Experimental Design and Sample Preparation in Forest Tree Metabolomics




{kind=link}
Abstract
1. Introduction
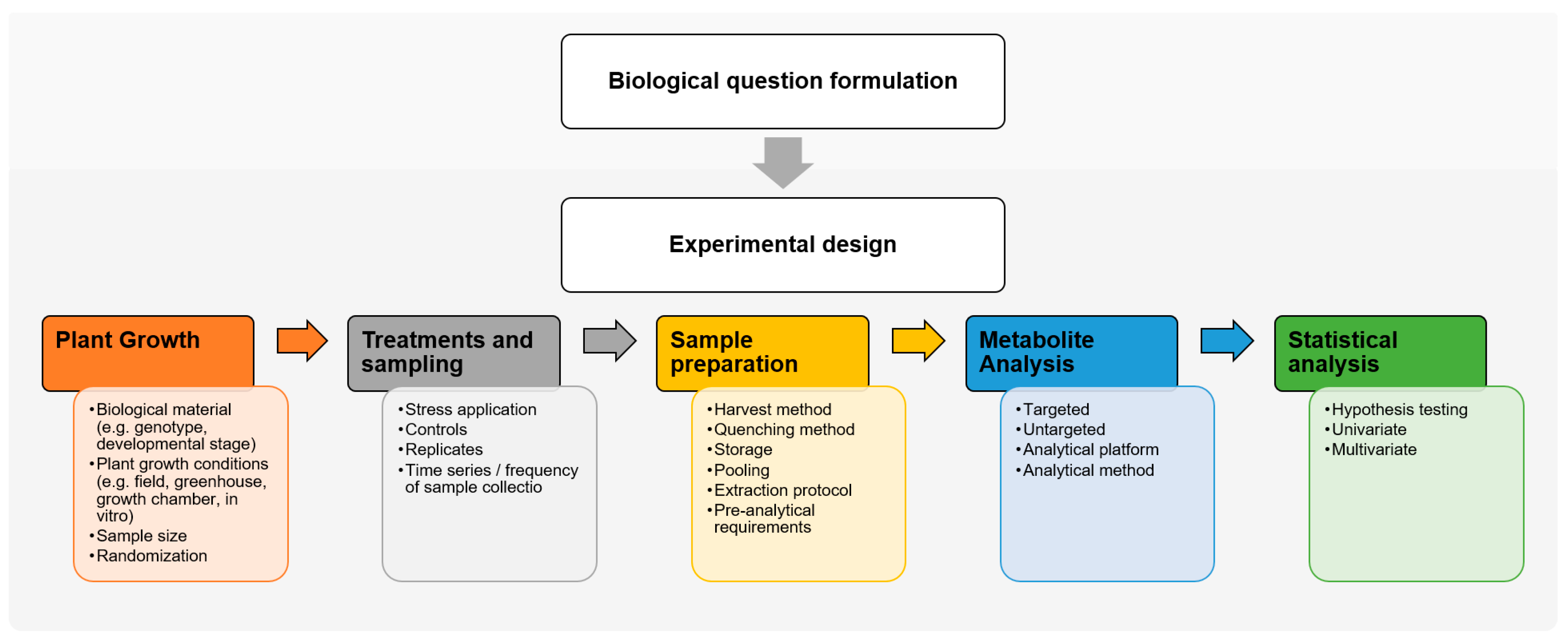
2. Experimental Design for Forest Tree Metabolomics
2.1. Biological Question Formulation
2.2. Experimental Design
2.2.1. Experimental Conditions
2.2.2. Replicates and Randomization
3. Sample Preparation for Forest Tree Metabolomics
3.1. Harvest and Quenching
3.2. Metabolite Extraction
3.2.1. GC-MS Metabolite Profiling
3.2.2. LC-MS Metabolite Profiling
3.3. Pre-Analytical Requirements
4. The Importance of Forest Tree Metadata Standardization
5. Conclusion
Author Contributions
Funding
Conflicts of Interest
References
- Oliver, S.G.; Winson, M.K.; Kell, D.B.; Baganz, F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998, 16, 373–378. [Google Scholar] [CrossRef]
- Jorge, T.F.; Rodrigues, J.A.; Caldana, C.; Schmidt, R.; van Dongen, J.T.; Thomas-Oates, J.; António, C. Mass spectrometry-based plant metabolomics: Metabolite responses to abiotic stress. Mass Spectrom. Rev. 2016, 35, 620–649. [Google Scholar] [CrossRef] [PubMed]
- Alseekh, S.; Wu, S.; Brotman, Y.; Fernie, A.R. Guidelines for sample normalization to minimize batch variation for large-scale metabolic profiling of plant natural genetic variance. In Plant Metabolomics; António, C., Ed.; Humana Press: New York, NY, USA, 2018. [Google Scholar] [CrossRef]
- Fernie, A.R.; Stitt, M. On the discordance of metabolomics with proteomics and transcriptomics: Coping with increasing complexity in logic, chemistry, and network interactions scientific correspondence. Plant Physiol. 2012, 158, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry based-metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Verpoorte, R. Sample preparation for plant metabolomics. Phytochem. Anal. 2010, 21, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Allwood, J.W.; De Vos, R.C.H.; Moing, A.; Deborde, C.; Erban, A.; Kopka, J.; Goodacre, R.; Hall, R.D. Plant metabolomics and its potential for systems biology research: Background concepts, technology, and methodology. Methods Enzymol. 2011, 500, 299–336. [Google Scholar] [CrossRef]
- Rodrigues, A.M.; Miguel, C.; Chaves, I.; António, C. Mass spectrometry-based forest tree metabolomics. Mass Spectrom. Rev. 2019. [Google Scholar] [CrossRef]
- Martins, M.C.M.; Caldana, C.; Wolf, L.D.; de Abreu, L.G.F. The Importance of Experimental Design, Quality Assurance, and Control in Plant Metabolomics Experiments. In Plant Metabolomics; António, C., Ed.; Humana Press: New York, NY, USA, 2018. [Google Scholar] [CrossRef]
- Baumgartner, R.J. Sustainable development goals and the forest sector—A complex relationship. Forests 2019, 10, 152. [Google Scholar] [CrossRef]
- Bončina, A.; Simončič, T.; Rosset, C. Assessment of the concept of forest functions in Central European forestry. Environ. Sci. Policy 2019, 99, 123–135. [Google Scholar] [CrossRef]
- Loomis, J.J.; Knaus, M.; Dziedzic, M. Integrated quantification of forest total economic value. Land Use Policy 2019, 84, 335–346. [Google Scholar] [CrossRef]
- IPCC Core Writing Team. Climate Change 2014: Synthesis Report. In Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; IPCC: Geneva, Switzerland, 2014. [Google Scholar]
- Janz, D.; Behnke, K.; Schnitzler, J.P.; Kanawati, B.; Schmitt-Kopplin, P.; Polle, A. Pathway analysis of the transcriptome and metabolome of salt sensitive and tolerant poplar species reveals evolutionary adaption of stress tolerance mechanisms. BMC Plant Biol. 2010, 10, 150. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.R.; Aranda, I.; Cano, F.J. Metabolomics demonstrates divergent responses of two Eucalyptus species to water stress. Metabolomics 2012, 8, 186–200. [Google Scholar] [CrossRef]
