Combination Therapy with Vitamin C Could Eradicate Cancer Stem Cells



Abstract
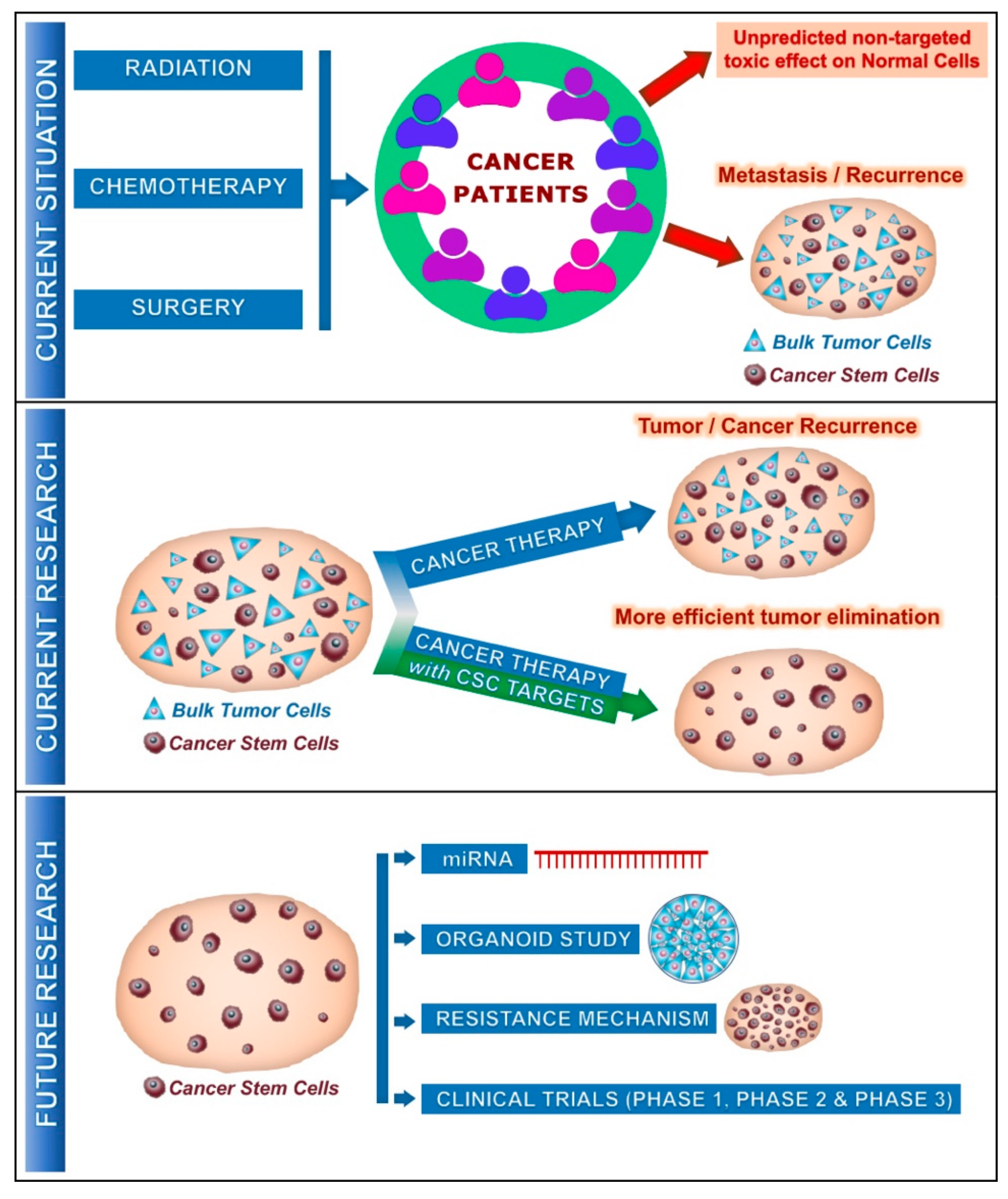
1. Introduction
2. Cancer Stem Cell Metabolism
3. Vitamin C/Ascorbic Acid—Metabolism and Chemistry of Vitamin C
4. Transporters of Vit.C
5. Evidence for Vit.C Effect in Cancer Treatment
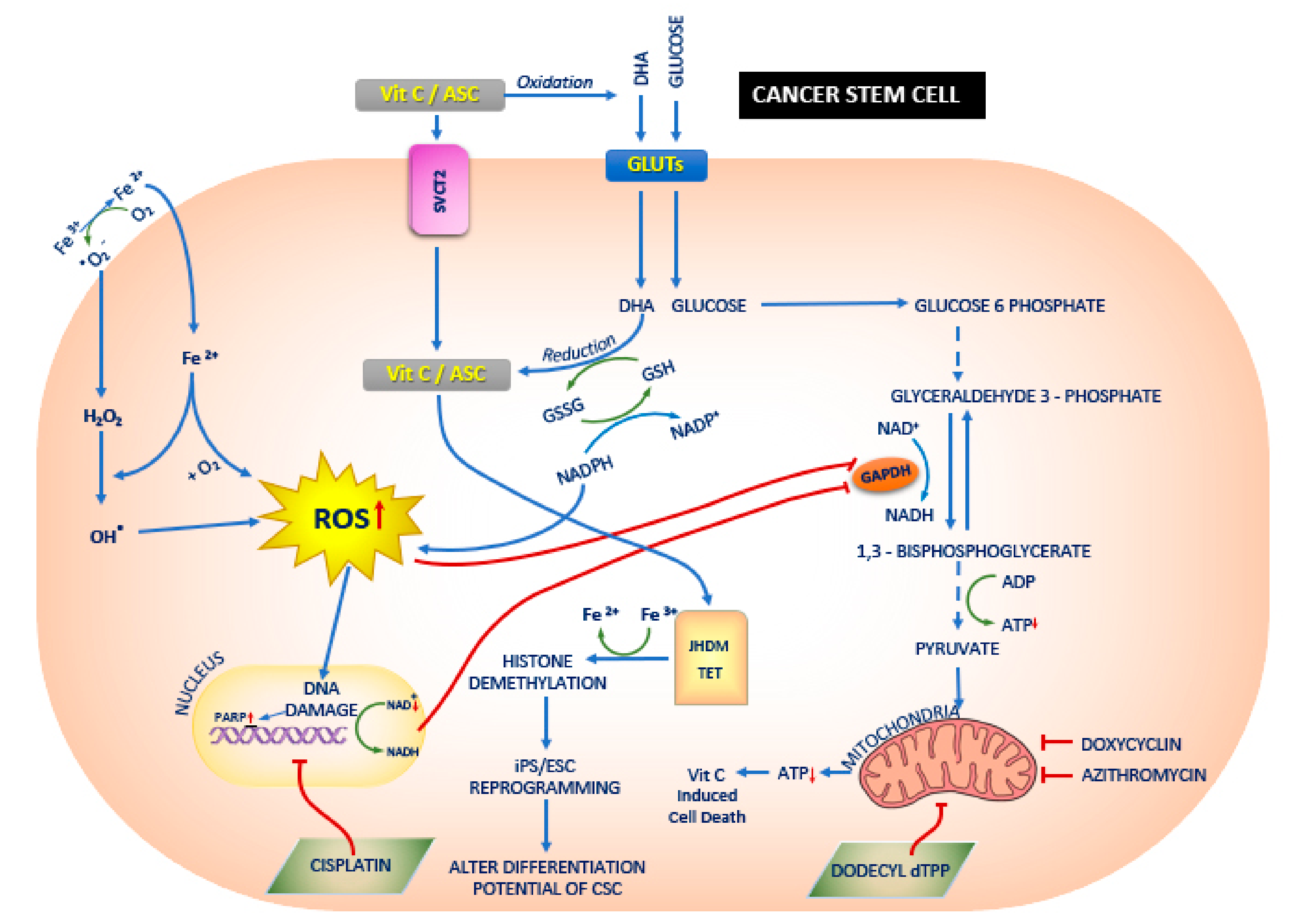
6. Vit.C and Its Anticancer Mechanism
6.1. Redox Imbalance
6.1.1. Increased Levels of Labile Metals Like Iron
6.1.2. DHA Uptake Instead of Glucose
6.2. Ten Eleven Translocation (TET)—Cancer and Effect of Vit.C (Targeting Epigenetic Regulators)
6.3. Hypoxia-Inducible Factor (HIF)—Cancer and Effect of Vit.C
7. Synergetic Effect of Vit.C on Energy Metabolism in Cancer Stem Cells
8. Role of Vit.C in Cancer Epigenome Regulation
9. Future Studies on Vit.C on Cancer Stem Cells
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| OH | Hydroxyl radical |
| α-KGDDs | alpha-ketoglutarate dependent dioxygenases |
| 5-caC | 5-carboxycytosine |
| 5-fC | 5-formylcytosine |
| 5-hmC | 5-hydroxymethylcytosine |
| 5-mC | 5-methylcytosine |
| AKT | Protein kinase B |
| ALDH | Aldehyde dehydrogenase |
| BRAF | B-Raf proto-oncogene, serine/threonine kinase |
| CD | Cluster of differentiation |
| CSCs | Cancer stem cells |
| Cu2+ | Cupric ion |
| CXCR4 | C-X-C chemokine receptor type 4 |
| d-TPP | Dodecyl—tri-phenyl-phosphonium |
| DHA | Dehydroascorbic acid |
| DNMT | DNA methyltransferase |
| EMT | Epithelial-mesenchymal transition |
| ESCs | Embryonic stem cells |
| Fe2+ | Ferrous ion |
| GAPDH | Glyceraldehyde-phosphate dehydrogenase |
| GLUT | Glucose transporter |
| H2O2 | Hydrogen peroxide |
| HCC | Hepatocellular carcinoma |
| HIF1α | Hypoxia-inducible factor 1 alpha |
| IC-50 | Inhibitory concentration |
| IDH | Isocitrate dehydrogenase |
| iPS | Induced pluripotent stem cells |
| JAK | Janus kinase |
| JHDM | Jumonji-C domain-containing histone demethylases |
| KRAS | KRAS proto-oncogene, GTPase |
| LGR5 | Leucine-rich repeat-containing G-protein coupled receptor 5 |
| miRNA | MicroRNA |
| mTOR | Mammalian target of rapamycin |
| NAD+ | Nicotinamide adenine dinucleotide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| PARP | Poly (ADP-ribose) polymerase |
| PBMC | Peripheral blood mononuclear cells |
| PI3K | Phosphatidylinositol-4,5-bisphosphate 3-kinase |
| ROS | Reactive oxygen species |
| SCVT1/2 | Sodium dependent Vit.C transporter 1/2 |
| STAT | Signal transducer and activator of transcription |
| TET | Ten eleven translocation |
| TPP | Tri-phenyl-phosphonium |
| Vit.C | Vitamin C |
References
- Ghosh, D.; Venkataramani, P.; Nandi, S.; Bhattacharjee, S. CRISPR-Cas9 a boon or bane: The bumpy road ahead to cancer therapeutics. Cancer Cell Int. 2019, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Fu, L. Targeting cancer stem cells: A new therapy to cure cancer patients. Am. J. Cancer Res. 2012, 2, 340–356. [Google Scholar] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, G.; Amiji, M.M. Cancer stem cell-targeted therapeutics and delivery strategies. Expert Opin. Drug Deliv. 2017, 14, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C.; Kim, J.H. Cancer stem cell metabolism: Target for cancer therapy. BMB Rep. 2018, 51, 319–326. [Google Scholar] [CrossRef]
