, P. Integration of superoxide formation and cristae morphology for mitochondrial redox signaling. Int. J. Biochem. Cell Biol. 2016, 80, 31–50. [Google Scholar] [CrossRef] [PubMed]
- Ježek, P.; Jabůrek, M.; Porter, R.K. Uncoupling mechanism and redox regulation of mitochondrial uncoupling protein 1 (UCP1). Biochim. Biophys. Acta BBA Bioenerg. 2019, 1860, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Jezek, P.; Holendova, B.; Garlid, K.D.; Jaburek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid. Redox Signal. 2018, 29, 667–714. [Google Scholar] [CrossRef] [PubMed]
- Ježek, J.; Engstová, H.; Ježek, P. Antioxidant mechanism of mitochondria-targeted plastoquinone SkQ1 is suppressed in aglycemic HepG2 cells dependent on oxidative phosphorylation. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 750–762. [Google Scholar] [CrossRef] [PubMed]
- Pryde, K.R.; Hirst, J. Superoxide is produced by the reduced flavin in mitochondrial complex I: A single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 2011, 286, 18056–18065. [Google Scholar] [CrossRef]
- Treberg, J.R.; Quinlan, C.L.; Brand, M.D. Evidence for Two Sites of Superoxide Production by Mitochondrial NADH-Ubiquinone Oxidoreductase (Complex I). J. Biol. Chem. 2011, 286, 27103–27110. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Brune, B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef]
- Zepeda, A.B.; Pessoa, A., Jr.; Castillo, R.L.; Figueroa, C.A.; Pulgar, V.M.; Farias, J.G. Cellular and molecular mechanisms in the hypoxic tissue: Role of HIF-1 and ROS. Cell Biochem. Funct. 2013, 31, 451–459. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Smolková, K.; Plecitá-Hlavatá, L.; Bellance, N.; Benard, G.; Rossignol, R.; Ježek, P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int. J. Biochem. Cell Biol. 2011, 43, 950–968. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Jezek, P. 2-hydroxyglutarate in cancer cells. Antioxid. Redox Signal. 2019. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Nazarewicz, R.R.; Bikineyeva, A.; Hilenski, L.; Lassegue, B.; Griendling, K.K.; Harrison, D.G.; Dikalova, A.E. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid. Redox Signal. 2014, 20, 281–294. [Google Scholar] [CrossRef]
- Plecitá-Hlavatá, L.; D’alessandro, A.; El Kasmi, K.; Li, M.; Zhang, H.; Ježek, P.; Stenmark, K.R. Metabolic Reprogramming and Redox Signaling in Pulmonary Hypertension. In Pulmonary Vasculature Redox Signaling in Health and Disease; Springer: Berlin, Germany, 2017; pp. 241–260. [Google Scholar] [CrossRef]
- Ježek, P.; Holendová, B.; Jabůrek, M.; Tauber, J.; Plecitá-Hlavatá, L. Mitochondrial Redox Signaling Upon 2-keto-isocaproate Stimulated Insulin Secretion. Free Radic. Biol. Med. 2019, 145, S88. [Google Scholar]
- Ježek, P.; Jabůrek, M.; Plecitá-Hlavatá, L. Contribution of Oxidative Stress and Impaired Biogenesis of Pancreatic β-Cells to Type 2 Diabetes. Antioxid. Redox Signal. 2019, 31, 722–751. [Google Scholar] [CrossRef]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef]
- Horn, A.; Van der Meulen, J.H.; Defour, A.; Hogarth, M.; Sreetama, S.C.; Reed, A.; Scheffer, L.; Chandel, N.S.; Jaiswal, J.K. Mitochondrial redox signaling enables repair of injured skeletal muscle cells. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Carter, H.N.; Pauly, M.; Tryon, L.D.; Hood, D.A. Effect of contractile activity on PGC-1alpha transcription in young and aged skeletal muscle. J. Appl. Physiol. 2018, 124, 1605–1615. [Google Scholar] [CrossRef]
- Garcia, S.; Nissanka, N.; Mareco, E.A.; Rossi, S.; Peralta, S.; Diaz, F.; Rotundo, R.L.; Carvalho, R.F.; Moraes, C.T. Overexpression of PGC-1alpha in aging muscle enhances a subset of young-like molecular patterns. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Halling, J.F.; Ringholm, S.; Olesen, J.; Prats, C.; Pilegaard, H. Exercise training protects against aging-induced mitochondrial fragmentation in mouse skeletal muscle in a PGC-1alpha dependent manner. Exp. Gerontol. 2017, 96, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Kazak, L.; Jedrychowski, M.P.; Lu, G.Z.; Erickson, B.K.; Szpyt, J.; Pierce, K.A.; Laznik-Bogoslavski, D.; Vetrivelan, R.; Clish, C.B.; et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 2016, 532, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef]
- Kaminski, M.M.; Sauer, S.W.; Klemke, C.D.; Suss, D.; Okun, J.G.; Krammer, P.H.; Gulow, K. Mitochondrial reactive oxygen species control T cell activation by regulating IL-2 and IL-4 expression: Mechanism of ciprofloxacin-mediated immunosuppression. J. Immunol. 2010, 184, 4827–4841. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Bleier, L.; Dröse, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta BBA Bioenergy 2013, 1827, 1320–1331. [Google Scholar] [CrossRef]
- Dröse, S.; Brandt, U. Molecular Mechanisms of Superoxide Production by the Mitochondrial Respiratory Chain. In Advances in Experimental Medicine and Biology; Springer: Berlin, Germany, 2012; Volume 748, pp. 145–169. [Google Scholar]
- Chouchani, E.T.; Methner, C.; Buonincontri, G.; Hu, C.H.; Logan, A.; Sawiak, S.J.; Murphy, M.P.; Krieg, T. Complex I deficiency due to selective loss of Ndufs4 in the mouse heart results in severe hypertrophic cardiomyopathy. PLoS ONE 2014, 9, e94157. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Kazak, L.; Spiegelman, B.M. Mitochondrial reactive oxygen species and adipose tissue thermogenesis: Bridging physiology and mechanisms. J. Biol. Chem. 2017, 292, 16810–16816. [Google Scholar] [CrossRef]
- Mills, E.L.; Pierce, K.A.; Jedrychowski, M.P.; Garrity, R.; Winther, S.; Vidoni, S.; Yoneshiro, T.; Spinelli, J.B.; Lu, G.Z.; Kazak, L.; et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 2018, 560, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Ježek, J.; Dlasková, A.; Zelenka, J.; Jabůrek, M.; Ježek, P. H2O2-Activated Mitochondrial Phospholipase iPLA2γ Prevents Lipotoxic Oxidative Stress in Synergy with UCP2, Amplifies Signaling via G-Protein–Coupled Receptor GPR40, and Regulates Insulin Secretion in Pancreatic β-Cells. Antioxid. Redox Signal. 2015, 23, 958–972. [Google Scholar] [CrossRef] [PubMed]
- O-Uhi, J.; Ryu, S.Y.; Jhun, B.S.; Hurst, S.; Sheu, S.S. Mitochondrial ion channels/transporters as sensors and regulators of cellular redox signaling. Antioxid. Redox Signal. 2014, 21, 987–1006. [Google Scholar] [CrossRef] [PubMed]