- Correia, B.; Pintó-Marijuan, M.; Castro, B.B.; Brossa, R.; López-Carbonell, M.; Pinto, G. Hormonal dynamics during recovery from drought in two Eucalyptus globulus genotypes: From root to leaf. Plant Physiol. Biochem. 2014, 82, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Budzinski, I.G.; Moon, D.H.; Morosini, J.S.; Lindén, P.; Bragatto, J.; Moritz, T.; Labate, C.A. Integrated analysis of gene expression from carbon metabolism, proteome and metabolome, reveals altered primary metabolism in Eucalyptus grandis bark, in response to seasonal variation. BMC Plant Biol. 2016, 16, 149. [Google Scholar] [CrossRef]
- Correia, B.; Valledor, L.; Hancock, R.D.; Renaut, J.; Pascual, J.; Soares, A.M.V.M.; Pinto, G. Integrated proteomics and metabolomics to unlock global and clonal responses of Eucalyptus globulus recovery from water deficit. Metabolomics 2016, 12, 141. [Google Scholar] [CrossRef]
- De Miguel, M.; Guevara, M.A.; Sánchez-Gómez, D.; María, N.; Díaz, L.M.; Mancha, J.A.; de Símon, B.F.; Cadahía, E.; Desai, N.; Aranda, I.; et al. Organ-specific metabolic responses to drought in Pinus pinaster Ait. Plant Physiol. Biochem. 2016, 102, 17–26. [Google Scholar] [CrossRef]
- Rivas-Ubach, A.; Barbeta, A.; Sardans, J.; Guenther, A.; Ogaya, R.; Oravec, M.; Urban, O.; Peñuelas, J. Topsoil depth substantially influences the responses to drought of the foliar metabolomes of Mediterranean forests. Perspect. Plant Ecol. Syst. 2016, 21, 41–54. [Google Scholar] [CrossRef]
- De Símon, B.F.; Sanz, M.; Cervera, M.T.; Pinto, E.; Aranda, I.; Cadahía, E. Leaf metabolic response to water deficit in Pinus pinaster Ait. relies upon ontogeny and genotype. Environ. Exp. Bot. 2017, 14, 41–55. [Google Scholar] [CrossRef]
- Correia, B.; Hancock, R.D.; Amaral, J.; Gomez-Cadenas, A.; Valledor, L.; Pinto, G. Combined drought and heat activates protective responses in Eucalyptus globulus that are not activated when subjected to drought or heat stress alone. Front Plant Sci. 2018, 9, 819. [Google Scholar] [CrossRef]
- Escandón, M.; Meijón, M.; Valledor, L.; Pascual, J.; Pinto, G.; Cañal, M.J. Metabolome integrated analysis of high-temperature response in Pinus radiata. Front. Plant Sci. 2018, 9, 485. [Google Scholar] [CrossRef]
- Mokochinski, J.B.; Mazzafera, P.; Sawaya, A.C.H.F.; Mumm, R.; de Vos, R.C.H.; Hall, R.D. Metabolic responses of Eucalyptus species to different temperature regimes. J. Integr. Plant Biol. 2018, 60, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Calcerrada, J.; Rodrigues, A.M.; Perdiguero, P.; António, C.; Altkin, O.K.; Li, M.; Colada, C.; Gil, L. A molecular approach to drought-induced reduction in leaf CO2 exchange in drought-resistant Quercus ilex. Physiol. Plant. 2018, 162, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Netzer, F.; Tohge, T.; Orf, I.; Brotman, Y.; Dubbert, D.; Fernie, A.R.; Rennenberg, H.; Hoefgen, R.; Herschbach, C. Metabolome and lipidome profiles of Populus canescens twig tissues during annual growth show phospholipid-linked storage and mobilization of C, N, and S. Front Plant Sci. 2018, 9, 1292. [Google Scholar] [CrossRef] [PubMed]
- Cadahía, E.; de Simón, B.F.; Aranda, I.; Sanz, M.; Sánchez-Gómez, D.; Pinto, E. Non-targeted metabolomic profile of Fagus sylvatica L. leaves using liquid chromatography with mass spectrometry and gas chromatography with mass spectrometry. Phytochem. Anal. 2015, 26, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Dhandapani, S.; Jin, J.; Sridhar, V.; Sarojam, R.; Chua, N.H.; Jang, I.C. Integrated metabolome and transcriptome analysis of Magnolia champaca identifies biosynthetic pathways for floral volatile organic compounds. BMC Genom. 2017, 18, 463. [Google Scholar] [CrossRef]