- Peiris-Pages, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Jones, R.J.; Matsui, W.H.; Smith, B.D. Cancer stem cells: Are we missing the target? J. Natl. Cancer Inst. 2004, 96, 583–585. [Google Scholar] [CrossRef]
- Carr, A.C.; Cook, J. Intravenous Vitamin C for Cancer Therapy—Identifying the Current Gaps in Our Knowledge. Front. Physiol. 2018, 9, 1182. [Google Scholar] [CrossRef]
- Liskova, A.; Kubatka, P.; Samec, M.; Zubor, P.; Mlyncek, M.; Bielik, T.; Samuel, S.M.; Zulli, A.; Kwon, T.K.; Busselberg, D. Dietary Phytochemicals Targeting Cancer Stem Cells. Molecules 2019, 24, 899. [Google Scholar] [CrossRef]
- Rafalski, V.A.; Mancini, E.; Brunet, A. Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J. Cell Sci. 2012, 125, 5597–5608. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.Q.; Ahmed, E.I.; Elareer, N.R.; Junejo, K.; Steinhoff, M.; Uddin, S. Role of miRNA-Regulated Cancer Stem Cells in the Pathogenesis of Human Malignancies. Cells 2019, 8, 840. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.U.; Miyazaki, H.; Ochiya, T. The role of microRNAs in the regulation of cancer stem cells. Front. Genet. 2014, 4, 295. [Google Scholar] [CrossRef]
- Nechuta, S.; Lu, W.; Chen, Z.; Zheng, Y.; Gu, K.; Cai, H.; Zheng, W.; Shu, X.O. Vitamin supplement use during breast cancer treatment and survival: A prospective cohort study. Cancer Epidemiol. Biomark. Prev. 2011, 20, 262–271. [Google Scholar] [CrossRef]
- Hao, J.; Zhao, S.; Zhang, Y.; Zhao, Z.; Ye, R.; Wen, J.; Li, J. Emerging role of microRNAs in cancer and cancer stem cells. J. Cell. Biochem. 2014, 115, 605–610. [Google Scholar] [CrossRef]
- Croker, A.K.; Goodale, D.; Chu, J.; Postenka, C.; Hedley, B.D.; Hess, D.A.; Allan, A.L. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J. Cell. Mol. Med. 2009, 13, 2236–2252. [Google Scholar] [CrossRef]
- Garg, M. Emerging role of microRNAs in cancer stem cells: Implications in cancer therapy. World J. Stem Cells 2015, 7, 1078–1089. [Google Scholar] [CrossRef]
- Hwang-Verslues, W.W.; Chang, P.H.; Wei, P.C.; Yang, C.Y.; Huang, C.K.; Kuo, W.H.; Shew, J.Y.; Chang, K.J.; Lee, E.Y.; Lee, W.H. miR-495 is upregulated by E12/E47 in breast cancer stem cells, and promotes oncogenesis and hypoxia resistance via downregulation of E-cadherin and REDD1. Oncogene 2011, 30, 2463–2474. [Google Scholar] [CrossRef]
- Fiorillo, M.; Toth, F.; Sotgia, F.; Lisanti, M.P. Doxycycline, Azithromycin and Vitamin C (DAV): A potent combination therapy for targeting mitochondria and eradicating cancer stem cells (CSCs). Aging 2019, 11, 2202–2216. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Asadzadeh, Z.; Mansoori, B.; Mohammadi, A.; Aghajani, M.; Haji-Asgarzadeh, K.; Safarzadeh, E.; Mokhtarzadeh, A.; Duijf, P.H.G.; Baradaran, B. microRNAs in cancer stem cells: Biology, pathways, and therapeutic opportunities. J. Cell. Physiol. 2019, 234, 10002–10017. [Google Scholar] [CrossRef] [PubMed]