- Borecky, J.; Jezek, P.; Siemen, D. 108-pS channel in brown fat mitochondria might Be identical to the inner membrane anion channel. J. Biol. Chem. 1997, 272, 19282–19289. [Google Scholar] [PubMed]
- Jezek, P.; Borecky, J. Inner membrane anion channel and dicarboxylate carrier in brown adipose tissue mitochondria. Int. J. Biochem. Cell Biol. 1996, 28, 659–666. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: An update and review. Biochim. Biophys. Acta 2006, 1757, 509–517. [Google Scholar] [CrossRef]
- Brady, N.R.; Hamacher-Brady, A.; Westerhoff, H.V.; Gottlieb, R.A. A wave of reactive oxygen species (ROS)-induced ROS release in a sea of excitable mitochondria. Antioxid. Redox Signal. 2006, 8, 1651–1665. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; Marban, E.; O’Rourke, B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 2003, 278, 44735–44744. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; O’Rourke, B. Redox-optimized ROS balance: A unifying hypothesis. Biochim. Biophys. Acta 2010, 1797, 865–877. [Google Scholar] [CrossRef]
- Urbani, A.; Giorgio, V.; Carrer, A.; Franchin, C.; Arrigoni, G.; Jiko, C.; Abe, K.; Maeda, S.; Shinzawa-Itoh, K.; Bogers, J.F.M.; et al. Purified F-ATP synthase forms a Ca(2+)-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat. Commun. 2019, 10, 4341. [Google Scholar] [CrossRef]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Buntinas, L.; Gunter, K.K.; Sparagna, G.C.; Gunter, T.E. The rapid mode of calcium uptake into heart mitochondria (RaM): Comparison to RaM in liver mitochondria. Biochim. Biophys. Acta 2001, 1504, 248–261. [Google Scholar] [CrossRef]
- Sparagna, G.C.; Gunter, K.K.; Sheu, S.S.; Gunter, T.E. Mitochondrial calcium uptake from physiological-type pulses of calcium. A description of the rapid uptake mode. J. Biol. Chem. 1995, 270, 27510–27515. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Sci. N. Y. 2009, 326, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Bogeski, I.; Gulaboski, R.; Kappl, R.; Mirceski, V.; Stefova, M.; Petreska, J.; Hoth, M. Calcium binding and transport by coenzyme Q. J. Am. Chem. Soc. 2011, 133, 9293–9303. [Google Scholar] [CrossRef] [PubMed]
- Beutner, G.; Sharma, V.K.; Giovannucci, D.R.; Yule, D.I.; Sheu, S.S. Identification of a ryanodine receptor in rat heart mitochondria. J. Biol. Chem. 2001, 276, 21482–21488. [Google Scholar] [CrossRef] [PubMed]
- Beutner, G.; Sharma, V.K.; Lin, L.; Ryu, S.Y.; Dirksen, R.T.; Sheu, S.S. Type 1 ryanodine receptor in cardiac mitochondria: Transducer of excitation-metabolism coupling. Biochim. Biophys. Acta 2005, 1717, 1–10. [Google Scholar] [CrossRef]
- Ryu, S.Y.; Beutner, G.; Kinnally, K.W.; Dirksen, R.T.; Sheu, S.S. Single channel characterization of the mitochondrial ryanodine receptor in heart mitoplasts. J. Biol. Chem. 2011, 286, 21324–21329. [Google Scholar] [CrossRef]
- Chaudhuri, D.; Sancak, Y.; Mootha, V.K.; Clapham, D.E. MCU encodes the pore conducting mitochondrial calcium currents. eLife 2013, 2, e00704. [Google Scholar] [CrossRef]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef]
- Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial calcium uptake in organ physiology: From molecular mechanism to animal models. Pflug. Arch. Eur. J. Physiol. 2018, 470, 1165–1179. [Google Scholar] [CrossRef]
- Dong, Z.; Shanmughapriya, S.; Tomar, D.; Siddiqui, N.; Lynch, S.; Nemani, N.; Breves, S.L.; Zhang, X.; Tripathi, A.; Palaniappan, P.; et al. Mitochondrial Ca(2+) Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol. Cell 2017, 65, 1014–1028.e1017. [Google Scholar] [CrossRef]
- Laskowski, M.; Augustynek, B.; Kulawiak, B.; Koprowski, P.; Bednarczyk, P.; Jarmuszkiewicz, W.; Szewczyk, A. What do we not know about mitochondrial potassium channels? Biochim. Biophys. Acta 2016, 1857, 1247–1257. [Google Scholar] [CrossRef]
- Checchetto, V.; Azzolini, M.; Peruzzo, R.; Capitanio, P.; Leanza, L. Mitochondrial potassium channels in cell death. Biochem. Biophys. Res. Commun. 2018, 500, 51–58. [Google Scholar] [CrossRef]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef]
- Li, X.Q.; Hegazy, M.G.; Mahdi, F.; Jezek, P.; Lane, R.D.; Garlid, K.D. Purification of a reconstitutively active K+/H+ antiporter from rat liver mitochondria. J. Biol. Chem. 1990, 265, 15316–15322. [Google Scholar]
- Jezek, P.; Mahdi, F.; Garlid, K.D. Reconstitution of the beef heart and rat liver mitochondrial K+/H+ (Na+/H+) antiporter. Quantitation of K+ transport with the novel fluorescent probe, PBFI. J. Biol. Chem. 1990, 265, 10522–10526. [Google Scholar]
- Zotova, L.; Aleschko, M.; Sponder, G.; Baumgartner, R.; Reipert, S.; Prinz, M.; Schweyen, R.J.; Nowikovsky, K. Novel components of an active mitochondrial K(+)/H(+) exchange. J. Biol. Chem. 2010, 285, 14399–14414. [Google Scholar] [CrossRef]
- Nazarewicz, R.R.; Dikalova, A.E.; Bikineyeva, A.; Dikalov, S.I. Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1131–H1140. [Google Scholar] [CrossRef]
- Queliconi, B.B.; Wojtovich, A.P.; Nadtochiy, S.M.; Kowaltowski, A.J.; Brookes, P.S. Redox regulation of the mitochondrial K(ATP) channel in cardioprotection. Biochim. Biophys. Acta 2011, 1813, 1309–1315. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Zakar, M.; Rosenthal, K.; Abu-Hamad, S. Key regions of VDAC1 functioning in apoptosis induction and regulation by hexokinase. Biochim. Biophys. Acta 2009, 1787, 421–430. [Google Scholar] [CrossRef]
- Maldonado, E.N.; Sheldon, K.L.; DeHart, D.N.; Patnaik, J.; Manevich, Y.; Townsend, D.M.; Bezrukov, S.M.; Rostovtseva, T.K.; Lemasters, J.J. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: Regulation by free tubulin and erastin. J. Biol. Chem. 2013, 288, 11920–11929. [Google Scholar] [CrossRef]
- Sheldon, K.L.; Gurnev, P.A.; Bezrukov, S.M.; Sackett, D.L. Tubulin tail sequences and post-translational modifications regulate closure of mitochondrial voltage-dependent anion channel (VDAC). J. Biol. Chem. 2015, 290, 26784–26789. [Google Scholar] [CrossRef]
- Schwarzer, C.; Barnikol-Watanabe, S.; Thinnes, F.P.; Hilschmann, N. Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74. Int. J. Biochem. Cell Biol. 2002, 34, 1059–1070. [Google Scholar] [CrossRef]