- Dean, L.L. Targeted and non-targeted analyses of secondary metabolites in nut and seed processing. Eur. J. Lipid Sci. Technol. 2018, 120, 1700479. [Google Scholar] [CrossRef]
- Li, S.S.; Wu, Q.; Yin, D.D.; Feng, C.Y.; Liu, Z.A.; Wang, L.S. Phytochemical variation among the traditional Chinese medicine Mu Dan Pi from Paeonia suffruticosa (tree peony). Phytochemistry 2018, 146, 16–24. [Google Scholar] [CrossRef]
- Moura, I.; Duvane, J.A.; Silva, M.J.; Ribeiro, N.; Ribeiro-Barros, A.I. Woody species from the Mozambican Miombo woodlands: A review on their ethnomedicinal uses and pharmacological potential. J. Med. Plant Res. 2018, 12, 15–31. [Google Scholar] [CrossRef]
- Farag, M.A.; El-Kersh, D.M.; Ehrlich, A.; Choucry, M.A.; El-Seedi, H.; Frolov, A.; Wessjohann, L.A. Variation in Ceratonia siliqua pod metabolome in context of its different geographical origin, ripening stage and roasting process. Food Chem. 2019, 283, 675–687. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, C.; Liu, S.; He, C.; Chen, F.; Xiao, P. Comprehensive metabolic profile analysis of the root bark of different species of tree peonies (Paeonia Sect. Moutan). Phytochemistry 2019, 163, 118–125. [Google Scholar] [CrossRef]
- Álvarez-Sánchez, B.; Priego-Capote, F.; de Castro, M.D.L. Metabolomics analysis II. Preparation of biological samples prior to detection. Trends Anal. Chem. 2010, 29, 120–127. [Google Scholar] [CrossRef]
- Broadhurst, D.I.; Kell, D.B. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2006, 2, 171–196. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Stitt, M.; Fernie, A.R. From measurements of metabolites to metabolomics: An ‘on the fly’ perspective illustrated by recent studies of carbon–nitrogen interactions. Curr. Opin. Biotechnol. 2003, 14, 136–144. [Google Scholar] [CrossRef]
- Hall, R.D. Plant metabolomics: From holistic hope, to hype, to hot topic. New Phytol. 2006, 169, 453–468. [Google Scholar] [CrossRef]
- Ghimenti, S.; Tabucchi, S.; Lomonaco, T.; Di Francesco, F.; Fuoco, R.; Onor, M.; Lenzi, M.; Trivella, M.G. Monitoring breath during oral glucose tolerance tests. J. Breath Res. 2013, 7, 017115. [Google Scholar] [CrossRef]
- Lomonaco, T.; Ghimenti, S.; Piga, I.; Biagini, D.; Onor, M.; Fuoco, R.; Paolicchi, A.; Ruocco, L.; Pellegrini, G.; Trivella, M.G.; et al. Monitoring of warfarin therapy: Preliminary results from a longitudinal pilot study. Microchem. J. 2018, 136, 170–176. [Google Scholar] [CrossRef]
- Neale, D.B.; Kremer, A. Forest tree genomics: Growing resources and applications. Nat. Rev. Genet. 2011, 12, 111–122. [Google Scholar] [CrossRef]
- Asbjornsen, H.; Campbell, J.; Jennings, K.; Vadeboncoeur, M.; McIntire, C.; Templer, P.; Phillips, R.; Bauerle, T.; Dietze, M.; Frey, S.; et al. Guidelines and considerations for designing field experiments for simulating precipitation extremes in forest ecosystems. Methods Ecol. Evol. 2018, 9, 2310–2325. [Google Scholar] [CrossRef]
- Morgenstern, E.K. Geographic Variation in Forest Trees. In Genetic Basis and Application of Knowledge in Silviculture; UBC Press: Vancouver, BC, Canada, 1996; ISBN 0-7748-0579-X. [Google Scholar]
- Faria, J.M.; Sena, I.; Vieira da Silva, I.; Ribeiro, B.; Barbosa, P.; Ascensão, L.; Bennett, R.N.; Mota, M.; Figueiredo, A.C. In vitro co-cultures of Pinus pinaster with Bursaphelenchus xylophilus: A biotechnological approach to study pine wilt disease. Planta 2015, 241, 1325–1336. [Google Scholar] [CrossRef]
- Confalonieri, M.; Balestrazzi, A.; Bisoffi, S.; Carbonera, D. In vitro culture and genetic engineering of Populus spp.: Synergy for forest tree improvement. Plant Cell Tissue Org. Cult. 2003, 72, 109. [Google Scholar] [CrossRef]