- Okuda, H.; Xing, F.; Pandey, P.R.; Sharma, S.; Watabe, M.; Pai, S.K.; Mo, Y.Y.; Iiizumi-Gairani, M.; Hirota, S.; Liu, Y.; et al. miR-7 suppresses brain metastasis of breast cancer stem-like cells by modulating KLF4. Cancer Res. 2013, 73, 1434–1444. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Xiao, G.G.; Mao, J.; Lu, Y.; Song, B.; Wang, L.; Fan, S.; Fan, P.; Hou, Z.; Li, J.; et al. Dysregulation of the miR-34a-SIRT1 axis inhibits breast cancer stemness. Oncotarget 2015, 6, 10432–10444. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, Y.; Tsuyada, A.; Ren, X.; Wu, X.; Stubblefield, K.; Rankin-Gee, E.K.; Wang, S.E. Transforming growth factor-beta regulates the sphere-initiating stem cell-like feature in breast cancer through miRNA-181 and ATM. Oncogene 2011, 30, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Ph 2 Trial of Vitamin C & G-FLIP (Low Doses Gemcitabine, 5FU, Leucovorin, Irinotecan, Oxaliplatin) for Pancreatic Cancer). Available online: https://clinicaltrials.gov/ct2/show/NCT01905150?term=vitamin+c&recrs=ade&cond=Pancreatic+Cancer&cntry=US&rank=1 (accessed on 2 October 2019).
- Lechner, A.; Leech, C.A.; Abraham, E.J.; Nolan, A.L.; Habener, J.F. Nestin-positive progenitor cells derived from adult human pancreatic islets of Langerhans contain side population (SP) cells defined by expression of the ABCG2 (BCRP1) ATP-binding cassette transporter. Biochem. Biophys. Res. Commun. 2002, 293, 670–674. [Google Scholar] [CrossRef]
- Hasegawa, S.; Eguchi, H.; Nagano, H.; Konno, M.; Tomimaru, Y.; Wada, H.; Hama, N.; Kawamoto, K.; Kobayashi, S.; Nishida, N.; et al. MicroRNA-1246 expression associated with CCNG2-mediated chemoresistance and stemness in pancreatic cancer. Br. J. Cancer 2014, 111, 1572–1580. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Trial of Ascorbic Acid (AA) + Nanoparticle Paclitaxel Protein Bound + Cisplatin + Gemcitabine (AA NABPLAGEM) (AA NABPLAGEM). Available online: https://clinicaltrials.gov/ct2/show/NCT03410030?term=vitamin+c&recrs=ade&cond=Pancreatic+Cancer&cntry=US&phase=123&rank=2 (accessed on 2 October 2019).
- Tomuleasa, C.; Mosteanu, O.; Susman, S.; Cristea, V. ALDH as a tumor marker for pancreatic cancer. J. Gastrointest. Liver Dis. 2011, 20, 443–444, author reply 444. [Google Scholar]
- Bao, B.; Ali, S.; Ahmad, A.; Azmi, A.S.; Li, Y.; Banerjee, S.; Kong, D.; Sethi, S.; Aboukameel, A.; Padhye, S.B.; et al. Hypoxia-induced aggressiveness of pancreatic cancer cells is due to increased expression of VEGF, IL-6 and miR-21, which can be attenuated by CDF treatment. PLoS ONE 2012, 7, e50165. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. High Dose Vitamin C Intravenous Infusion in Patients with Resectable or Metastatic Solid Tumor Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT03146962?term=vitamin+c&recrs=ade&cond=Pancreatic+Cancer&cntry=US&phase=123&rank=3 (accessed on 2 October 2019).