- Xu, X.; Forbes, J.G.; Colombini, M. Actin modulates the gating of Neurospora crassa VDAC. J. Membr. Biol. 2001, 180, 73–81. [Google Scholar] [CrossRef]
- Saletti, R.; Reina, S.; Pittala, M.G.G.; Magri, A.; Cunsolo, V.; Foti, S.; De Pinto, V. Post-translational modifications of VDAC1 and VDAC2 cysteines from rat liver mitochondria. Biochim. Biophys. Acta. Bioenerg. 2018, 1859, 806–816. [Google Scholar] [CrossRef]
- Shimizu, S.; Narita, M.; Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 1999, 399, 483–487. [Google Scholar] [CrossRef]
- Cheng, E.H.; Sheiko, T.V.; Fisher, J.K.; Craigen, W.J.; Korsmeyer, S.J. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Sci. N. Y. 2003, 301, 513–517. [Google Scholar] [CrossRef]
- Reina, S.; Checchetto, V.; Saletti, R.; Gupta, A.; Chaturvedi, D.; Guardiani, C.; Guarino, F.; Scorciapino, M.A.; Magri, A.; Foti, S.; et al. VDAC3 as a sensor of oxidative state of the intermembrane space of mitochondria: The putative role of cysteine residue modifications. Oncotarget 2016, 7, 2249–2268. [Google Scholar] [CrossRef]
- Martel, C.; Wang, Z.; Brenner, C. VDAC phosphorylation, a lipid sensor influencing the cell fate. Mitochondrion 2014, 19, 69–77. [Google Scholar] [CrossRef]
- Yu, H.; Diao, H.; Wang, C.; Lin, Y.; Yu, F.; Lu, H.; Xu, W.; Li, Z.; Shi, H.; Zhao, S.; et al. Acetylproteomic analysis reveals functional implications of lysine acetylation in human spermatozoa (sperm). Mol. Cell. Proteom. MCP 2015, 14, 1009–1023. [Google Scholar] [CrossRef]
- Yang, M.; Camara, A.K.; Wakim, B.T.; Zhou, Y.; Gadicherla, A.K.; Kwok, W.M.; Stowe, D.F. Tyrosine nitration of voltage-dependent anion channels in cardiac ischemia-reperfusion: Reduction by peroxynitrite scavenging. Biochim. Biophys. Acta 2012, 1817, 2049–2059. [Google Scholar] [CrossRef]
- Fu, L.; Liu, K.; Ferreira, R.B.; Carroll, K.S.; Yang, J. Proteome-Wide Analysis of Cysteine S-Sulfenylation Using a Benzothiazine-Based Probe. Curr. Protoc. Protein Sci. 2019, 95, e76. [Google Scholar] [CrossRef]
- Van der Reest, J.; Lilla, S.; Zheng, L.; Zanivan, S.; Gottlieb, E. Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat. Commun. 2018, 9, 1581. [Google Scholar] [CrossRef]
- Boronat, S.; Domènech, A.; Hidalgo, E. Proteomic Characterization of Reversible Thiol Oxidations in Proteomes and Proteins. Antioxid. Redox Signal. 2017, 26, 329–344. [Google Scholar] [CrossRef]
- Bak, D.W.; Weerapana, E. Interrogation of Functional Mitochondrial Cysteine Residues by Quantitative Mass Spectrometry. Methods Mol. Biol. Clifton 2019, 1967, 211–227. [Google Scholar] [CrossRef]
- Gould, N.S. Site-Specific Proteomic Mapping of Modified Cysteine Residues. Methods Mol. Biol. Clifton 2019, 1967, 183–195. [Google Scholar] [CrossRef]
- Abreu, I.A.; Cabelli, D.E. Superoxide dismutases-a review of the metal-associated mechanistic variations. Biochim. Biophys. Acta 2010, 1804, 263–274. [Google Scholar] [CrossRef]
- Bielski, B.H.J.; Allen, A.O. Mechanism of the disproportionation of superoxide radicals. J. Phys. Chem. 1977, 81, 1048–1050. [Google Scholar] [CrossRef]
- Jones, C.M.; Lawrence, A.; Wardman, P.; Burkitt, M.J. Kinetics of superoxide scavenging by glutathione: An evaluation of its role in the removal of mitochondrial superoxide. Biochem. Soc. Trans. 2003, 31, 1337–1339. [Google Scholar] [CrossRef]
- Mikkelsen, R.B.; Wardman, P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene 2003, 22, 5734–5754. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; Di Stefano, L.; Di Meo, S. Mitochondrial metabolism of reactive oxygen species. Mitochondrion 2013, 13, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Mishina, N.M.; Bogdanova, Y.A.; Ermakova, Y.G.; Panova, A.S.; Kotova, D.A.; Bilan, D.S.; Steinhorn, B.; Arner, E.S.J.; Michel, T.; Belousov, V.V. Which Antioxidant System Shapes Intracellular H2O2 Gradients? Antioxid. Redox Signal. 2019, 31, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef]
- Peskin, A.V.; Low, F.M.; Paton, L.N.; Maghzal, G.J.; Hampton, M.B.; Winterbourn, C.C. The high reactivity of peroxiredoxin 2 with H(2)O(2) is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 2007, 282, 11885–11892. [Google Scholar] [CrossRef]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. CCS 2015, 13, 39. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef]
- Hohn, A.; Jung, T.; Grune, T. Pathophysiological importance of aggregated damaged proteins. Free Radic. Biol. Med. 2014, 71, 70–89. [Google Scholar] [CrossRef]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [CrossRef]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Sci. N. Y. 2003, 300, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Chae, H.Z.; Seo, M.S.; Kim, K.; Baines, I.C.; Rhee, S.G. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J. Biol. Chem. 1998, 273, 6297–6302. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.C.; Choi, H.I.; Park, Y.S.; Nam, H.W.; Woo, H.A.; Kwon, K.S.; Kim, Y.S.; Rhee, S.G.; Kim, K.; Chae, H.Z. Irreversible oxidation of the active-site cysteine of peroxiredoxin to cysteine sulfonic acid for enhanced molecular chaperone activity. J. Biol. Chem. 2008, 283, 28873–28880. [Google Scholar] [CrossRef] [PubMed]
- Knoops, B.; Goemaere, J.; Van der Eecken, V.; Declercq, J.P. Peroxiredoxin 5: Structure, mechanism, and function of the mammalian atypical 2-Cys peroxiredoxin. Antioxid. Redox Signal. 2011, 15, 817–829. [Google Scholar] [CrossRef]
- Rhee, S.G.; Kang, S.W.; Chang, T.S.; Jeong, W.; Kim, K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life 2001, 52, 35–41. [Google Scholar] [CrossRef]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A(2) activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef]
- Hall, A.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Structural evidence that peroxiredoxin catalytic power is based on transition-state stabilization. J. Mol. Biol. 2010, 402, 194–209. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Peskin, A.V. Kinetic Approaches to Measuring Peroxiredoxin Reactivity. Mol. Cells 2016, 39, 26–30. [Google Scholar] [CrossRef]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef]
