
Fine-Tuning of Atomic Energies in Relativistic Multiconfiguration Calculations




Abstract
1. Introduction
2. Multiconfiguration Methods
Expansions over Configuration State Functions in - and -Coupling
3. Hamiltonian Matrix in - and -Coupling
4. Transition Parameters in - and -Coupling
5. Transformation between Coupling Schemes
6. Fine-Tuning of Eigenvalues
7. Fine-Tuning of Eigenvalues in -Coupling
8. Fine-Tuning of Eigenvalues in -Coupling
- arrange the full CSF expansion so that the CSFs in the multireference (MR) come first.
- compute the coupling transformation matrix between the -coupled CSFs in the MR and the corresponding -coupled CSFs.
- perform a relativistic CI calculation for the full CSF expansion. Save the Hamiltonian on disk in sparse format.
- read and transform the Hamiltonian submatrix corresponding to the CSFs in the MR from -coupling () to -coupling ( according to
- allow the user to fine-tune the diagonal elements of to yield .
- transform the fine-tuned Hamiltonian matrix back to -coupling according toand merge it into the Hamiltonian saved on disk.
- perform a relativistic CI calculation for the full CSF expansion based on the Hamiltonian for which the submatrix corresponding to the CSFs in the MR was modified.
9. Program Implementation
10. Test-Run
11. Applications
11.1. Transition Rates in C III
11.2. Lifetimes of the Rydberg States in B I
12. Heavy and Complex Systems
13. Summary and Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hibbert, A. Estimation of Inaccuracies in Oscillator Strength Calculations. Phys. Scr. 1996, T65, 104–109. [Google Scholar] [CrossRef]
- Brage, T.; Hibbert, A.; Leckrone, D.S. Transition rates of the intercombination UV0.01 multiplet in N III. Astrophys. J. 1997, 478, 423–429. [Google Scholar] [CrossRef]
- Hibbert, A. Problem transitions in Na III. J. Phys. B At. Mol. Opt. Phys. 2003, 36, 4703–4715. [Google Scholar] [CrossRef]
- Hibbert, A. Successes and Difficulties in Calculating Atomic Oscillator Strengths and Transition Rates. Galaxies 2018, 6, 77. [Google Scholar] [CrossRef]
- Froese Fischer, C.; Brage, T.; Jönsson, P. Computational Atomic Structure—An MCHF Approach; Institute of Physics Publishing (IoP): Bristol, UK, 1997. [Google Scholar]
- Froese Fischer, C.; Tachiev, C. Breit–Pauli energy levels, lifetimes, and transition probabilities for the beryllium-like to neon-like sequences. At. Data Nucl. Data Tables 2004, 87, 1–184. [Google Scholar] [CrossRef]
- Carlsson, J. Configuration interaction in the neutral copper atom. Phys. Rev. A 1998, 38, 1702. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, H.; Li, Z.S.; Jönsson, P. Experimental and theoretical investigations of radiative lifetimes in the s and d sequences of neutral boron. Phys. Rev. A 2001, 63, 032505. [Google Scholar] [CrossRef]