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Sureban, S.M.; May, R.; Qu, D.; Weygant, N.; Chandrakesan, P.; Ali, N.; Lightfoot, S.A.; Pantazis, P.; Rao, C.V.; Postier, R.G.; et al. DCLK1 regulates pluripotency and angiogenic factors via microRNA-dependent mechanisms in pancreatic cancer. PLoS ONE 2013, 8, e73940. [Google Scholar] [CrossRef]
- Immervoll, H.; Hoem, D.; Sakariassen, P.O.; Steffensen, O.J.; Molven, A. Expression of the “stem cell marker” CD133 in pancreas and pancreatic ductal adenocarcinomas. BMC Cancer 2008, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Lu, J.; Li, X.; Zhu, H.; Fan, X.; Zhu, S.; Wang, Y.; Guo, Q.; Wang, L.; Huang, Y.; et al. MiR-200a inhibits epithelial-mesenchymal transition of pancreatic cancer stem cell. BMC Cancer 2014, 14, 85. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, D.; Campbell, N.R.; Karikari, C.; Chivukula, R.; Kent, O.A.; Mendell, J.T.; Maitra, A. Restitution of tumor suppressor microRNAs using a systemic nanovector inhibits pancreatic cancer growth in mice. Mol. Cancer Ther. 2011, 10, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Corney, D.C.; Flesken-Nikitin, A.; Godwin, A.K.; Wang, W.; Nikitin, A.Y. MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res. 2007, 67, 8433–8438. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Treatment of Newly Diagnosed Ovarian Cancer with Antioxidants. Available online: https://clinicaltrials.gov/ct2/show/NCT00228319?term=vitamin+c&recrs=ade&cond=Ovarian+cancer&cntry=US&phase=123&rank=1 (accessed on 2 October 2019).
- Dou, J.; Jiang, C.; Wang, J.; Zhang, X.; Zhao, F.; Hu, W.; He, X.; Li, X.; Zou, D.; Gu, N. Using ABCG2-molecule-expressing side population cells to identify cancer stem-like cells in a human ovarian cell line. Cell Biol. Int. 2011, 35, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.M.; Shaw, P.A.; Gedye, C.; Bernardini, M.Q.; Neel, B.G.; Ailles, L.E. Phenotypic heterogeneity and instability of human ovarian tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6468–6473. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Phase 2 Trial of High-Dose Ascorbate in Glioblastoma Multiforme. Available online: https://clinicaltrials.gov/ct2/show/NCT02344355?term=vitamin+c&recrs=ade&cond=Glioblastoma&cntry=US&phase=123&rank=1 (accessed on 2 October 2019).
- Cui, S.Y.; Wang, R.; Chen, L.B. MicroRNA-145: A potent tumour suppressor that regulates multiple cellular pathways. J. Cell. Mol. Med. 2014, 18, 1913–1926. [Google Scholar] [CrossRef]
- Shang, C.; Guo, Y.; Hong, Y.; Liu, Y.H.; Xue, Y.X. MiR-21 up-regulation mediates glioblastoma cancer stem cells apoptosis and proliferation by targeting FASLG. Mol. Biol. Rep. 2015, 42, 721–727. [Google Scholar] [CrossRef]
- Turchi, L.; Debruyne, D.N.; Almairac, F.; Virolle, V.; Fareh, M.; Neirijnck, Y.; Burel-Vandenbos, F.; Paquis, P.; Junier, M.P.; Van Obberghen-Schilling, E.; et al. Tumorigenic potential of miR-18A* in glioma initiating cells requires NOTCH-1 signaling. Stem Cells 2013, 31, 1252–1265. [Google Scholar] [CrossRef]
- Ying, Z.; Li, Y.; Wu, J.; Zhu, X.; Yang, Y.; Tian, H.; Li, W.; Hu, B.; Cheng, S.Y.; Li, M. Loss of miR-204 expression enhances glioma migration and stem cell-like phenotype. Cancer Res. 2013, 73, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Williams, S.; Otsuki, A.; Nuovo, G.; Raychaudhury, A.; Newton, H.B.; Chiocca, E.A.; Lawler, S. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008, 68, 9125–9130. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Luo, H.; Pu, Y.; Zhou, Z.; Wu, X.; Xu, W.; Yang, Z. Methylation mediated silencing of miR-23b expression and its role in glioma stem cells. Neurosci. Lett. 2012, 528, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Summer, R.; Kotton, D.N.; Sun, X.; Ma, B.; Fitzsimmons, K.; Fine, A. Side population cells and Bcrp1 expression in lung. Am. J. Physiol Lung Cell. Mol. Physiol. 2003, 285, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Qiu, M.; Jiang, F.; Zhang, S.; Yang, X.; Wang, J.; Xu, L.; Yin, R. MiR-145 regulates cancer stem-like properties and epithelial-to-mesenchymal transition in lung adenocarcinoma-initiating cells. Tumour Biol. 2014, 35, 8953–8961. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Pharmacological Ascorbate for Lung Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02420314?term=vitamin+c&recrs=ade&cond=lung+cancer&cntry=US&phase=123&rank=1 (accessed on 2 October 2019).
- Jiang, F.; Qiu, Q.; Khanna, A.; Todd, N.W.; Deepak, J.; Xing, L.; Wang, H.; Liu, Z.; Su, Y.; Stass, S.A.; et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol. Cancer Res. 2009, 7, 330–338. [Google Scholar] [CrossRef]
- Xu, W.; Ji, J.; Xu, Y.; Liu, Y.; Shi, L.; Liu, Y.; Lu, X.; Zhao, Y.; Luo, F.; Wang, B.; et al. MicroRNA-191, by promoting the EMT and increasing CSC-like properties, is involved in neoplastic and metastatic properties of transformed human bronchial epithelial cells. Mol. Carcinog. 2015, 54, 148–161. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Phase 2 Study Adding Ascorbate to Chemotherapy and Radiation Therapy for NSCLC (XACT-LUNG). Available online: https://clinicaltrials.gov/ct2/show/NCT02905591?term=vitamin+c&recrs=ade&cond=lung+cancer&cntry=US&phase=123&rank=2 (accessed on 2 October 2019).