- Mishra, M.; Jiang, H.; Wu, L.; Chawsheen, H.A.; Wei, Q. The sulfiredoxin-peroxiredoxin (Srx-Prx) axis in cell signal transduction and cancer development. Cancer Lett. 2015, 366, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells 2018, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.H.; Lee, K.O.; Chi, Y.H.; Jung, B.G.; Park, S.K.; Park, J.H.; Lee, J.R.; Lee, S.S.; Moon, J.C.; Yun, J.W.; et al. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 2004, 117, 625–635. [Google Scholar] [CrossRef]
- Neumann, C.A.; Cao, J.; Manevich, Y. Peroxiredoxin 1 and its role in cell signaling. Cell Cycle Georget. Tex. 2009, 8, 4072–4078. [Google Scholar] [CrossRef]
- Stocker, S.; Van Laer, K.; Mijuskovic, A.; Dick, T.P. The Conundrum of Hydrogen Peroxide Signaling and the Emerging Role of Peroxiredoxins as Redox Relay Hubs. Antioxid. Redox Signal. 2018, 28, 558–573. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A.; Kang, D. The Role of Peroxiredoxins in the Transduction of H2O2 Signals. Antioxid. Redox Signal. 2018, 28, 537–557. [Google Scholar] [CrossRef]
- Sobotta, M.C.; Liou, W.; Stocker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015, 11, 64–70. [Google Scholar] [CrossRef]
- Jarvis, R.M.; Hughes, S.M.; Ledgerwood, E.C. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic. Biol. Med. 2012, 53, 1522–1530. [Google Scholar] [CrossRef]
- Stocker, S.; Maurer, M.; Ruppert, T.; Dick, T.P. A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 2018, 14, 148–155. [Google Scholar] [CrossRef]
- Go, Y.M.; Roede, J.R.; Walker, D.I.; Duong, D.M.; Seyfried, N.T.; Orr, M.; Liang, Y.; Pennell, K.D.; Jones, D.P. Selective targeting of the cysteine proteome by thioredoxin and glutathione redox systems. Mol. Cell. Proteom. MCP 2013, 12, 3285–3296. [Google Scholar] [CrossRef]
- Soga, M.; Matsuzawa, A.; Ichijo, H. Oxidative Stress-Induced Diseases via the ASK1 Signaling Pathway. Int. J. Cell Biol. 2012, 2012, 439587. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.C.; Papaconstantinou, J. Thioredoxin-ASK1 complex levels regulate ROS-mediated p38 MAPK pathway activity in livers of aged and long-lived Snell dwarf mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Tavender, T.J.; Springate, J.J.; Bulleid, N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010, 29, 4185–4197. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.C.; Hsieh, Y.H.; Su, M.I.; Jiang, X.; Hsu, P.H.; Lo, W.T.; Weng, J.Y.; Jeng, Y.M.; Wang, J.M.; Chen, P.L.; et al. Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol. Cell 2012, 48, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Lindsay, J.G.; Isaacs, N.W. Mitochondrial peroxiredoxins. Sub. Cell. Biochem. 2007, 44, 295–315. [Google Scholar] [CrossRef]
- Seo, M.S.; Kang, S.W.; Kim, K.; Baines, I.C.; Lee, T.H.; Rhee, S.G. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 2000, 275, 20346–20354. [Google Scholar] [CrossRef]
- Kropotov, A.; Usmanova, N.; Serikov, V.; Zhivotovsky, B.; Tomilin, N. Mitochondrial targeting of human peroxiredoxin V protein and regulation of PRDX5 gene expression by nuclear transcription factors controlling biogenesis of mitochondria. FEBS J. 2007, 274, 5804–5814. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Herbette, S.; Roeckel-Drevet, P.; Drevet, J.R. Seleno-independent glutathione peroxidases. More than simple antioxidant scavengers. FEBS J. 2007, 274, 2163–2180. [Google Scholar] [CrossRef]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Marsboom, G.; Kim, G.H.; Zhang, H.J.; Toth, P.T.; Svensson, E.C.; Dyck, J.R.; Gomberg-Maitland, M.; Thebaud, B.; Husain, A.N.; et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation 2010, 121, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Salazar, G. NADPH Oxidases and Mitochondria in Vascular Senescence. Int. J. Mol. Sci. 2018, 19, 1327. [Google Scholar] [CrossRef]
- Xiong, S.; Salazar, G.; San Martin, A.; Ahmad, M.; Patrushev, N.; Hilenski, L.; Nazarewicz, R.R.; Ma, M.; Ushio-Fukai, M.; Alexander, R.W. PGC-1 alpha serine 570 phosphorylation and GCN5-mediated acetylation by angiotensin II drive catalase down-regulation and vascular hypertrophy. J. Biol. Chem. 2010, 285, 2474–2487. [Google Scholar] [CrossRef]
- Mistry, Y.; Poolman, T.; Williams, B.; Herbert, K.E. A role for mitochondrial oxidants in stress-induced premature senescence of human vascular smooth muscle cells. Redox Biol. 2013, 1, 411–417. [Google Scholar] [CrossRef]
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kroller-Schon, S.; Steven, S.; Schulz, E.; Munzel, T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2017, 174, 1670–1689. [Google Scholar] [CrossRef]
- Salazar, G.; Huang, J.; Feresin, R.G.; Zhao, Y.; Griendling, K.K. Zinc regulates Nox1 expression through a NF-kappaB and mitochondrial ROS dependent mechanism to induce senescence of vascular smooth muscle cells. Free Radic. Biol. Med. 2017, 108, 225–235. [Google Scholar] [CrossRef]
- Appelhoff, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef]
- Lu, H.; Dalgard, C.L.; Mohyeldin, A.; McFate, T.; Tait, A.S.; Verma, A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J. Biol. Chem. 2005, 280, 41928–41939. [Google Scholar] [CrossRef]
- Gerald, D.; Berra, E.; Frapart, Y.M.; Chan, D.A.; Giaccia, A.J.; Mansuy, D.; Pouyssegur, J.; Yaniv, M.; Mechta-Grigoriou, F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell 2004, 118, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Briggs, K.J.; Koivunen, P.; Cao, S.; Backus, K.M.; Olenchock, B.A.; Patel, H.; Zhang, Q.; Signoretti, S.; Gerfen, G.J.; Richardson, A.L.; et al. Paracrine Induction of HIF by Glutamate in Breast Cancer: EglN1 Senses Cysteine. Cell 2016, 166, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Won, H.S.; Lee, Y.M.; Choi, J.W.; Oh, T.I.; Jang, J.H.; Choi, D.K.; Lim, B.O.; Kim, Y.J.; Park, J.W.; et al. Oxidative Dimerization of PHD2 is Responsible for its Inactivation and Contributes to Metabolic Reprogramming via HIF-1alpha Activation. Sci. Rep. 2016, 6, 18928. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Flashman, E.; Mecinovic, J.; Kramer, H.B.; Kessler, B.M.; Frapart, Y.M.; Boucher, J.L.; Clifton, I.J.; McDonough, M.A.; Schofield, C.J. Studies on the reaction of nitric oxide with the hypoxia-inducible factor prolyl hydroxylase domain 2 (EGLN1). J. Mol. Biol. 2011, 410, 268–279. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Sci. N. Y. 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Sci. N. Y. 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Schodel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef]