- Hibbert, A. CIV3—A general program to calculate configuration interaction wave functions and electric-dipole oscillator strengths. Comput. Phys. Commun. 1975, 9, 141–172. [Google Scholar] [CrossRef]
- Froese Fischer, C.; Tachiev, G.; Gaigalas, G.; Godefroid, M. An MCHF atomic-structure package for large-scale calculations. Comput. Phys. Commun. 2007, 176, 559. [Google Scholar] [CrossRef]
- Froese Fischer, C.; Godefroid, M.; Brage, T.; Jönsson, P.; Gaigalas, G. Advanced multiconfiguration methods for complex atoms: I. Energies and wave functions. J. Phys. B At. Mol. Opt. Phys. 2016, 49, 182004. [Google Scholar] [CrossRef]
- Jönsson, P.; Godefroid, M.; Gaigalas, G.; Ekman, J.; Grumer, J.; Li, W.; Li, J.; Brage, T.; Grant, I.P.; Bieroń, J.; et al. An Introduction to Relativistic Theory as Implemented in GRASP. Atoms 2023, 11, 7. [Google Scholar] [CrossRef]
- Grant, I.P. Relativistic Quantum Theory of Atoms and Molecules: Theory and Computation; Springer Science and Business Media, LLC: New York, NY, USA, 2007. [Google Scholar]
- Fano, U. Interaction between Configurations with Several Open Shells. Phys. Rev. A 1965, 67, 140. [Google Scholar] [CrossRef]
- Armstrong, L., Jr.; Feneuille, S. Relativistic Effects in The Many-Electron Atom. Adv. At. Mol. Phys. 1974, 10, 1. [Google Scholar]
- Boualili, F.Z.; Nemouchi, M.; Godefroid, M.; Jönsson, P. Weak correlation and strong relativistic effects on the hyperfine interaction in fluorine. Phys. Rev. A 2021, 104, 062813. [Google Scholar] [CrossRef]
- Froese Fischer, C.; Gaigalas, G.; Jönsson, P.; Bieroń, J. GRASP2018—A Fortran 95 version of the General Relativistic Atomic Structure Package. Comput. Phys. Commun. 2019, 237, 184. [Google Scholar] [CrossRef]
- Grant, I.P. Gauge invariance and relativistic radiative transitions. J. Phys. B At. Mol. Opt. 1974, 7, 1458. [Google Scholar] [CrossRef]
- Ynnerman, A.; Froese Fischer, C. Multiconfigurational-Dirac-Fock calculation of the 2s21S0-2s2p3P1 spin-forbidden transition for the Be-like isoelectronic sequence. Phys. Rev. A 1995, 51, 2020. [Google Scholar] [CrossRef] [PubMed]
- Cowan, R.D. The Theory of Atomic Structure and Spectra; University of California Press: Berkeley, CA, USA, 1981. [Google Scholar]
- Gaigalas, G.; Froese Fischer, C.; Rynkun, P.; Jönsson, P. JJ2LSJ Transformation and Unique Labeling for Energy Levels. Atoms 2017, 5, 6. [Google Scholar] [CrossRef]
- Žalandauskas, T.; Gaigalas, G. Routine for transformation of atomic state functions from LS- to jj-coupled basis. Lith. J. Phys. 2003, 43, 15–26. [Google Scholar]
- Gaigalas, G.; Rudzikas, Z.; Žalandauskas, T. Analytical expressions for special cases of LS-jj transformation matrices for a shell of equivalent electrons. Lith. J. Phys. 2001, 41, 226–231. [Google Scholar]