- Allwood, J.W.; Heald, J.; Lloyd, A.J.; Goodacre, R.; Mur, L.A.J. Separating the inseparable: The metabolomic analysis of plant-pathogen interactions. In Plant Metabolomics; Nigel, W.H., Robert, D.H., Eds.; Humana Press: New York, NY, USA, 2012; Volume 860, pp. 31–49. [Google Scholar] [CrossRef]
- Boughton, B.A.; Thinagaran, D.; Sarabia, D.; Bacic, A.; Roessner, U. Mass spectrometry imaging for plant biology: A review. Phytochem. Rev. 2016, 15, 445–488. [Google Scholar] [CrossRef] [PubMed]
- Blainey, P.; Krzywinski, M.; Altman, N. Points of significance: Replication. Nat. Methods 2014, 11, 879–880. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O.; Wohlgemuth, G.; Scholz, M. Setup and Annotation of Metabolomic Experiments by Integrating Biological and Mass Spectrometric Metadata. In Data Integration in the Life Sciences; Ludäscher, B., Raschid, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 3615. [Google Scholar] [CrossRef]
- Lisec, J.; Schauer, N.; Kopka, J.; Willmitzer, L.; Fernie, A.R. Gas chromatography mass spectrometry–Based metabolite profiling in plants. Nat. Protoc. 2006, 1, 387–396. [Google Scholar] [CrossRef]
- Blaise, B.J.; Correia, G.; Tin, A.; Young, J.H.; Vergnaud, A.-C.; Lewis, M.; Pearce, J.T.M.; Elliott, P.; Nicholson, J.K.; Holmes, E.; et al. Power analysis and sample size determination in metabolic phenotyping. Anal. Chem. 2016, 88, 5179–5188. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. Using MetaboAnalyst 3.0 for comprehensive metabolomics data analysis. Curr. Protoc. Bioinform. 2016, 55, 14.10.1–14.10.91. [Google Scholar] [CrossRef]
- Fukusaki, E.; Kobayashi, A. Plant Metabolomics: Potential for Practical Operation. J. Biosci. Bioeng. 2005, 100, 347–354. [Google Scholar] [CrossRef]
- Hannemann, J.; Poorter, H.; Usadel, B.; Bläsing, O.E.; Finck, A.; Tardieu, F.; Atkin, O.K.; Pons, T.; Stitt, M.; Gibon, Y. Xeml Lab: A tool that supports the design of experiments at a graphical interface and generates computer-readable metadata files, which capture information about genotypes, growth conditions, environmental perturbations and sampling strategy. Plant Cell Environ. 2009, 32, 1185–2000. [Google Scholar] [CrossRef]
- Van Es, H.M.; Gomes, C.P.; Sellmann, M.; van Es, C.L. Spatially-Balanced Complete Block designs for field experiments. Geoderma 2007, 140, 346–352. [Google Scholar] [CrossRef]
- Gezan, S.A.; White, T.L.; Huber, D.A. Comparison of Experimental Designs for Clonal Forestry Using Simulated Data. For. Sci. 2006, 52, 108–116. [Google Scholar] [CrossRef]
- Fiehn, O.; Wohlgemuth, G.; Scholz, M.; Kind, T.; Lee, D.Y.; Lu, Y.; Moon, S.; Nikolau, B. Quality control for plant metabolomics: Reporting MSI-compliant studies. Plant J. 2008, 53, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Duvane, J.A.; Jorge, T.F.; Maquia, I.; Ribeiro, N.; Ribeiro-Barros, A.I.F.; António, C. Characterization of the primary metabolome of Brachystegia boehmii and Colophospermum mopane under different fire regimes in Miombo and Mopane African Woodlands. Front. Plant Sci. 2017, 8, 2130. [Google Scholar] [CrossRef] [PubMed]
- T’Kindt, R.; Morreel, K.; Deforce, D.; Boerjan, W.; Van Bocxlaer, J. Joint GC–MS and LC–MS platforms for comprehensive plant metabolomics: Repeatability and sample pre-treatment. J. Chromatogr. B. 2009, 877, 3572–3580. [Google Scholar] [CrossRef] [PubMed]
- Forcat, S.; Bennett, M.H.; Mansfield, J.W.; Grant, M.R. A rapid and robust method for simultaneously measuring changes in the phytohormones ABA, JA and SA in plants following biotic and abiotic stress. Plant Methods 2008, 4, 16. [Google Scholar] [CrossRef]
- Delatorre, C.; Rodríguez, A.; Rodríguez, L.; Majada, J.P.; Ordás, R.J.; Feito, I. Hormonal profiling: Development of a simple method to extract and quantify phytohormones in complex matrices by UHPLC–MS/MS. J. Chromatogr. B 2017, 1040, 239–249. [Google Scholar] [CrossRef]