- Brocato, J.; Chervona, Y.; Costa, M. Molecular responses to hypoxia-inducible factor 1alpha and beyond. Mol. Pharmacol. 2014, 85, 651–657. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem. J. 2007, 405, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Mansfield, K.D.; Bertozzi, C.C.; Rudenko, V.; Chan, D.A.; Giaccia, A.J.; Simon, M.C. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol. Cell. Biol. 2007, 27, 912–925. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, K.S.; Lienard, B.M.; McDonough, M.A.; Clifton, I.J.; Butler, D.; Soares, A.S.; Oldham, N.J.; McNeill, L.A.; Schofield, C.J. Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates. J. Biol. Chem. 2007, 282, 3293–3301. [Google Scholar] [CrossRef]
- Koivunen, P.; Hirsila, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: Possible links between cell metabolism and stabilization of HIF. J. Biol. Chem. 2007, 282, 4524–4532. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell 2016, 61, 199–209. [Google Scholar] [CrossRef]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Burr, S.P.; Costa, A.S.; Grice, G.L.; Timms, R.T.; Lobb, I.T.; Freisinger, P.; Dodd, R.B.; Dougan, G.; Lehner, P.J.; Frezza, C.; et al. Mitochondrial Protein Lipoylation and the 2-Oxoglutarate Dehydrogenase Complex Controls HIF1alpha Stability in Aerobic Conditions. Cell Metab. 2016, 24, 740–752. [Google Scholar] [CrossRef]
- Wen, Y.A.; Xiong, X.; Scott, T.; Li, A.T.; Wang, C.; Weiss, H.L.; Tan, L.; Bradford, E.; Fan, T.W.M.; Chandel, N.S.; et al. The mitochondrial retrograde signaling regulates Wnt signaling to promote tumorigenesis in colon cancer. Cell Death Differ. 2019, 26, 1955–1969. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Plecitá-Hlavatá, L.; Ježek, J.; Ježek, P. Aglycemia keeps mitochondrial oxidative phosphorylation under hypoxic conditions in HepG2 cells. J. Bioenerg. Biomembr. 2015, 47, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.K.; Cavadas, M.A.; Scholz, C.C.; Fitzpatrick, S.F.; Bruning, U.; Cummins, E.P.; Tambuwala, M.M.; Manresa, M.C.; Kholodenko, B.N.; Taylor, C.T.; et al. A dynamic model of the hypoxia-inducible factor 1alpha (HIF-1alpha) network. J. Cell Sci. 2013, 126, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Hernansanz-Agustin, P.; Ramos, E.; Navarro, E.; Parada, E.; Sanchez-Lopez, N.; Pelaez-Aguado, L.; Cabrera-Garcia, J.D.; Tello, D.; Buendia, I.; Marina, A.; et al. Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol. 2017, 12, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Schroedl, C.; McClintock, D.S.; Budinger, G.R.; Chandel, N.S. Hypoxic but not anoxic stabilization of HIF-1alpha requires mitochondrial reactive oxygen species. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L922–L931. [Google Scholar] [CrossRef]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef]
- Waypa, G.B.; Marks, J.D.; Guzy, R.; Mungai, P.T.; Schriewer, J.; Dokic, D.; Schumacker, P.T. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ. Res. 2010, 106, 526–535. [Google Scholar] [CrossRef]
- Comito, G.; Calvani, M.; Giannoni, E.; Bianchini, F.; Calorini, L.; Torre, E.; Migliore, C.; Giordano, S.; Chiarugi, P. HIF-1alpha stabilization by mitochondrial ROS promotes Met-dependent invasive growth and vasculogenic mimicry in melanoma cells. Free Radic. Biol. Med. 2011, 51, 893–904. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Waypa, G.B.; Marks, J.D.; Schumacker, P.T. Peroxiredoxin-5 targeted to the mitochondrial intermembrane space attenuates hypoxia-induced reactive oxygen species signalling. Biochem. J. 2013, 456, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.A.; Lafleur, V.N.; Robitaille, G.A.; Chan, D.A.; Giaccia, A.J.; Richard, D.E. Hypoxia-inducible factor-1 activation in nonhypoxic conditions: The essential role of mitochondrial-derived reactive oxygen species. Mol. Biol. Cell 2010, 21, 3247–3257. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.L.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 2015, 11, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Jezek, P.; Plecita-Hlavata, L. Mitochondrial reticulum network dynamics in relation to oxidative stress, redox regulation, and hypoxia. Int. J. Biochem. Cell Biol. 2009, 41, 1790–1804. [Google Scholar] [CrossRef]
- Yue, X.; Zhao, P.; Wu, K.; Huang, J.; Zhang, W.; Wu, Y.; Liang, X.; He, X. GRIM-19 inhibition induced autophagy through activation of ERK and HIF-1alpha not STAT3 in Hela cells. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 9789–9796. [Google Scholar] [CrossRef]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell. Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef]
- Saito, Y.; Ishii, K.A.; Aita, Y.; Ikeda, T.; Kawakami, Y.; Shimano, H.; Hara, H.; Takekoshi, K. Loss of SDHB Elevates Catecholamine Synthesis and Secretion Depending on ROS Production and HIF Stabilization. Neurochem. Res. 2016, 41, 696–706. [Google Scholar] [CrossRef]
- Diaz, F.; Enriquez, J.A.; Moraes, C.T. Cells lacking Rieske iron-sulfur protein have a reactive oxygen species-associated decrease in respiratory complexes I and IV. Mol. Cell. Biol. 2012, 32, 415–429. [Google Scholar] [CrossRef]
- Bastian, A.; Matsuzaki, S.; Humphries, K.M.; Pharaoh, G.A.; Doshi, A.; Zaware, N.; Gangjee, A.; Ihnat, M.A. AG311, a small molecule inhibitor of complex I and hypoxia-induced HIF-1alpha stabilization. Cancer Lett. 2017, 388, 149–157. [Google Scholar] [CrossRef]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordonez, A.; Corral-Escariz, M.; Soro, I.; Lopez-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef]
- Lai, R.K.; Xu, I.M.; Chiu, D.K.; Tse, A.P.; Wei, L.L.; Law, C.T.; Lee, D.; Wong, C.M.; Wong, M.P.; Ng, I.O.; et al. NDUFA4L2 Fine-tunes Oxidative Stress in Hepatocellular Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3105–3117. [Google Scholar] [CrossRef] [PubMed]
