Electron-Induced Chemistry in the Condensed Phase


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. The Ionization Potential
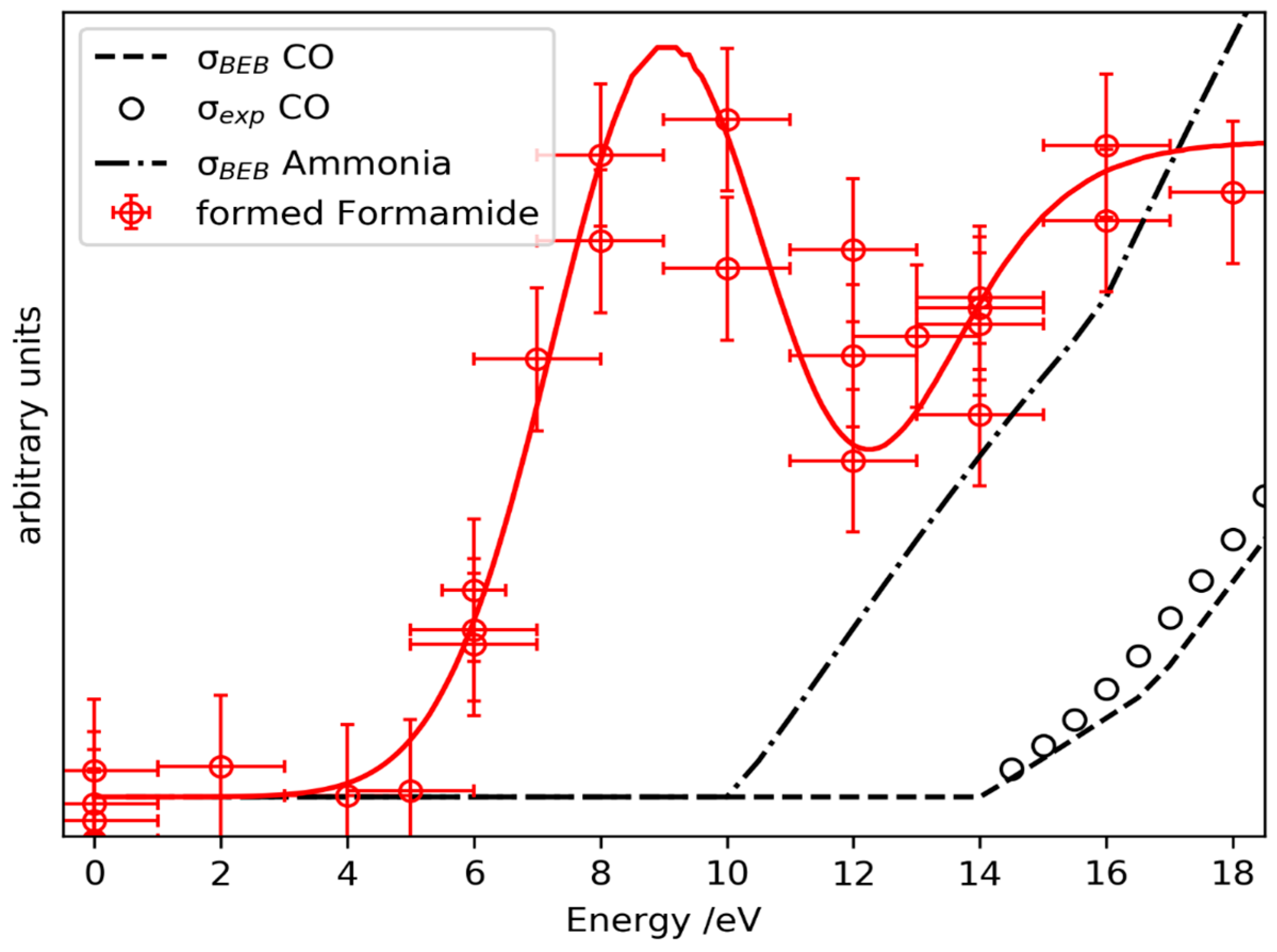
2.2. DEA Cross-Sections
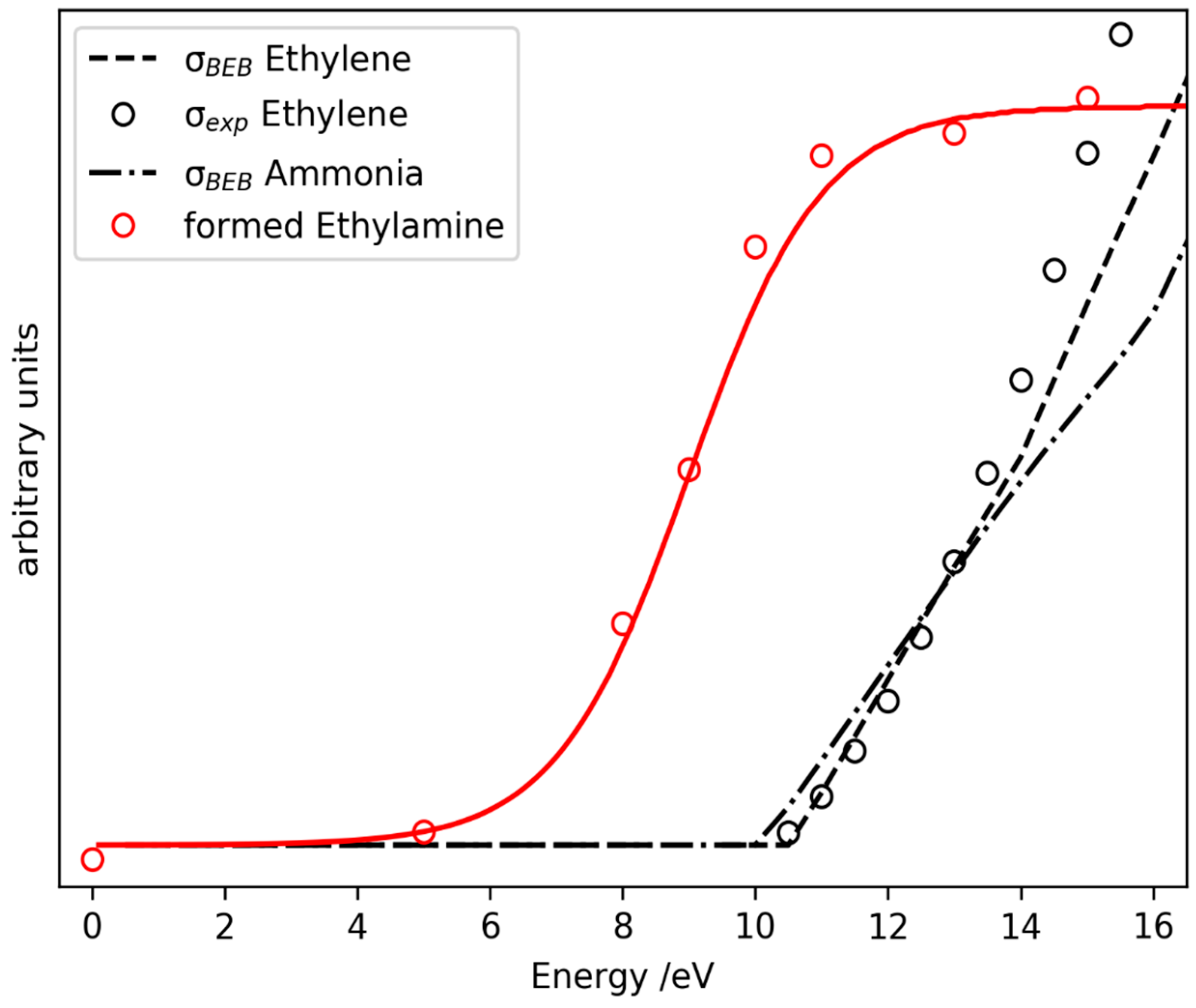
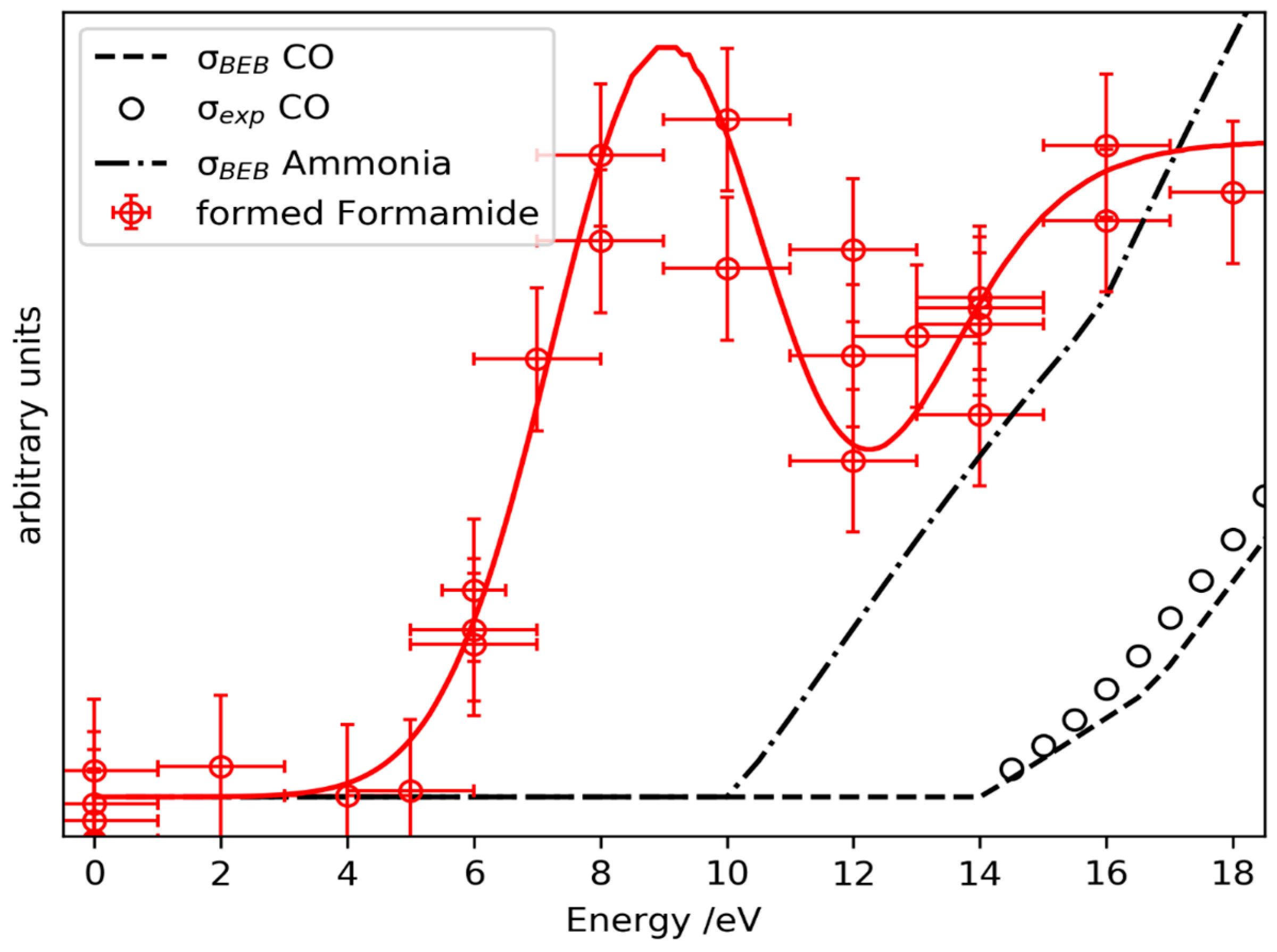
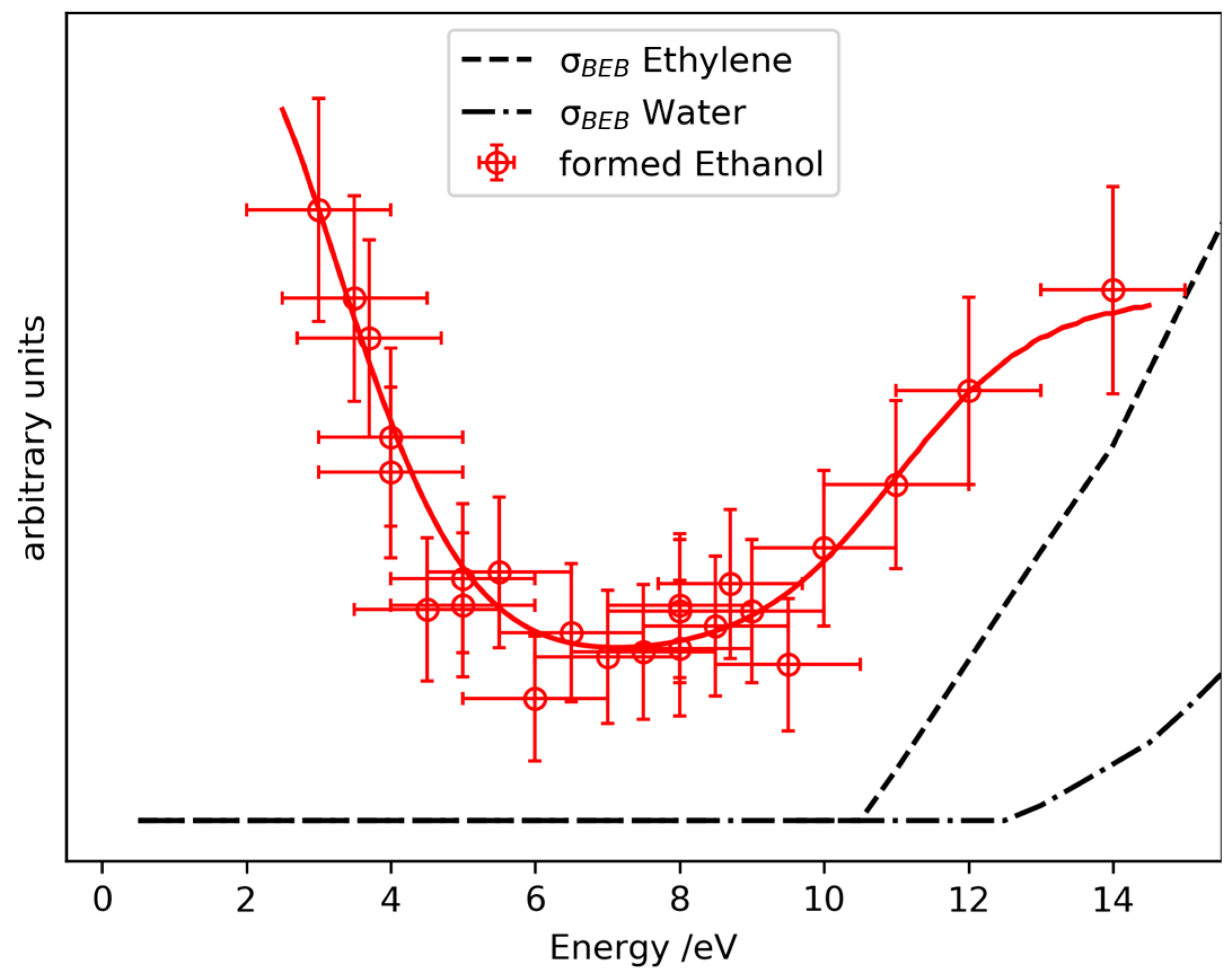
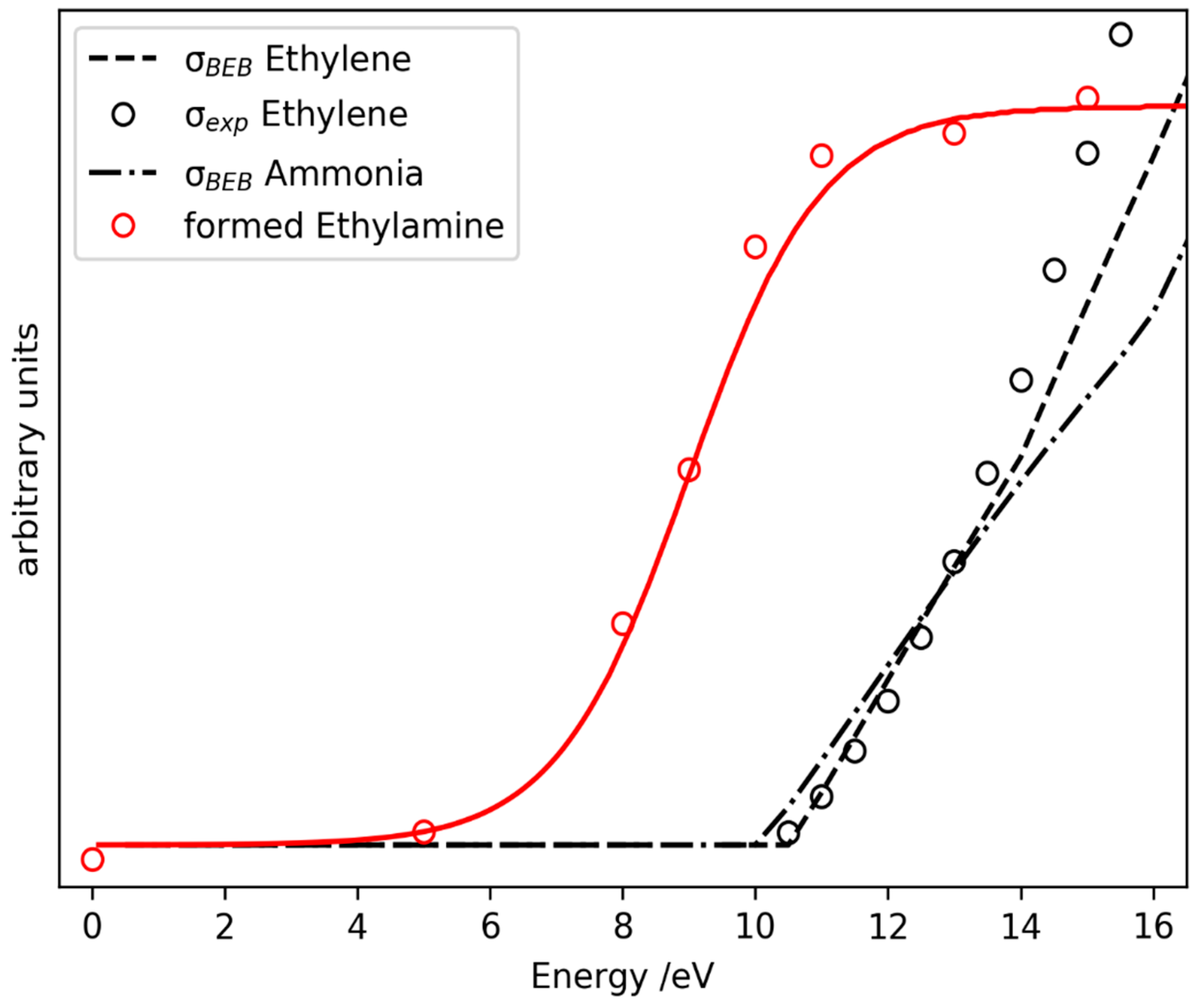
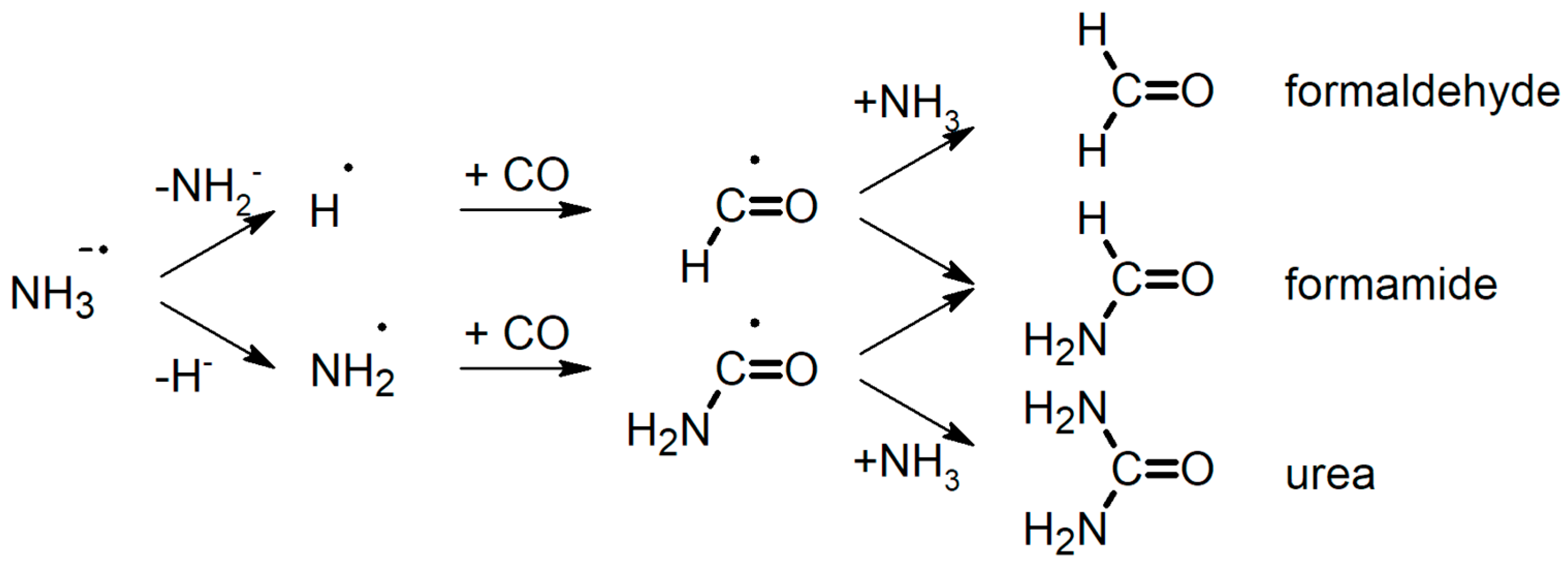