- Vuckovic, D. Current trends and challenges in sample preparation for global metabolomics using liquid chromatography–mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 1523–1548. [Google Scholar] [CrossRef]
- Gullberg, J.; Jonsson, P.; Nordström, A.; Sjöström, M.; Moritz, T. Design of experiments: An efficient strategy to identify factors influencing extraction and derivatization of Arabidopsis thaliana samples in metabolomic studies with gas chromatography/mass spectrometry. Anal. Biochem. 2004, 331, 283–295. [Google Scholar] [CrossRef]
- Rodrigues, A.M.; António, C. Standard Key Steps in Mass Spectrometry-Based Plant Metabolomics Experiments: Instrument Performance and Analytical Method Validation. In Plant Metabolomics; António, C., Ed.; Humana Press: New York, NY, USA, 2018; Volume 1778. [Google Scholar] [CrossRef]
- Jorge, T.F.; Mata, A.T.; António, C. Mass spectrometry as a quantitative tool in plant metabolomics. Philos. Trans. R. Soc. A 2016, 374, 20150370. [Google Scholar] [CrossRef]
- Harfouche, A.; Meilan, R.; Altman, A. Molecular and physiological responses to abiotic stress in forest trees and their relevance to tree improvement. Tree Physiol. 2014, 34, 1181–1198. [Google Scholar] [CrossRef]
- Srivastava, V.; Obudulu, O.; Bygdell, J.; Löfstedt, T.; Rydén, P.; Nilsson, R.; Ahnlund, M.; Johansson, A.; Jonsson, P.; Freyhult, E.; et al. OnPLS integration of transcriptomic, proteomic and metabolomic data shows multi-level oxidative stress responses in the cambium of transgenic hipI- superoxide dismutase Populus plants. BMC Genom. 2013, 14, 893. [Google Scholar] [CrossRef]
- Angelcheva, L.; Mishra, Y.; Antti, H.; Kjellsen, T.; Funk, C.; Strimbeck, R.G.; Schröder, W.P. Metabolomic analysis of extreme freezing tolerance in Siberian spruce (Picea obovata). New Phytol. 2014, 204, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Qu, L.; Hu, J.; Zhang, L.; Tang, F.; Lu, M. Metabolomics reveals constitutive metabolites that contribute resistance to fall webworm (Hyphantria cunea) in Populus deltoides. Environm. Exp. Bot. 2017, 136, 31–40. [Google Scholar] [CrossRef]
- Andersson-Gunnerås, S.; Mellerowicz, E.J.; Love, J.; Segerman, B.; Ohmiya, Y.; Coutinho, P.M.; Nilsson, P.; Henrissat, B.; Moritz, T.; Sundberg, B. Biosynthesis of cellulose-enriched tension wood in Populus: Global analysis of transcripts and metabolites identifies biochemical and developmental regulators in secondary wall biosynthesis. Plant J. 2006, 45, 144–165. [Google Scholar] [CrossRef] [PubMed]
- Druart, N.; Johansson, A.; Baba, K.; Schrader, J.; Sjödin, A.; Bhalerao, R.R.; Resman, L.; Trygg, J.; Moritz, T.; Bhalerao, R.P. Environmental and hormonal regulation of the activity-dormancy cycle in the cambial meristem involves stage-specific modulation of transcriptional and metabolic networks. Plant J. 2007, 50, 557–573. [Google Scholar] [CrossRef]
- Hoffman, D.E.; Jonsson, P.; Bylesjö, M.; Trygg, J.; Antti, H.; Eriksson, M.E.; Moritz, T. Changes in diurnal patterns within the Populus transcriptome and metabolome in response to photoperiod variation. Plant Cell Environ. 2010, 33, 1298–1313. [Google Scholar] [CrossRef]
- Kusano, M.; Jonsson, P.; Fukushima, A.; Gullberg, J.; Sjöström, M.; Trygg, J.; Moritz, T. Metabolite signature during short-day induced growth cessation in Populus. Front. Plant Sci. 2011, 2, 29. [Google Scholar] [CrossRef]
- Businge, E.; Brackmann, K.; Moritz, T.; Egertsdotter, U. Metabolite profiling reveals clear metabolic changes during somatic embryo development of Norway spruce (Picea abies). Tree Physiol. 2012, 32, 232–244. [Google Scholar] [CrossRef]
- Li, Q.F.; Wang, J.H.; Pulkkinen, P.; Kong, L.S. Changes in the metabolome of Picea balfouriana embryogenic tissues that were linked to different levels of 6-BAP by gas chromatography-mass spectrometry approach. PLoS ONE 2015, 10, e0141841. [Google Scholar] [CrossRef]