- Guaras, A.; Perales-Clemente, E.; Calvo, E.; Acin-Perez, R.; Loureiro-Lopez, M.; Pujol, C.; Martinez-Carrascoso, I.; Nunez, E.; Garcia-Marques, F.; Rodriguez-Hernandez, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Pannell, B.K. Redox Characterization of Functioning Skeletal Muscle. Front. Physiol. 2015, 6, 338. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Kanatous, S.B.; Thurmond, F.A.; Gallardo, T.; Isotani, E.; Bassel-Duby, R.; Williams, R.S. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Sci. N. Y. 2002, 296, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Gowans, G.J.; Hawley, S.A.; Ross, F.A.; Hardie, D.G. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013, 18, 556–566. [Google Scholar] [CrossRef]
- Pogozelski, A.R.; Geng, T.; Li, P.; Yin, X.; Lira, V.A.; Zhang, M.; Chi, J.T.; Yan, Z. p38gamma mitogen-activated protein kinase is a key regulator in skeletal muscle metabolic adaptation in mice. PLoS ONE 2009, 4, e7934. [Google Scholar] [CrossRef]
- Irrcher, I.; Ljubicic, V.; Hood, D.A. Interactions between ROS and AMP kinase activity in the regulation of PGC-1alpha transcription in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 2009, 296, C116–C123. [Google Scholar] [CrossRef]
- Powers, S.K.; Talbert, E.E.; Adhihetty, P.J. Reactive oxygen and nitrogen species as intracellular signals in skeletal muscle. J. Physiol. 2011, 589, 2129–2138. [Google Scholar] [CrossRef]
- Saleem, A.; Hood, D.A. Acute exercise induces tumour suppressor protein p53 translocation to the mitochondria and promotes a p53-Tfam-mitochondrial DNA complex in skeletal muscle. J. Physiol. 2013, 591, 3625–3636. [Google Scholar] [CrossRef]
- Picard, M.; Gentil, B.J.; McManus, M.J.; White, K.; St Louis, K.; Gartside, S.E.; Wallace, D.C.; Turnbull, D.M. Acute exercise remodels mitochondrial membrane interactions in mouse skeletal muscle. J. Appl. Physiol. 2013, 115, 1562–1571. [Google Scholar] [CrossRef]
- Mishra, P.; Varuzhanyan, G.; Pham, A.H.; Chan, D.C. Mitochondrial Dynamics is a Distinguishing Feature of Skeletal Muscle Fiber Types and Regulates Organellar Compartmentalization. Cell Metab. 2015, 22, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.; Reidy, P.T.; Bhattarai, N.; Sidossis, L.S.; Rasmussen, B.B. Resistance Exercise Training Alters Mitochondrial Function in Human Skeletal Muscle. Med. Sci. Sports Exerc. 2015, 47, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Gejl, K.D.; Hey-Mogensen, M.; Holmberg, H.C.; Suetta, C.; Krustrup, P.; Elemans, C.P.H.; Ortenblad, N. Plasticity in mitochondrial cristae density allows metabolic capacity modulation in human skeletal muscle. J. Physiol. 2017, 595, 2839–2847. [Google Scholar] [CrossRef] [PubMed]
- Holloway, G.P. Nutrition and Training Influences on the Regulation of Mitochondrial Adenosine Diphosphate Sensitivity and Bioenergetics. Sports Med. 2017, 47, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Dahl, R.; Larsen, S.; Dohlmann, T.L.; Qvortrup, K.; Helge, J.W.; Dela, F.; Prats, C. Three-dimensional reconstruction of the human skeletal muscle mitochondrial network as a tool to assess mitochondrial content and structural organization. Acta Physiol. 2015, 213, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Hartnell, L.M.; Combs, C.A.; Femnou, A.; Sun, J.; Murphy, E.; Subramaniam, S.; Balaban, R.S. Power Grid Protection of the Muscle Mitochondrial Reticulum. Cell Rep. 2017, 19, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; White, K.; Turnbull, D.M. Mitochondrial morphology, topology, and membrane interactions in skeletal muscle: A quantitative three-dimensional electron microscopy study. J. Appl. Physiol. 2013, 114, 161–171. [Google Scholar] [CrossRef]
- Iqbal, S.; Hood, D.A. Cytoskeletal regulation of mitochondrial movements in myoblasts. Cytoskelet. 2014, 71, 564–572. [Google Scholar] [CrossRef]
- Goncalves, R.L.S.; Watson, M.A.; Wong, H.S.; Orr, A.L.; Brand, M.D. The use of site-specific suppressors to measure the relative contributions of different mitochondrial sites to skeletal muscle superoxide and hydrogen peroxide production. Redox Biol. 2019, 28, 101341. [Google Scholar] [CrossRef]
- Ježek, P.; Jabůrek, M.; Holendová, B.; Plecitá-Hlavatá, L. Fatty Acid-Stimulated Insulin Secretion vs. Lipotoxicity. Molecules 2018, 23, 1483. [Google Scholar] [CrossRef]
- Prentki, M.; Matschinsky, F.M.; Madiraju, S.R.M. Metabolic Signaling in Fuel-Induced Insulin Secretion. Cell Metab. 2013, 18, 162–185. [Google Scholar] [CrossRef] [PubMed]
- Carpinelli, A.R.; Picinato, M.C.; Stevanato, E.; Oliveira, H.R.; Curi, R. Insulin secretion induced by palmitate--a process fully dependent on glucose concentration. Diabetes Metab. 2002, 28, S37–S44. [Google Scholar]
- Gehrmann, W.; Elsner, M.; Lenzen, S. Role of metabolically generated reactive oxygen species for lipotoxicity in pancreatic β-cells. DiabetesObes. Metab. 2010, 12, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Graciano, M.F.R.; Valle, M.M.R.; Kowluru, A.; Curi, R.; Carpinelli, A.R. Regulation of insulin secretion and reactive oxygen species production by free fatty acids in pancreatic islets. Islets 2011, 3, 213–223. [Google Scholar] [CrossRef]
- Cen, J.; Sargsyan, E.; Bergsten, P. Fatty acids stimulate insulin secretion from human pancreatic islets at fasting glucose concentrations via mitochondria-dependent and -independent mechanisms. Nutr. Metab. 2016, 13, 59. [Google Scholar] [CrossRef]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; et al. Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef]
- Latour, M.G.; Alquier, T.; Oseid, E.; Tremblay, C.; Jetton, T.L.; Luo, J.; Lin, D.C.; Poitout, V. GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes 2007, 56, 1087–1094. [Google Scholar] [CrossRef]
- Sabrautzki, S.; Kaiser, G.; Przemeck, G.K.H.; Gerst, F.; Lorza-Gil, E.; Panse, M.; Sartorius, T.; Hoene, M.; Marschall, S.; Haring, H.U.; et al. Point mutation of Ffar1 abrogates fatty acid-dependent insulin secretion, but protects against HFD-induced glucose intolerance. Mol. Metab. 2017, 6, 1304–1312. [Google Scholar] [CrossRef]
- Hauge, M.; Vestmar, M.A.; Husted, A.S.; Ekberg, J.P.; Wright, M.J.; Di Salvo, J.; Weinglass, A.B.; Engelstoft, M.S.; Madsen, A.N.; Luckmann, M.; et al. GPR40 (FFAR1)—Combined Gs and Gq signaling in vitro is associated with robust incretin secretagogue action ex vivo and in vivo. Mol. Metab. 2015, 4, 3–14. [Google Scholar] [CrossRef]