2.3. Prediction of Possible Reaction Routes
3. Conclusions
- must be corrected for the energy shift due to stabilization of ions in a matrix,
- should be taken with a great deal of caution with regards to absolute cross-sections and thus branching ratios, and
- are utterly unable to predict the formation of reactive species that relies on interaction between molecules as well as with electrons.
Funding
Conflicts of Interest
References
- International Commission on Radiation Units and Measurement; ICRU Report 31; ICRU: Washington, DC, USA, 1979.
- Ptasinska, S.; Denifl, S.; Scheier, P.; Illenberger, E. Bond- and Site-Selective Loss of H Atoms from Nucleobases by Very-Low-Energy Electrons (<3 eV). Angew. Chem. Int. Ed. 2005, 44, 6941–6943. [Google Scholar] [CrossRef]
- Utke, I.; Gölzhäuser, A. Small, Minimally Invasive, Direct: Electrons Induce Local Reactions of Adsorbed Functional Molecules on the Nanoscale. Angew. Chem. Int. Ed. 2010, 49. [Google Scholar] [CrossRef] [PubMed]
- Swiderek, P.; Jolondz, E.; Bredehöft, J.H.; Borrmann, T.; Dölle, C.; Ott, M.; Schmüser, C.; Hartwig, A.; Danilov, V.; Wagner, H.-E.; et al. Modification of Polydimethylsiloxane Coatings by H2 RF Plasma, Xe2* Excimer VUV Radiation, and Low-Energy Electron Beams. Macromol. Mater. Eng. 2012, 297, 1091–1101. [Google Scholar] [CrossRef]
- Öberg, K.I. Photochemistry and Astrochemistry: Photo-chemical Pathways to Interstellar Complex Organic Molecules. Chem. Rev. 2016, 116, 9631–9663. [Google Scholar] [CrossRef] [PubMed]
- Mason, N.J.; Nair, B.; Jheeta, S.; Szymańska, E. Electron induced chemistry: A new frontier in astrochemistry. Faraday Discuss. 2014, 168, 235–247. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Standards and Technology. Mass Spectra. In NIST Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom, P.J., Mallard, W.G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2016; p. 20899. Available online: http://webbook.nist.gov (accessed on 30 November 2018).