- Xi, S.; Xu, H.; Shan, J.; Tao, Y.; Hong, J.A.; Inchauste, S.; Zhang, M.; Kunst, T.F.; Mercedes, L.; Schrump, D.S. Cigarette smoke mediates epigenetic repression of miR-487b during pulmonary carcinogenesis. J. Clin. Investig. 2013, 123, 1241–1261. [Google Scholar] [CrossRef]
- Bertolini, G.; Roz, L.; Perego, P.; Tortoreto, M.; Fontanella, E.; Gatti, L.; Pratesi, G.; Fabbri, A.; Andriani, F.; Tinelli, S.; et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 16281–16286. [Google Scholar] [CrossRef] [PubMed]
- King, C.E.; Cuatrecasas, M.; Castells, A.; Sepulveda, A.R.; Lee, J.S.; Rustgi, A.K. LIN28B promotes colon cancer progression and metastasis. Cancer Res. 2011, 71, 4260–4268. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; Appelman, H.; Fields, J.Z.; Wicha, M.S.; Boman, B.M. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009, 69, 3382–3389. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Jaksch, M.; Munera, J.; Bajpai, R.; Terskikh, A.; Oshima, R.G. Cell cycle-dependent variation of a CD133 epitope in human embryonic stem cell, colon cancer, and melanoma cell lines. Cancer Res. 2008, 68, 7882–7886. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. TET2 Mutations in Myelodysplastic Syndromes and Acute Myeloid Leukemia with Azacitidine + Ascorbic Acid. Available online: https://clinicaltrials.gov/ct2/show/NCT03397173?term=vitamin+c&recrs=ade&cond=Leukemia&cntry=US&phase=123&rank=1 (accessed on 2 October 2019).
- Scheibner, K.A.; Teaboldt, B.; Hauer, M.C.; Chen, X.; Cherukuri, S.; Guo, Y.; Kelley, S.M.; Liu, Z.; Baer, M.R.; Heimfeld, S.; et al. MiR-27a functions as a tumor suppressor in acute leukemia by regulating 14-3-3theta. PLoS ONE 2012, 7, e50895. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Therapeutic Use of Intravenous Vitamin C in Allogeneic Stem Cell Transplant Recipients. Available online: https://clinicaltrials.gov/ct2/show/NCT03613727?term=vitamin+c&recrs=ade&cond=Leukemia&cntry=US&phase=123&rank=2 (accessed on 2 October 2019).
- ClinicalTrials.gov. Ascorbic Acid and Combination Chemotherapy in Treating Patients with Relapsed or Refractory Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03418038?term=vitamin+c&recrs=ade&cond=Lymphoma&cntry=US&phase=123&rank=1 (accessed on 2 October 2019).
- Ma, S.; Tang, K.H.; Chan, Y.P.; Lee, T.K.; Kwan, P.S.; Castilho, A.; Ng, I.; Man, K.; Wong, N.; To, K.F.; et al. miR-130b Promotes CD133+ liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear protein 1. Cell Stem Cell 2010, 7, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.C.; Park, C.Y.; Bhagat, G.; Zhang, J.; Wang, Y.; Fan, J.B.; Liu, M.; Zou, Y.; Weissman, I.L.; Gu, H. microRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. J. Exp. Med. 2010, 207, 475–489. [Google Scholar] [CrossRef]
- Babashah, S.; Sadeghizadeh, M.; Hajifathali, A.; Tavirani, M.R.; Zomorod, M.S.; Ghadiani, M.; Soleimani, M. Targeting of the signal transducer Smo links microRNA-326 to the oncogenic Hedgehog pathway in CD34+ CML stem/progenitor cells. Int. J. Cancer 2013, 133, 579–589. [Google Scholar] [CrossRef]
- Morris, V.A.; Zhang, A.; Yang, T.; Stirewalt, D.L.; Ramamurthy, R.; Meshinchi, S.; Oehler, V.G. MicroRNA-150 expression induces myeloid differentiation of human acute leukemia cells and normal hematopoietic progenitors. PLoS ONE 2013, 8, e75815. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Docetaxel with or without Ascorbic Acid in Treating Patients with Metastatic Prostate Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02516670?term=vitamin+c&recrs=ade&cond=Prostate+cancer&cntry=US&phase=123&rank=1 (accessed on 2 October 2019).