- Guerra, F.P.; Richards, J.H.; Fiehn, O.; Famula, R.; Stanton, B.J.; Shuren, R.; Skykes, R.; Davis, M.F.; Neale, D.B. Analysis of the genetic variation in growth, ecophysiology, and chemical and metabolomic composition of wood of Populus trichocarpa provenances. Tree Genet. Genomes 2016, 12, 6. [Google Scholar] [CrossRef]
- Dobrowolska, I.; Businge, E.; Abreu, I.N.; Moritz, T.; Egertsdotter, U. Metabolome and transcriptome profiling reveal new insights into somatic embryo germination in Norway spruce (Picea abies). Tree Physiol. 2017, 37, 1752–1766. [Google Scholar] [CrossRef]
- Dudareva, N.; Negre, F.; Nagegowda, D.; Orlova, I. Plant volatiles: Recent advances and future perspectives. Crit. Rev. Plant Sci. 2006, 25, 417–440. [Google Scholar] [CrossRef]
- Figueiredo, A.C. Biological properties of essential oils and volatiles: Sources of variability. Nat. Volatiles Essent. Oils 2017, 4, 1–13. [Google Scholar]
- Council of Europe. European Pharmacopoeia, 7th ed.; European Directorate for the Quality of Medicines and Healthcare: Strasbourg, France, 2010. [Google Scholar]
- Keszei, A.; Brubaker, C.L.; Foley, W.J. A molecular perspective on terpene variation in Australian Myrtaceae. Aust. J. Bot. 2008, 56, 197–213. [Google Scholar] [CrossRef]
- Keszei, A.; Brubaker, C.L.; Carter, R.; Köllner, T.; Degenhardt, J.; Foley, W.J. Functional and evolutionary relationships between terpene synthases from Australian Myrtaceae. Phytochemistry 2010, 71, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Arrabal, C.; García-Vallejo, M.C.; Cadahia, E.; Cortijo, M.; de Símon, B.F. Characterization of two chemotypes of Pinus pinaster by their terpene and acid patterns in needles. Plant Syst. Evol. 2012, 298, 511–522. [Google Scholar] [CrossRef]
- Rodrigues, A.M.; Mendes, M.D.; Lima, A.S.; Barbosa, P.M.; Ascensão, L.; Barroso, J.G.; Pedro, L.G.; Mota, M.M.; Figueiredo, A.C. Pinus halepensis, P. pinaster, P. pinea and P. sylvestris essential oils chemotypes and monoterpene hydrocarbon enantiomers, before and after inoculation with the pinewood nematode Bursaphelenchus xylophilus. Chem. Biodivers. 2017, 14, e1600153. [Google Scholar] [CrossRef] [PubMed]
- Szmigielski, R.; Cieslak, M.; Rudziński, K.J.; Maciejewska, B. Identification of volatiles from Pinus silvestris attractive for Monochamus galloprovincialis using a SPME-GC/MS platform. Environ. Sci. Pollut. Res. Int. 2011, 9, 2860–2869. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gonçalves, E.; Figueiredo, A.C.; Barroso, J.G.; Henriques, J.; Sousa, E.; Bonifácio, L. Effect of Monochamus galloprovincialis feeding on Pinus pinaster and Pinus pinea, oleoresin and insect volatiles. Phytochemistry 2020, 169, 112159. [Google Scholar] [CrossRef]
- Schäfer, B.; Hennig, P.; Engewald, W. Analysis of monoterpenes from conifer needles using solid phase microextraction. J. High Res. Chromatogr. 1995, 18, 587–592. [Google Scholar] [CrossRef]
- Riikonen, J.; Kontunen-Soppela, S.; Ossipov, V.; Tervahauta, A.; Tuomainen, M.; Oksanen, E.; Vapaavouri, E.; Heinonen, J.; Kivimäenpää, M. Needle metabolome, freezing tolerance and gas exchange in Norway spruce seedlings exposed to elevated temperature and ozone concentration. Tree Physiol. 2012, 32, 1102–1112. [Google Scholar] [CrossRef]
- De Símon, B.F.; Cadahía, E.; Aranda, I. Metabolic response to elevated CO2 levels in Pinus pinaster Aiton needles in an ontogenetic and genotypic-dependent way. Plant Physiol. Biochem. 2018, 132, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Hantao, L.W.; Ribeiro, F.A.L.; Passador, M.M.; Furtado, E.L.; Poppi, R.J.; Gozzo, F.C.; Augusto, F. Metabolic profiling by ultra-performance liquid chromatography mass spectrometry and parallel factor analysis for the determination of disease biomarkers in Eucalyptus. Metabolomics 2014, 13, 1318–1325. [Google Scholar] [CrossRef]