- Kim, Y.-K.; Rudd, M.E. Binary-encounter-dipole model for electron-impact ionization. Phys. Rev. A 1994, 50, 3954–3967. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.; Kim, Y.-K.; Rudd, M.E. New model for elctron-impact ionization cross sections of molecules. J. Chem. Phys. 1996, 104, 2956–2966. [Google Scholar] [CrossRef]
- Rapp, D.; Englander-Golden, P. Total Cross Sections for Ionization and Attachment in Gases by Electron Impact. I. Positive Ionization. J. Chem. Phys. 1965, 43, 1464–1479. [Google Scholar] [CrossRef]
- Böhler, E.; Bredehöft, J.H.; Swiderek, P. Low-energy electron-induced hydroamination reactions between different amines and olefins. J. Phys. Chem. C 2014, 118, 6922–6933. [Google Scholar] [CrossRef]
- Arumainayagam, C.R.; Lee, H.L.; Nelson, R.B.; Haines, D.R.; Gunawardane, R.P. Low-energy electron-induced reactions in condensed matter. Surf. Sci. Rep. 2010, 65, 1–44. [Google Scholar] [CrossRef]
- Balog, R.; Langer, J.; Gohlke, S.; Stano, M.; Abdoul-Carime, H.; Illenberger, E. Low energy electron driven reactions in free and bound molecules: From unimolecular processes in the gas phase to complex reactions in a condensed environment. Int. J. Mass Spectrom. 2004, 233, 267–291. [Google Scholar] [CrossRef]
- Bald, I.; Langer, J.; Tegeder, P.; Ingólfsson, O. From isolated molecules through clusters and condensates to the building blocks of life. Int. J. Mass Spectrom. 2008, 277, 4–25. [Google Scholar] [CrossRef]
- McConkey, J.W.; Malone, C.P.; Johnson, P.V.; Winstead, C.; McKoy, V.; Kanik, I. Electron impact dissociation of oxygen-containing molecules—A critical review. Phys. Rep. 2008, 466, 1–103. [Google Scholar] [CrossRef]
- Bhargava Ram, N.; Krishnakumar, E. Dissociative electron attachment resonances in ammonia: A velocity slice imaging based study. J. Chem. Phys. 2012, 136, 164308. [Google Scholar] [CrossRef] [PubMed]
- Nag, P.; Nandi, D. Fragmentation dynamics in dissociative electron attachment to CO probed by velocity slice imaging. Phys. Chem. Chem. Phys. 2015, 17, 7130–7137. [Google Scholar] [CrossRef] [PubMed]
- Itikawa, Y. Cross Sections for Electron Collisions with Carbon Dioxide. J. Phys. Chem. Ref. Data 2002, 31, 749–767. [Google Scholar] [CrossRef]
- Rawat, P.; Prabhudesai, V.S.; Rahman, M.A.; Bhargava Ram, N.; Krishnakumar, E. Absolute cross sections for dissociative electron attachment to NH3 and CH4. Int. J. Mass Spectrom. 2008, 277, 96–102. [Google Scholar] [CrossRef]
- Bredehöft, J.H.; Böhler, E.; Schmidt, F.; Borrmann, T.; Swiderek, P. Electron-Induced Synthesis of Formamide in Condensed Mixtures of Carbon Monoxide and Ammonia. ACS Earth Space Chem. 2017, 1, 50–59. [Google Scholar] [CrossRef]
- Simpson, W.C.; Orlando, T.M.; Parenteau, L.; Nagesha, K.; Sanche, L. Dissociative electron attachment in nanoscale ice films: Thickness and charge trapping effects. J. Chem. Phys. 1998, 108, 5027–5034. [Google Scholar] [CrossRef]
- Warneke, J.; Wang, Z.; Swiderek, P.; Bredehöft, J.H. Electron-induced hydration of an alkene: Alternative reaction pathways. Angew. Chem. Int. Ed. 2015, 54, 4397–4400. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.G.; Walker, I.C. Dissociative Electron Attachment in Water and Methanol (5–14 eV). J. Chem. Soc. Faraday Trans. 1992, 88, 2805–2810. [Google Scholar] [CrossRef]
- Szymańska, E.; Mason, N.J.; Krishnakumar, E.; Matias, C.; Mauracher, A.; Scheier, P.; Denifl, S. Dissociative electron attachment and dipolar dissociation in ethylene. Int. J. Mass. Spectrom. 2014, 365–366, 356–364. [Google Scholar] [CrossRef]

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bredehöft, J.H. Electron-Induced Chemistry in the Condensed Phase. Atoms 2019, 7, 33. https://doi.org/10.3390/atoms7010033
Bredehöft JH. Electron-Induced Chemistry in the Condensed Phase. Atoms. 2019; 7(1):33. https://doi.org/10.3390/atoms7010033
Chicago/Turabian StyleBredehöft, Jan Hendrik. 2019. "Electron-Induced Chemistry in the Condensed Phase" Atoms 7, no. 1: 33. https://doi.org/10.3390/atoms7010033
APA StyleBredehöft, J. H. (2019). Electron-Induced Chemistry in the Condensed Phase. Atoms, 7(1), 33. https://doi.org/10.3390/atoms7010033