- Hellsten, R.; Johansson, M.; Dahlman, A.; Sterner, O.; Bjartell, A. Galiellalactone inhibits stem cell-like ALDH-positive prostate cancer cells. PLoS ONE 2011, 6, e22118. [Google Scholar] [CrossRef]
- Chang, Y.L.; Zhou, P.J.; Wei, L.; Li, W.; Ji, Z.; Fang, Y.X.; Gao, W.Q. MicroRNA-7 inhibits the stemness of prostate cancer stem-like cells and tumorigenesis by repressing KLF4/PI3K/Akt/p21 pathway. Oncotarget 2015, 6, 24017–24031. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Vander Griend, D.J.; Karthaus, W.L.; Dalrymple, S.; Meeker, A.; DeMarzo, A.M.; Isaacs, J.T. The role of CD133 in normal human prostate stem cells and malignant cancer-initiating cells. Cancer Res. 2008, 68, 9703–9711. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, K.; Okada, M. Retarded nuclear migration in Drosophila embryos with aberrant F-actin reorganization caused by maternal mutations and by cytochalasin treatment. Development 1991, 111, 909–920. [Google Scholar]
- Padayatty, S.J.; Levine, M. Vitamin C: The known and the unknown and Goldilocks. Oral Dis. 2016, 22, 463–493. [Google Scholar] [CrossRef] [PubMed]
- Svirbely, J.L.; Szent-Gyorgyi, A. The chemical nature of vitamin C. Biochem. J. 1932, 26, 865–870. [Google Scholar] [CrossRef]
- King, C.G.; Waugh, W.A. The Chemical Nature of Vitamin C. Science 1932, 75, 357–358. [Google Scholar] [CrossRef]
- Parrow, N.L.; Leshin, J.A.; Levine, M. Parenteral ascorbate as a cancer therapeutic: A reassessment based on pharmacokinetics. Antioxid. Redox Signal. 2013, 19, 2141–2156. [Google Scholar] [CrossRef]
- Burzle, M.; Hediger, M.A. Functional and physiological role of vitamin C transporters. Curr. Top. Membr. 2012, 70, 357–375. [Google Scholar] [CrossRef]
- Englard, S.; Seifter, S. The biochemical functions of ascorbic acid. Annu. Rev. Nutr. 1986, 6, 365–406. [Google Scholar] [CrossRef]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature 1999, 399, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dutta, B.; Huang, W.; Devoe, L.D.; Leibach, F.H.; Ganapathy, V.; Prasad, P.D. Human Na+-dependent vitamin C transporter 1 (hSVCT1): Primary structure, functional characteristics and evidence for a non-functional splice variant. Biochim. Biophys. Acta Biomembr. 1999, 1461, 1–9. [Google Scholar] [CrossRef]
- Daruwala, R.; Song, J.; Koh, W.S.; Rumsey, S.C.; Levine, M. Cloning and functional characterization of the human sodium-dependent vitamin C transporters hSVCT1 and hSVCT2. FEBS Lett. 1999, 460, 480–484. [Google Scholar] [CrossRef]
- Wang, Y.; Mackenzie, B.; Tsukaguchi, H.; Weremowicz, S.; Morton, C.C.; Hediger, M.A. Human vitamin C (L-ascorbic acid) transporter SVCT1. Biochem. Biophys. Res. Commun. 2000, 267, 488–494. [Google Scholar] [CrossRef]
- Corpe, C.P.; Eck, P.; Wang, J.; Al-Hasani, H.; Levine, M. Intestinal dehydroascorbic acid (DHA) transport mediated by the facilitative sugar transporters, GLUT2 and GLUT8. J. Biol. Chem. 2013, 288, 9092–9101. [Google Scholar] [CrossRef]
- Rumsey, S.C.; Kwon, O.; Xu, G.W.; Burant, C.F.; Simpson, I.; Levine, M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J. Biol. Chem. 1997, 272, 18982–18989. [Google Scholar] [CrossRef]
- Rumsey, S.C.; Daruwala, R.; Al-Hasani, H.; Zarnowski, M.J.; Simpson, I.A.; Levine, M. Dehydroascorbic acid transport by GLUT4 in Xenopus oocytes and isolated rat adipocytes. J. Biol. Chem. 2000, 275, 28246–28253. [Google Scholar] [CrossRef]
- Vera, J.C.; Rivas, C.I.; Fischbarg, J.; Golde, D.W. Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature 1993, 364, 79–82. [Google Scholar] [CrossRef]
- Tu, H.; Li, H.; Wang, Y.; Niyyati, M.; Wang, Y.; Leshin, J.; Levine, M. Low Red Blood Cell Vitamin C Concentrations Induce Red Blood Cell Fragility: A Link to Diabetes Via Glucose, Glucose Transporters, and Dehydroascorbic Acid. EBioMedicine 2015, 2, 1735–1750. [Google Scholar] [CrossRef]
- Gillberg, L.; Orskov, A.D.; Liu, M.; Harslof, L.B.S.; Jones, P.A.; Gronbaek, K. Vitamin C—A new player in regulation of the cancer epigenome. Semin. Cancer Biol. 2018, 51, 59–67. [Google Scholar] [CrossRef]
- Mayland, C.R.; Bennett, M.I.; Allan, K. Vitamin C deficiency in cancer patients. Palliat. Med. 2005, 19, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Klimant, E.; Wright, H.; Rubin, D.; Seely, D.; Markman, M. Intravenous vitamin C in the supportive care of cancer patients: A review and rational approach. Curr. Oncol. 2018, 25, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Kagohara, L.T.; Stein-O’Brien, G.L.; Kelley, D.; Flam, E.; Wick, H.C.; Danilova, L.V.; Easwaran, H.; Favorov, A.V.; Qian, J.; Gaykalova, D.A.; et al. Epigenetic regulation of gene expression in cancer: Techniques, resources and analysis. Brief. Funct. Genom. 2018, 17, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.H.; Chen, S.; Corpe, C.; Dutta, A.; Dutta, S.K.; et al. Vitamin C as an antioxidant: Evaluation of its role in disease prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef]
- Wilson, M.K.; Baguley, B.C.; Wall, C.; Jameson, M.B.; Findlay, M.P. Review of high-dose intravenous vitamin C as an anticancer agent. Asia Pac. J. Clin. Oncol. 2014, 10, 22–37. [Google Scholar] [CrossRef]
- Lee, W.J. The prospects of vitamin C in cancer therapy. Immune Netw. 2009, 9, 147–152. [Google Scholar] [CrossRef]
- Lv, H.; Wang, C.; Fang, T.; Li, T.; Lv, G.; Han, Q.; Yang, W.; Wang, H. Vitamin C preferentially kills cancer stem cells in hepatocellular carcinoma via SVCT-2. NPJ Precis. Oncol. 2018, 2, 1. [Google Scholar] [CrossRef]