- Rivas-Ubach, A.; Sardans, J.; Hódar, J.A.; Garcia-Porta, J.; Guenther, A.; Paša-Tolić, L.; Oravec, M.; Urban, O.; Peñuelas, J. Close and distant: Contrasting the metabolism of two closely related subspecies of Scots pine under the effects of folivory and summer drought. Ecol. Evol. 2017, 7, 8976–8988. [Google Scholar] [CrossRef] [PubMed]
- Meijón, M.; Feito, I.; Oravec, M.; Delatorre, C.; Weckwerth, W.; Majada, J.; Valledor, L. Exploring natural variation of Pinus pinaster Aiton using metabolomics: Is it possible to identify the region of origin of a pine from its metabolites? Mol. Ecol. 2016, 25, 959–976. [Google Scholar] [CrossRef]
- Obudulu, O.; Mähler, N.; Skotare, T.; Bygdell, J.; Abreu, I.N.; Ahnlund, M.; Latha Gandla, M.; Petterle, A.; Moritz, T.; Hvidsten, T.R.; et al. A multi-omics approach reveals function of Secretory Carrier-Associated Membrane Proteins in wood formation of Populus trees. BMC Genom. 2018, 19, 11. [Google Scholar] [CrossRef]
- Pan, X.; Wang, X. Profiling of plant hormones by mass spectrometry. J. Chromatogr. B 2009, 877, 280–2813. [Google Scholar] [CrossRef]
- Pan, X.; Welti, R.; Wang, X. Quantitative analysis of major plant hormones in crude plant extracts by high-performance liquid chromatography-mass spectrometry. Nat Protoc. 2010, 5, 986–992. [Google Scholar] [CrossRef]
- Bieleski, R.L. The problem of halting enzyme action when extracting plant tissues. Anal. Biochem. 1964, 9, 431–442. [Google Scholar] [CrossRef]
- Kang, J.W.; Lee, H.; Lim, H.; Lee, W.Y. Identification of potential metabolic markers for the selection of a high-yield clone of Quercus acutissima in clonal seed orchard. Forests 2018, 9, 116. [Google Scholar] [CrossRef]
- De Diego, N.; Pérez-Alfocea, F.; Cantero, E.; Lacuesta, M.; Moncaleán, P. Physiological response to drought in radiata pine: Phytohormone implication at leaf level. Tree Physiol. 2012, 32, 435–449. [Google Scholar] [CrossRef]
- Ryu, M.; Mishra, R.C.; Jeon, J.; Lee, S.K.; Bae, H. Drought-induced susceptibility for Cenangium ferruginosum leads to progression of Cenangium-dieback disease in Pinus koraiensis. Sci. Rep. 2018, 8, 16368. [Google Scholar] [CrossRef] [PubMed]
- Novák, O.; Hauserová, E.; Amakorová, P.; Dolezal, K.; Strnad, M. Cytokinin profiling in plant tissues using ultra-performance liquid chromatography-electrospray tandem mass spectrometry. Phytochemistry 2008, 69, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Raterink, R.J.; Lindenburg, P.W.; Vreeken, R.J.; Ramautar, R.; Hankemeier, T. Recent developments in sample-pretreatment techniques for mass spectrometry-based metabolomics. Trends Anal. Chem. 2014, 61, 157–167. [Google Scholar] [CrossRef]
- Fiehn, O.; Robertson, D.; Griffin, J.; van der Werf, M.; Nikolau, B.; Morrison, N.; Sumner, L.W.; Goodacre, R.; Hardy, N.W.; Taylor, C.; et al. The metabolomics standards initiative (MSI). Metabolomics 2007, 3, 175–178. [Google Scholar] [CrossRef]
- Fiehn, O.; Sumner, L.W.; Rhee, S.Y.; Ward, J.; Dickerson, J.; Lange, B.M.; Lane, G.; Roessner, U.; Last, R.; Nikolau, B. Minimum reporting standards for plant biology context information in metabolomic studies. Metabolomics 2007, 3, 195–201. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef]
- Van Rijswijk, M.; Beirnaert, C.; Caron, C.; Cascante, M.; Dominguez, V.; Dunn, W.B.; Ebbels, T.M.D.; Giacomoni, F.; Gonzalez-Beltran, A.; Hankemeier, T.; et al. The future of metabolomics in ELIXIR. F1000Res 2017, 6, 1649. [Google Scholar] [CrossRef]