- Gaigalas, G.; Fritzsche, S. Maple procedures for the coupling of angular momenta. VI. LS-jj transformations. Comput. Phys. Commun. 2002, 49, 39–60. [Google Scholar] [CrossRef]
- Gaigalas, G.; Žalandauskas, T.; Rudzikas, Z. LS-jj transformation matrices for a shell of equivalent electrons. At. Data Nucl. Data Tables 2003, 84, 99–190. [Google Scholar] [CrossRef]
- Gaigalas, G.; Žalandauskas, T.; Fritzsche, S. Spectroscopic LSJ notation for atomic levels obtained from relativistic calculations. Comput. Phys. Commun. 2004, 157, 239–253. [Google Scholar] [CrossRef]
- Jönsson, P.; Gaigalas, G.; Froese Fischer, C.; Bieroń, J.; Grant, I.P.; Brage, T.; Ekman, J.; Godefroid, M.; Grumer, J.; Li, J.; et al. GRASP Manual for Users. Atoms 2023, 11, 68. [Google Scholar] [CrossRef]
- Kramida, A.; Ralchenko, Y.; Reader, J.; NIST ASD Team. NIST Atomic Spectra Database (ver. 5.10); National Institute of Standards and Technology: Gaithersburg, MD, USA, 2022. Available online: https://physics.nist.gov/asd (accessed on 5 March 2023).
- Doerfert, J.; Träbert, E.; Wolf, A.; Schwalm, D.; Uwira, O. Precision Measurement of the Electric Dipole Intercombination Rate in C2+. Phys. Rev. Lett. 1997, 78, 4355. [Google Scholar] [CrossRef]
- O’Brien, T.R.; Lawler, J.E. Radiative lifetimes in B I using ultraviolet and vacuum-ultraviolet laser-induced fluorescence. Astron. Astrophys. 1992, 255, 420–426. [Google Scholar]
- Martinson, I.; Bickel, W.S.; Ölme, A. Beam-Foil Spectra of Boron 450–5000 Å. J. Opt. Soc. Am. 1970, 60, 1213–1220. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Song, C.; Zhang, C.; Si, R.; Wang, K.; Godefroid, M.; Gaigalas, G.; Jönsson, P.; Chen, C. Performance Tests and Improvements on the rmcdhf and rci Programs of GRASP. Atoms 2023, 11, 12. [Google Scholar] [CrossRef]
- Gaigalas, G. Coupling: The program for searching optimal coupling scheme in atomic theory. Comput. Phys. Commun. 2020, 247, 106960. [Google Scholar] [CrossRef]
- Dennis, J.E.; Gay, D.M.; Welsch, R.E. Algorithm 573: NL2SOL—An Adaptive Nonlinear Least-Squares Algorithm [E4]. Acm Trans. Math. Softw. 1981, 7, 369–383. [Google Scholar] [CrossRef]
- Ekman, J.; Jönsson, P.; Gustafsson, S.; Hartmann, H.; Gaigalas, G.; Godefroid, M.R.; Froese Fischer, C. Calculations with spectroscopic accuracy: Energies, transition rates, and Landé gJ-factors in the carbon isoelectronic sequence from Ar XIII to Zn XXV. Astron. Astrophys. 2014, 564, A24. [Google Scholar] [CrossRef]


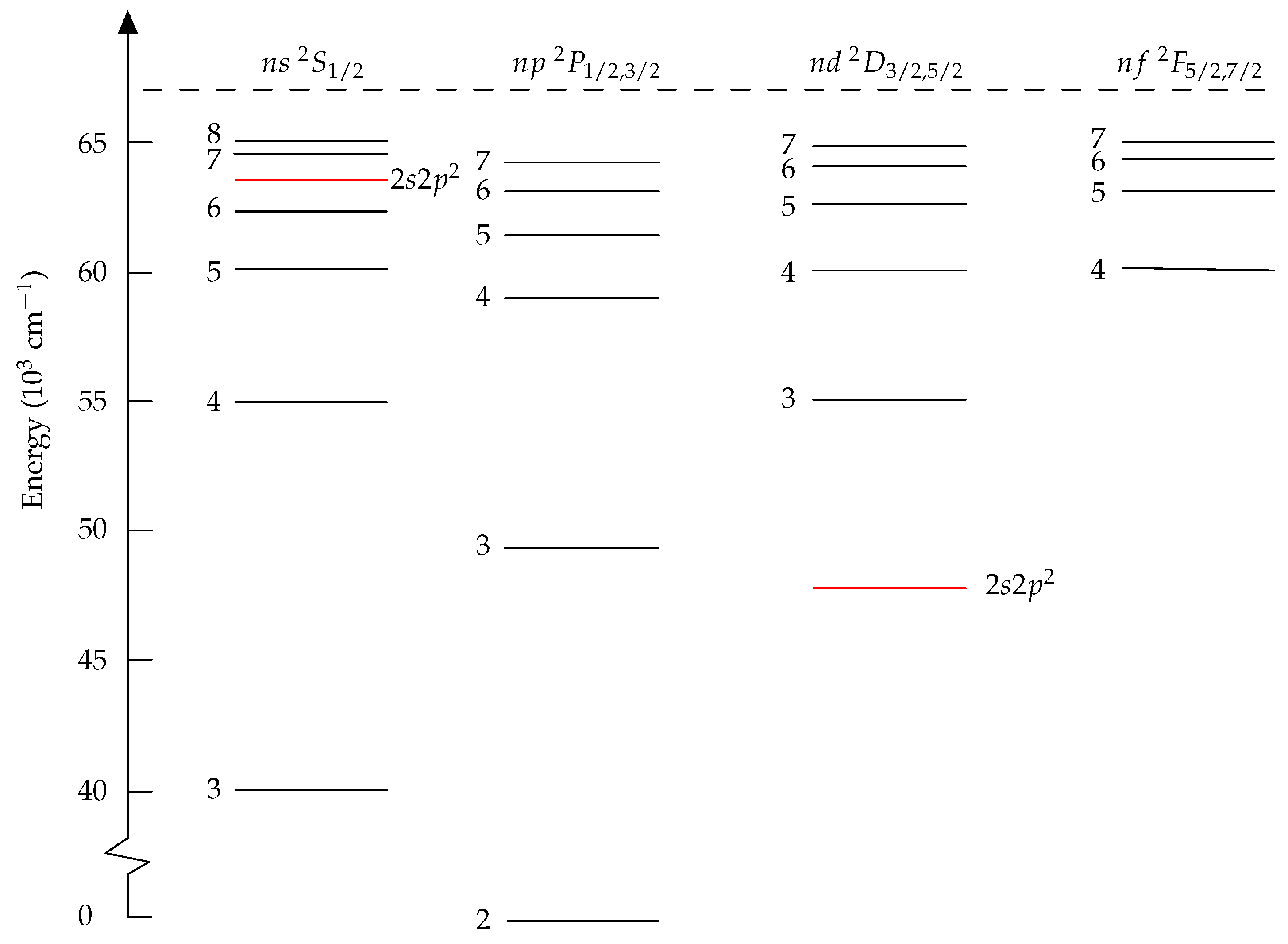{kind=link}
| No | Pos | J | Parity | Configuration | RCI | NIST | fine-tune | |
| (cm^−1) | (cm^−1) | (cm^−1) | ||||||
| 1 | 1 | 0 | + | 2s(2)_1S | 0.00 | 0.00 | ||
| 2 | 1 | 0 | − | 2s_2S.2p_3P | 52463.43 | 52367.06 | −96 | |
| 3 | 1 | 1 | − | 2s_2S.2p_3P | 52486.91 | 52390.75 | −96 | |
| 4 | 1 | 2 | − | 2s_2S.2p_3P | 52543.19 | 52447.11 | −96 | |