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 2019, 19, 271–282. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, 453–462. [Google Scholar] [CrossRef]
- Chio, I.I.C.; Tuveson, D.A. ROS in Cancer: The Burning Question. Trends Mol. Med. 2017, 23, 411–429. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Omenn, G.S.; Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Glass, A.; Keogh, J.P.; Meyskens, F.L.; Valanis, B.; Williams, J.H.; et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N. Engl. J. Med. 1996, 334, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative stress, prooxidants, and antioxidants: The interplay. BioMed Res. Int. 2014, 2014, 761264. [Google Scholar] [CrossRef] [PubMed]
- Wondrak, G.T. Redox-directed cancer therapeutics: Molecular mechanisms and opportunities. Antioxid. Redox Signal. 2009, 11, 3013–3069. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Rychtarcikova, Z.; Lettlova, S.; Tomkova, V.; Korenkova, V.; Langerova, L.; Simonova, E.; Zjablovskaja, P.; Alberich-Jorda, M.; Neuzil, J.; Truksa, J. Tumor-initiating cells of breast and prostate origin show alterations in the expression of genes related to iron metabolism. Oncotarget 2017, 8, 6376–6398. [Google Scholar] [CrossRef]
- Kiessling, M.K.; Klemke, C.D.; Kaminski, M.M.; Galani, I.E.; Krammer, P.H.; Gulow, K. Inhibition of constitutively activated nuclear factor-kappaB induces reactive oxygen species- and iron-dependent cell death in cutaneous T-cell lymphoma. Cancer Res. 2009, 69, 2365–2374. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. Correction to: ‘The Warburg Effect: How Does it Benefit Cancer Cells?’: [Trends in Biochemical Sciences, 41 (2016) 211]. Trends Biochem. Sci. 2016, 41, 287. [Google Scholar] [CrossRef]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Nandi, S. Rare Genetic Diseases with Defects in DNA Repair: Opportunities and Challenges in Orphan Drug Development for Targeted Cancer Therapy. Cancers 2018, 10, 298. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. Synthetic lethality in DNA repair network: A novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life 2017, 69, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Melamed, P.; Yosefzon, Y.; David, C.; Tsukerman, A.; Pnueli, L. Tet Enzymes, Variants, and Differential Effects on Function. Front. Cell Dev. Biol. 2018, 6, 22. [Google Scholar] [CrossRef]
- Mastrangelo, D.; Pelosi, E.; Castelli, G.; Lo-Coco, F.; Testa, U. Mechanisms of anti-cancer effects of ascorbate: Cytotoxic activity and epigenetic modulation. Blood Cells Mol. Dis. 2018, 69, 57–64. [Google Scholar] [CrossRef]
- Bonuccelli, G.; De Francesco, E.M.; de Boer, R.; Tanowitz, H.B.; Lisanti, M.P. NADH autofluorescence, a new metabolic biomarker for cancer stem cells: Identification of Vitamin C and CAPE as natural products targeting “stemness”. Oncotarget 2017, 8, 20667–20678. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef]
- Hore, T.A.; von Meyenn, F.; Ravichandran, M.; Bachman, M.; Ficz, G.; Oxley, D.; Santos, F.; Balasubramanian, S.; Jurkowski, T.P.; Reik, W. Retinol and ascorbate drive erasure of epigenetic memory and enhance reprogramming to naive pluripotency by complementary mechanisms. Proc. Natl. Acad. Sci. USA 2016, 113, 12202–12207. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Bonuccelli, G.; Maggiolini, M.; Sotgia, F.; Lisanti, M.P. Vitamin C and Doxycycline: A synthetic lethal combination therapy targeting metabolic flexibility in cancer stem cells (CSCs). Oncotarget 2017, 8, 67269–67286. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Ozsvari, B.; Sotgia, F.; Lisanti, M.P. Dodecyl-TPP Targets Mitochondria and Potently Eradicates Cancer Stem Cells (CSCs): Synergy With FDA-Approved Drugs and Natural Compounds (Vitamin C and Berberine). Front. Oncol. 2019, 9, 615. [Google Scholar] [CrossRef]
- An, J.; Rao, A.; Ko, M. TET family dioxygenases and DNA demethylation in stem cells and cancers. Exp. Mol. Med. 2017, 49, e323. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, L.; Abdel-Wahab, O.; Levine, R.L.; Aifantis, I. TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell 2011, 9, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Rao, A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet. 2014, 30, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; An, J.; Pastor, W.A.; Koralov, S.B.; Rajewsky, K.; Rao, A. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol. Rev. 2015, 263, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Guillamot, M.; Cimmino, L.; Aifantis, I. The Impact of DNA Methylation in Hematopoietic Malignancies. Trends Cancer 2016, 2, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.L.; Brena, R.M.; Kolle, G.; Grimmond, S.M.; Berman, B.P.; Laird, P.W.; Pera, M.F.; Wolvetang, E.J. Vitamin C promotes widespread yet specific DNA demethylation of the epigenome in human embryonic stem cells. Stem Cells 2010, 28, 1848–1855. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Yin, R.; Mao, S.Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 2013, 135, 10396–10403. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- Gustafson, C.B.; Yang, C.; Dickson, K.M.; Shao, H.; Van Booven, D.; Harbour, J.W.; Liu, Z.J.; Wang, G. Epigenetic reprogramming of melanoma cells by vitamin C treatment. Clin. Epigenet. 2015, 7, 51. [Google Scholar] [CrossRef]
- Mustafi, S.; Sant, D.W.; Liu, Z.J.; Wang, G. Ascorbate induces apoptosis in melanoma cells by suppressing Clusterin expression. Sci. Rep. 2017, 7, 3671. [Google Scholar] [CrossRef] [PubMed]