- Qian, J.; Gu, Y.; Wu, C.; Yu, F.; Chen, Y.; Zhu, J.; Yao, X.; Bei, C.; Zhu, Q. Agonist-induced activation of human FFA1 receptor signals to extracellular signal-regulated kinase 1 and 2 through Gq- and Gi-coupled signaling cascades. Cell. Mol. Biol. Lett. 2017, 22, 13. [Google Scholar] [CrossRef]
- Husted, A.S.; Trauelsen, M.; Rudenko, O.; Hjorth, S.A.; Schwartz, T.W. GPCR-Mediated Signaling of Metabolites. Cell Metab. 2017, 25, 777–796. [Google Scholar] [CrossRef] [PubMed]
- Graciano, M.F.; Valle, M.M.; Curi, R.; Carpinelli, A.R. Evidence for the involvement of GPR40 and NADPH oxidase in palmitic acid-induced superoxide production and insulin secretion. Islets 2013, 5, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Kristinsson, H.; Bergsten, P.; Sargsyan, E. Free fatty acid receptor 1 (FFAR1/GPR40) signaling affects insulin secretion by enhancing mitochondrial respiration during palmitate exposure. Biochim. Biophys. Acta 2015, 1853, 3248–3257. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Li, F.; Li, Y.; Tang, Y.; Kong, X.; Feng, Z.; Anthony, T.G.; Watford, M.; Hou, Y.; Wu, G.; et al. The role of leucine and its metabolites in protein and energy metabolism. Amino Acids 2016, 48, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, L.M.; Valtat, B.; Medina, A.; Andersson, L.; Abels, M.; Mollet, I.G.; Jain, D.; Eliasson, L.; Wierup, N.; Fex, M.; et al. Mitochondrial transcription factor B2 is essential for mitochondrial and cellular function in pancreatic beta-cells. Mol. Metab. 2017, 6, 651–663. [Google Scholar] [CrossRef]
- Benmoussa, K.; Garaude, J.; Acin-Perez, R. How Mitochondrial Metabolism Contributes to Macrophage Phenotype and Functions. J. Mol. Biol. 2018, 430, 3906–3921. [Google Scholar] [CrossRef]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A.; Mills, E.L. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab. 2019, 1, 16–33. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Singer, B.D.; Steinert, E.M.; Martinez, C.A.; Mehta, M.M.; Martinez-Reyes, I.; Gao, P.; Helmin, K.A.; Abdala-Valencia, H.; Sena, L.A.; et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 2019, 565, 495–499. [Google Scholar] [CrossRef]
- Mehta, M.M.; Weinberg, S.E.; Chandel, N.S. Mitochondrial control of immunity: Beyond ATP. Nat. Rev. Immunol. 2017, 17, 608–620. [Google Scholar] [CrossRef]
- Kong, H.; Chandel, N.S. Regulation of redox balance in cancer and T cells. J. Biol. Chem. 2018, 293, 7499–7507. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Danielski, L.G.; Giustina, A.D.; Bonfante, S.; Barichello, T.; Petronilho, F. The NLRP3 Inflammasome and Its Role in Sepsis Development. Inflammation 2019. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.; Reis e Sousa, C. Myeloid C-type lectin receptors in pathogen recognition and host defense. Immunity 2011, 34, 651–664. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef]
- Sonoda, J.; Laganiere, J.; Mehl, I.R.; Barish, G.D.; Chong, L.W.; Li, X.; Scheffler, I.E.; Mock, D.C.; Bataille, A.R.; Robert, F.; et al. Nuclear receptor ERR alpha and coactivator PGC-1 beta are effectors of IFN-gamma-induced host defense. Genes Dev. 2007, 21, 1909–1920. [Google Scholar] [CrossRef]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Rousset, S.; Emre, Y.; Join-Lambert, O.; Hurtaud, C.; Ricquier, D.; Cassard-Doulcier, A.M. The uncoupling protein 2 modulates the cytokine balance in innate immunity. Cytokine 2006, 35, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Onuma, H.; Bai, X.; Medvedev, A.V.; Misukonis, M.; Weinberg, J.B.; Cao, W.; Robidoux, J.; Floering, L.M.; Daniel, K.W.; et al. Persistent nuclear factor-kappa B activation in Ucp2-/- mice leads to enhanced nitric oxide and inflammatory cytokine production. J. Biol. Chem. 2005, 280, 19062–19069. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef]
- Vazquez, C.; Horner, S.M. MAVS Coordination of Antiviral Innate Immunity. J. Virol. 2015, 89, 6974–6977. [Google Scholar] [CrossRef]
- Moon, J.S.; Lee, S.; Park, M.A.; Siempos, I.I.; Haslip, M.; Lee, P.J.; Yun, M.; Kim, C.K.; Howrylak, J.; Ryter, S.W.; et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J. Clin. Investig. 2015, 125, 665–680. [Google Scholar] [CrossRef]
- Ma, M.W.; Wang, J.; Dhandapani, K.M.; Brann, D.W. NADPH Oxidase 2 Regulates NLRP3 Inflammasome Activation in the Brain after Traumatic Brain Injury. Oxidative Med. Cell. Longev. 2017, 2017, 6057609. [Google Scholar] [CrossRef]
- Moon, J.S.; Nakahira, K.; Chung, K.P.; DeNicola, G.M.; Koo, M.J.; Pabon, M.A.; Rooney, K.T.; Yoon, J.H.; Ryter, S.W.; Stout-Delgado, H.; et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat. Med. 2016, 22, 1002–1012. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Wu, J.; Yan, Z.; Schwartz, D.E.; Yu, J.; Malik, A.B.; Hu, G. Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. J. Immunol. 2013, 190, 3590–3599. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.Y.; Park, H.H. Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J. Biol. Chem. 2011, 286, 39528–39536. [Google Scholar] [CrossRef]
- Ichinohe, T.; Yamazaki, T.; Koshiba, T.; Yanagi, Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. USA 2013, 110, 17963–17968. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Franchina, D.G.; Dostert, C.; Brenner, D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol. 2018, 39, 489–502. [Google Scholar] [CrossRef]
- Kaminski, M.M.; Roth, D.; Sass, S.; Sauer, S.W.; Krammer, P.H.; Gulow, K. Manganese superoxide dismutase: A regulator of T cell activation-induced oxidative signaling and cell death. Biochim. Biophys. Acta 2012, 1823, 1041–1052. [Google Scholar] [CrossRef]
- Kaminski, M.M.; Sauer, S.W.; Kaminski, M.; Opp, S.; Ruppert, T.; Grigaravicius, P.; Grudnik, P.; Grone, H.J.; Krammer, P.H.; Gulow, K. T cell activation is driven by an ADP-dependent glucokinase linking enhanced glycolysis with mitochondrial reactive oxygen species generation. Cell Rep. 2012, 2, 1300–1315. [Google Scholar] [CrossRef]
- Rashida Gnanaprakasam, J.N.; Wu, R.; Wang, R. Metabolic Reprogramming in Modulating T Cell Reactive Oxygen Species Generation and Antioxidant Capacity. Front. Immunol. 2018, 9, 1075. [Google Scholar] [CrossRef]
- Previte, D.M.; O’Connor, E.C.; Novak, E.A.; Martins, C.P.; Mollen, K.P.; Piganelli, J.D. Reactive oxygen species are required for driving efficient and sustained aerobic glycolysis during CD4+ T cell activation. PLoS ONE 2017, 12, e0175549. [Google Scholar] [CrossRef]