| 5 | 2 | 1 | − | 2s_2S.2p_1P | 102530.94 | 102352.04 | −179 |
| State | Ab Initio | Fine-Tuned | NIST | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 4 | 5 | 6 | 7 | 3 | 4 | 5 | 6 | 7 | ||
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 52,321.81 | 52,445.12 | 52,411.16 | 52,407.97 | 52,463.43 | 52,366.94 | 52,367.32 | 52,367.24 | 52,367.23 | 52,367.82 | 52,367.06 | |
| 52,344.14 | 52,468.08 | 52,434.34 | 52,431.32 | 52,486.91 | 52,390.57 | 52,391.08 | 52,390.92 | 52,390.89 | 52,391.30 | 52,390.75 | |
| 52,398.05 | 52,523.24 | 52,490.00 | 52,487.35 | 52,543.19 | 52,446.97 | 52,447.43 | 52,447.27 | 52,447.31 | 52,447.57 | 52,447.11 | |
| 104,112.00 | 102,962.02 | 102,613.81 | 102,517.46 | 102,530.94 | 102,395.09 | 102,366.93 | 102,358.33 | 102,356.05 | 102,356.24 | 102,352.04 | |
| 880.92 | 312.23 | 136.21 | 89.86 | 122.24 | 21.53 | 7.45 | 3.15 | 2.01 | 2.16 | ||
| Method | Orbitals | ||
|---|---|---|---|
| 3 | |||
| 4 | |||
| ab initio | 5 | ||
| 6 | |||
| 7 | |||
| 3 | |||
| 4 | |||
| fine-tuned | 5 | ||
| 6 | |||
| 7 | |||
| experiment |
| State | Ab Initio | Fine-Tuned | NIST |
|---|---|---|---|
| 0 | 0 | 0 | |
| 14.51 | 15.287 | ||
| 39,800.91 | 40,038.65 | 40,039.6907 | |
| 48,520.93 | 48,611.8663 | ||
| 48,522.60 | 48,613.6486 | ||
| 54,707.61 | 55,012.88 | 55,010.2338 | |
| 57,669.26 | 57,786.4336 | ||
| 57,669.86 | 57,787.0683 | ||
| 59,860.42 | 60,146.56 | 60,146.414 | |
| 61,306.69 | 61,433.59 | ||
| 61,306.97 | 61,433.59 | ||
| 62,254.45 | 62,476.47 | 62,482.167 | |
| 63,129.71 | 63,263.24 | ||
| 63,129.87 | 63,263.24 | ||
| 65,102.64 | 63,464.94 | 63,560.638 | |
| 63,526.70 | 64,166.09 | 64,156.017 |
| State | Ab Initio | Fine-Tuned | Experiment | MCHF |
|---|---|---|---|---|
| 4.11 | 3.99 | 4.0 (2) a | 3.97 b | |
| 9.86 | 8.67 | 8.7 (4) a | 8.59 b | |
| 17.6 | 12.2 | 11.0 (6) b | 11.3 b | |
| 25.1 | 8.40 | 7.7 (4) b | 7.65 b | |
| 3.24 | 3.29 | 3.3 (2) b | 3.65 b | |
| 3.6 (3) c | ||||
| 23.6 | 11.1 | 8.3 (4) b | 8.01 b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Gaigalas, G.; Li, W.; Chen, C.; Jönsson, P. Fine-Tuning of Atomic Energies in Relativistic Multiconfiguration Calculations. Atoms 2023, 11, 70. https://doi.org/10.3390/atoms11040070
Li Y, Gaigalas G, Li W, Chen C, Jönsson P. Fine-Tuning of Atomic Energies in Relativistic Multiconfiguration Calculations. Atoms. 2023; 11(4):70. https://doi.org/10.3390/atoms11040070
Chicago/Turabian StyleLi, Yanting, Gediminas Gaigalas, Wenxian Li, Chongyang Chen, and Per Jönsson. 2023. "Fine-Tuning of Atomic Energies in Relativistic Multiconfiguration Calculations" Atoms 11, no. 4: 70. https://doi.org/10.3390/atoms11040070
APA StyleLi, Y., Gaigalas, G., Li, W., Chen, C., & Jönsson, P. (2023). Fine-Tuning of Atomic Energies in Relativistic Multiconfiguration Calculations. Atoms, 11(4), 70. https://doi.org/10.3390/atoms11040070