- Knowles, H.J.; Raval, R.R.; Harris, A.L.; Ratcliffe, P.J. Effect of ascorbate on the activity of hypoxia-inducible factor in cancer cells. Cancer Res. 2003, 63, 1764–1768. [Google Scholar] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, C.; Vissers, M.C. Ascorbate as a co-factor for Fe- and 2-Oxoglutarate dependent dioxygenases: Physiological activity in tumor growth and progression. Front. Oncol. 2014, 4, 359. [Google Scholar] [CrossRef]
- Lee Chong, T.; Ahearn, E.L.; Cimmino, L. Reprogramming the Epigenome With Vitamin C. Front. Cell Dev. Biol. 2019, 7, 128. [Google Scholar] [CrossRef]
- Wang, F.; Li, Y.C.; Liu, L.P.; Zhang, H.M.; Tong, S. Circulating Tumor Cells and Tumor Stem Cells Detection in the Peripheral Blood Mononuclear Cells of Breast Cancer. J. Clin. Lab. Anal. 2016, 30, 616–622. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Type | In Vitro/In Vivo | Status/Month Year | Drugs | Reference | CSC Markers | Reference | miRNA in CSC | Reference |
|---|---|---|---|---|---|---|---|---|
| Breast Cancer | In vivo, Population based cohort | Completed/April 2006 | Chemotherapy, Radiation, Vit.C/E, Multivitamin | [15] | ALDH1 | [2,16,17] | miR-495 | [18,19] |
| In vitro, MCF-7 cells | Published/2019 | Doxycycline, Azithromycin, Vit.C | [20] | CD44 | [2,16,21] | miR-7 | [22,23] | |
| CD24 | [16] | miR-34a | [22,24] | |||||
| CD133 | [16] | miR-181 | [18,25] | |||||
| CD90 | [16] | |||||||
| α6-integrin | [16] | |||||||
| Hedgehog-Gli activity | [16] | |||||||
| Pancreatic Cancer | In vivo/Phase 2 (NCT01905150) | Completed/March 2019 | G-FLIP/G-FLIP-DM + Vit.C | [26] | ABCG2 | [2,16,27] | miR-1246 | [16,28] |
| In vivo, Phase 1/2 (NCT03410030) | Ongoing/July 2020 | Vit.C + Nanoparticle + Paclitaxel + Cisplatin + Gemcitabine | [29] | ALDH1 | [2,16,30] | miR-210 | [16,31] | |
| In vivo, Phase 2 (NCT03146962) | Ongoing/December 2021 | High dose Vit.C | [32] | CD24 | [16] | miR-21 | [16,31] | |
| CD44 | [2,16,33] | Let-7 | [16,34] | |||||
| CD133 | [2,16,35] | miR-200 family | [16,34] | |||||
| C-Met | [16] | miR-200a | [16,36] | |||||
| CXCR4 | [16] | miR-143/145 cluster | [16,37] | |||||
| Nestin | [16] | miR-145 | [16,34] | |||||
| Nodal-Activin | [16] | miR-34 family | [16,38] | |||||
| Ovarian Cancer | In vivo, Phase 1/2 (NCT00228319) | Completed/August 2007 | Paclitaxel + Carboplatin + Sodium Ascorbate + Vit.C, A & E | [39] | CD24 | [16] | ||
| CD44 | [2,16,40] | |||||||
| CD177 | [16] | |||||||
| CD133 | [2,16,41] | |||||||
| Glioma/Glioblastoma | In vivo, Phase 2 (NCT02344355) | Ongoing/December 2023 | Radiation + temozolomide + Vit.C | [42] | CD15 | [16] | miR-145 | [22,43] |
| CD90 | [16] | miR-21 | [22,44] | |||||
| CD133 | [16] | miR-18 | [18,45] | |||||
| Nestin | [16] | miR-204 | [18,46] | |||||
| α6-integrin | [16] | miR-128 | [18,47] | |||||
| miR-23b | [18,48] | |||||||
| Lung Cancer | In vivo, Phase 2 (NCT03146962) | Ongoing/December 2021 | High dose Vit.C | [32] | ABCG2 | [2,16,49] | miR-145 | [22,50] |
| In vivo, Phase 2 (NCT02420314) | Ongoing/December 2025 | Paclitaxel, Carboplatin + Vit.C | [51] | ALDH1 | [2,16,52] | miR-191 | [18,53] | |
| In vivo, Phase 2 (NCT02905591) | Ongoing/July 2026 | Radiation Therapy + Paclitaxel, Carboplatin + Vit.C | [54] | CD90 | [16] | miR-487b | [18,55] | |
| CD177 | [16] | |||||||
| CD133 | [2,16,56] | |||||||
| Colon Cancer | In vivo, Phase 2 (NCT03146962) | Ongoing/December 2021 | Vit.C | [32] | ABCB5 | [16] | Let-7 | [18,34,57] |
| ALDH1 | [2,16,58] | |||||||
| CD24 | [16] | |||||||
| CD26 | [16] | |||||||
| CD29 | [16] | |||||||
| CD44 | [2,16,59] | |||||||
| CD133 | [2,16,60] | |||||||
| CD166 | [16] | |||||||
| LGR5 | [16] | |||||||
| β-catenin activity | [16] | |||||||
| Leukemia | In vivo, Phase 2 (NCT03397173) | Ongoing/January 2020 | Azacitidine + Vit.C | [61] | miR-27a | [18,62] | ||
| In vivo, Phase 2 (NCT03613727) | Ongoing/October 2022 | Vit.C | [63] | |||||
| Lymphoma | In vivo, Phase 2 (NCT03418038) | Ongoing/March 2024 | Salvage Chemotherapy + Vit.C | [64] | ||||
| In vivo, Phase 2 (NCT03613727) | Ongoing/October 2022 | Vit.C | [63] | |||||
| Myeloid Leukemia | In vivo, Phase 2 (NCT03397173) | Ongoing/January 2020 | Azacitidine + Vit.C | [61] | miR-130b | [18,65] | ||
| In vivo, Phase 2 (NCT03613727) | Ongoing/October 2022 | Vit.C | [63] | miR-29a | [18,66] | |||
| miR-326 | [18,67] | |||||||
| miR-150 | [18,68] | |||||||
| Prostate Cancer | In vivo, Phase 2 (NCT02516670) | Ongoing/January 2030 | Docetaxel + Vit.C | [69] | ALDH1 | [2,16,70] | miR-7 | [22,71] |
| CD44 | [2,16,72] | miR-34a | [22,73] | |||||
| CD133 | [2,16,74] | |||||||
| CD166 | [16] | |||||||
| Trop2 | [16] | |||||||
| α2β1Integrin | [16] | |||||||
| α1Integrin | [16] | |||||||
| ABCG2 | [2,75] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Satheesh, N.J.; Samuel, S.M.; Büsselberg, D. Combination Therapy with Vitamin C Could Eradicate Cancer Stem Cells. Biomolecules 2020, 10, 79. https://doi.org/10.3390/biom10010079
Satheesh NJ, Samuel SM, Büsselberg D. Combination Therapy with Vitamin C Could Eradicate Cancer Stem Cells. Biomolecules. 2020; 10(1):79. https://doi.org/10.3390/biom10010079
Chicago/Turabian StyleSatheesh, Noothan Jyothi, Samson Mathews Samuel, and Dietrich Büsselberg. 2020. "Combination Therapy with Vitamin C Could Eradicate Cancer Stem Cells" Biomolecules 10, no. 1: 79. https://doi.org/10.3390/biom10010079
APA StyleSatheesh, N. J., Samuel, S. M., & Büsselberg, D. (2020). Combination Therapy with Vitamin C Could Eradicate Cancer Stem Cells. Biomolecules, 10(1), 79. https://doi.org/10.3390/biom10010079