- O’Sullivan, D.; van der Windt, G.J.W.; Huang, S.C.; Curtis, J.D.; Chang, C.H.; Buck, M.D.; Qiu, J.; Smith, A.M.; Lam, W.Y.; DiPlato, L.M.; et al. Memory CD8(+) T Cells Use Cell-Intrinsic Lipolysis to Support the Metabolic Programming Necessary for Development. Immunity 2018, 49, 375–376. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Staron, M.M.; Gray, S.M.; Ho, P.C.; Amezquita, R.A.; Wu, J.; Kaech, S.M. IL-7-Induced Glycerol Transport and TAG Synthesis Promotes Memory CD8+ T Cell Longevity. Cell 2015, 161, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Wofford, J.A.; Wieman, H.L.; Jacobs, S.R.; Zhao, Y.; Rathmell, J.C. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood 2008, 111, 2101–2111. [Google Scholar] [CrossRef] [PubMed]
- Angajala, A.; Lim, S.; Phillips, J.B.; Kim, J.H.; Yates, C.; You, Z.; Tan, M. Diverse Roles of Mitochondria in Immune Responses: Novel Insights into Immuno-Metabolism. Front. Immunol. 2018, 9, 1605. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.J.; Mano, H.; Aoki, K.; Hayashi, T.; Muto, A.; Nambu, Y.; Takahashi, K.; Itoh, K.; Taketani, S.; Nutt, S.L.; et al. Mitochondrial function provides instructive signals for activation-induced B-cell fates. Nat. Commun. 2015, 6, 6750. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, J.; Ballinger, S.W.; Darley-Usmar, V.M.; Landar, A. Free radicals, mitochondria, and oxidized lipids: The emerging role in signal transduction in vascular cells. Circ. Res. 2006, 99, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D.; Costa, A.D.; Quinlan, C.L.; Pierre, S.V.; Dos Santos, P. Cardioprotective signaling to mitochondria. J. Mol. Cell. Cardiol. 2009, 46, 858–866. [Google Scholar] [CrossRef]
- Corcoran, A.; Cotter, T.G. Redox regulation of protein kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef]
- Cosentino-Gomes, D.; Rocco-Machado, N.; Meyer-Fernandes, J.R. Cell signaling through protein kinase C oxidation and activation. Int. J. Mol. Sci. 2012, 13, 10697–10721. [Google Scholar] [CrossRef]
- Garlid, A.O.; Jaburek, M.; Jacobs, J.P.; Garlid, K.D. Mitochondrial reactive oxygen species: Which ROS signals cardioprotection? Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H960–H968. [Google Scholar] [CrossRef]
- Giorgi, C.; Agnoletto, C.; Baldini, C.; Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A.; et al. Redox control of protein kinase C: Cell- and disease-specific aspects. Antioxid. Redox Signal. 2010, 13, 1051–1085. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishna, R.; Jaken, S. Protein kinase C signaling and oxidative stress. Free Radic. Biol. Med. 2000, 28, 1349–1361. [Google Scholar] [CrossRef]
- Konishi, H.; Yamauchi, E.; Taniguchi, H.; Yamamoto, T.; Matsuzaki, H.; Takemura, Y.; Ohmae, K.; Kikkawa, U.; Nishizuka, Y. Phosphorylation sites of protein kinase C delta in H2O2-treated cells and its activation by tyrosine kinase in vitro. Proc. Natl. Acad. Sci. USA 2001, 98, 6587–6592. [Google Scholar] [CrossRef] [PubMed]
- Rybin, V.O.; Guo, J.; Sabri, A.; Elouardighi, H.; Schaefer, E.; Steinberg, S.F. Stimulus-specific differences in protein kinase C delta localization and activation mechanisms in cardiomyocytes. J. Biol. Chem. 2004, 279, 19350–19361. [Google Scholar] [CrossRef] [PubMed]
- Connor, K.M.; Subbaram, S.; Regan, K.J.; Nelson, K.K.; Mazurkiewicz, J.E.; Bartholomew, P.J.; Aplin, A.E.; Tai, Y.T.; Aguirre-Ghiso, J.; Flores, S.C.; et al. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J. Biol. Chem. 2005, 280, 16916–16924. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Zhang, G.X.; Nishiyama, A.; Shokoji, T.; Yao, L.; Fan, Y.Y.; Rahman, M.; Abe, Y. Mitochondria-derived reactive oxygen species and vascular MAP kinases: Comparison of angiotensin II and diazoxide. Hypertension 2005, 45, 438–444. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source/Event | Physiological Target/Function | Ref. | Source/Event | Pathology | Ref. |
|---|---|---|---|---|---|
| MitoROS = redox signaling/hypoxia | PHD/HIF-mediated transcriptome reprogramming | [19] [20] [21] | MitoROS - PHD-HIF - Warburg phenotype Oncogenes 2hydroxyglutarate- altered epigenetics | Cancer | [2,8,22] [23,24] [25] |
| RET, KATP opening => mito ROS | NOX2 in endothelial cells | [26] | RET, frequent KATP opening => mito ROS | Endothelial cell OX.STRESS Hypertension | [26] |
| MitoROS | NOX4 in pulmonary endothelial and recruited immune cells, fibroblasts | [6] [27] | MitoROS - PHD-HIF - Warburg phenotype | Pulmonary arthery remodelling Pulmonary hypertension | [6] [27] |
| MitoROS = redox signaling | Plasma membrane KATP closure => insulin release | [28] | Impaired Mito redox signaling OX.STRESS in pancreatic β-cells | Type 2 diabetes | [29] |
| MitoROS= redox signaling, skeletal muscle at excercise | PGC1α, skeletal muscle rejuvenation | [30] [31] | Impaired Mito redox signaling OX.STRESS, sedentary life-style | skeletal muscle senescence, weakness, athrophy | [32] [33,34] |
| Succinate accumulation => RET | Hypoxia/reperfusion Indry (heart) | [35] | |||
| Succinate accumulation => RET => intramitochondrial redox signaling | UCP1 in brown adipose tissue/thermogenesis | [14,36] | |||
| MitoROS = redox signaling in T cells | NFAT, NFκB/ Proximal T cell receptor signaling | [37] [38] | |||
| MitoROS = redox signaling, immune cells | NLRP3 inflammasome/IL-1β secretion | [39] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ježek, P.; Holendová, B.; Plecitá-Hlavatá, L. Redox Signaling from Mitochondria: Signal Propagation and Its Targets. Biomolecules 2020, 10, 93. https://doi.org/10.3390/biom10010093
Ježek P, Holendová B, Plecitá-Hlavatá L. Redox Signaling from Mitochondria: Signal Propagation and Its Targets. Biomolecules. 2020; 10(1):93. https://doi.org/10.3390/biom10010093
Chicago/Turabian StyleJežek, Petr, Blanka Holendová, and Lydie Plecitá-Hlavatá. 2020. "Redox Signaling from Mitochondria: Signal Propagation and Its Targets" Biomolecules 10, no. 1: 93. https://doi.org/10.3390/biom10010093
APA StyleJežek, P., Holendová, B., & Plecitá-Hlavatá, L. (2020). Redox Signaling from Mitochondria: Signal Propagation and Its Targets. Biomolecules, 10(1), 93. https://doi.org/10.3390/biom10010093