- Spicer, R.A.; Salek, R.; Steinbeck, C. Compliance with minimum information guidelines in public metabolomics repositories. Sci. Data 2017, 4, 170137. [Google Scholar] [CrossRef]
- Rocca-Serra, P.; Salek, R.M.; Arita, M.; Correa, E.; Dayalan, S.; Gonzalez-Beltran, A.; Ebbels, T.; Goodacre, R.; Hastings, J.; Haug, K.; et al. Data standards can boost metabolomics research, and if there is a will, there is a way. Metabolomics 2016, 12, 14. [Google Scholar] [CrossRef]
- Spicer, R.A.; Steinbeck, C. A lost opportunity for science: Journals promote data sharing in metabolomics but do not enforce it. Metabolomics 2018, 14, 16. [Google Scholar] [CrossRef]
- Yakovlev, I.A.; Fossdal, C.G.; Johnsen, Ø. MicroRNAs, the epigenetic memory and climatic adaptation in Norway spruce. New Phytol. 2010, 187, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Bräutigam, K.; Vining, K.J.; Lafon-Placette, C.; Fossdal, C.G.; Mirouze, M.; Marcos, J.G.; Fluch, S.; Fraga, M.F.; Guevara, M.A.; Abarca, D.; et al. Epigenetic regulation of adaptive responses of forest tree species to the environment. Ecol. Evol. 2013, 3, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Pieruschka, R.; Schurr, U. Plant Phenotyping: Past, Present, and Future. Plant Phenomics 2019, 7507131. [Google Scholar] [CrossRef]
- Krajewski, P.; Chen, D.; Ćwiek, H.; van Dijk, A.D.; Fiorani, F.; Kersey, P.; Klukas, C.; Lange, M.; Markiewicz, A.; Nap, J.P.; et al. Towards recommendations for metadata and data handling in plant phenotyping. J. Exp. Bot. 2015, 66, 5417–5427. [Google Scholar] [CrossRef]
- Cwiek-Kupczynska, H.; Altmann, T.; Arend, D.; Arnaud, E.; Chen, D.; Cornut, G.; Fiorani, F.; Frohmberg, W.; Junker, A.; Klukas, C.; et al. Measures for interoperability of phenotypic data: Minimum information requirements and formatting. Plant Methods 2016, 12, 44. [Google Scholar] [CrossRef]
- Pommier, C.; Michotey, C.; Cornut, G.; Roumet, P.; Duchêne, E.; Flores, R.; Lebreton, A.; Alaux, M.; Durand, S.; Kimmel, E.; et al. Applying FAIR Principles to Plant Phenotypic Data Management in GnpIS. Plant Phenomics 2019, 2019, 1671403. [Google Scholar] [CrossRef]
- Wegrzyn, J.L.; Staton, M.A.; Street, N.R.; Main, D.; Grau, E.; Herndon, N.; Buehler, S.; Falk, T.; Zaman, S.; Ramnath, R.; et al. Cyberinfrastructure to improve forest health and productivity: The role of tree databases in connecting genomes, phenomes, and the environment. Front. Plant Sci. 2019, 10, 813. [Google Scholar] [CrossRef]
- Michotey, C.; Chaves, I.; Anger, C.; Bastien, C.; Jorge, V.; Ehrenmann, F.; Adam-Blondon, A.-F.; Miguel, C. Woody Plant Ontology—A New Ontology for Describing Woody Plant Traits COSTFA1306 Meeting 2018—Plant Phenotyping for Future Climate Challenges; University of Leuven: Leuven, Belgium, 2018. [Google Scholar]
- Kindermann, G.E.; Kristöfel, F.; Neumann, M.; Rössler, G.; Ledermann, T.; Schueler, S. 109 years of forest growth measurements from individual Norway spruce trees. Sci. Data 2018, 5, 180077. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, A.M.; Ribeiro-Barros, A.I.; António, C. Experimental Design and Sample Preparation in Forest Tree Metabolomics. Metabolites 2019, 9, 285. https://doi.org/10.3390/metabo9120285
Rodrigues AM, Ribeiro-Barros AI, António C. Experimental Design and Sample Preparation in Forest Tree Metabolomics. Metabolites. 2019; 9(12):285. https://doi.org/10.3390/metabo9120285
Chicago/Turabian StyleRodrigues, Ana M., Ana I. Ribeiro-Barros, and Carla António. 2019. "Experimental Design and Sample Preparation in Forest Tree Metabolomics" Metabolites 9, no. 12: 285. https://doi.org/10.3390/metabo9120285
APA StyleRodrigues, A. M., Ribeiro-Barros, A. I., & António, C. (2019). Experimental Design and Sample Preparation in Forest Tree Metabolomics. Metabolites, 9(12), 285. https://doi.org/10.3390/metabo9120285