
Mendelian Randomization Analysis of Systemic Iron Status and Risk of Metabolic Dysfunction-Associated Steatotic Liver Disease

 , , , ,
, , , ,  ,
, 
Highlights
- Genetically elevated systemic iron status is causally associated with increased risks of hepatic steatosis and fibrosis/cirrhosis in MASLD.
- Iron homeostasis and ferroptosis represent potential targets for risk stratification and therapeutic intervention in MASLD.
Abstract
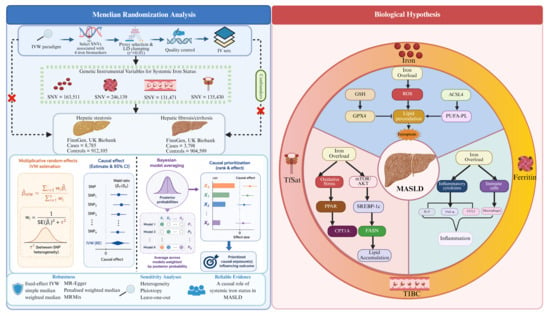
1. Introduction
2. Methods
2.1. MR Analysis Study Design
2.2. Data Sources from Three Genome-Wide Association Studies
2.3. Choosing the Instrumental Variables
2.4. Statistical Analysis
2.5. Sensitivity Analysis and Instrument Strength
3. Results
3.1. Features of SNVs Utilized as Genetic Tools
3.2. Main Analysis
3.3. Sensitivity Analysis
3.4. Analysis Using Different IV Selection Strategies
3.5. Analysis with Different MR Methods
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Fouad, Y.; Alboraie, M.; Shiha, G. Epidemiology and Diagnosis of Metabolic Dysfunction-Associated Fatty Liver Disease. Hepatol. Int. 2024, 18, 827–833. [Google Scholar] [CrossRef]
- Hagström, H.; Shang, Y.; Hegmar, H.; Nasr, P. Natural History and Progression of Metabolic Dysfunction-Associated Steatotic Liver Disease. Lancet Gastroenterol. Hepatol. 2024, 9, 944–956. [Google Scholar] [CrossRef]
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander Sharma, B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef]
- Wang, X.; Wu, Z.; Liu, Y.; Wu, C.; Jiang, J.; Hashimoto, K.; Zhou, X. The Role of Thyroid-Stimulating Hormone in Regulating Lipid Metabolism: Implications for Body–Brain Communication. Neurobiol. Dis. 2024, 201, 106658. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Wang, L. Dietary Pattern and Hepatic Lipid Metabolism. Liver Res. 2023, 7, 275–284. [Google Scholar] [CrossRef]
- WHO. Guideline on Use of Ferritin Concentrations to Assess Iron Status in Populations, 1st ed.; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Dale, J.C.; Burritt, M.F.; Zinsmeister, A.R. Diurnal Variation of Serum Iron, Iron-Binding Capacity, Transferrin Saturation, and Ferritin Levels. Am. J. Clin. Pathol. 2002, 117, 802–808. [Google Scholar] [CrossRef]
- Institute of Medicine. Dietary Reference Intakes: The Essential Guide to Nutrient Requirements; National Academies Press: Washington, DC, USA, 2006; p. 11537. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron Homeostasis and Oxidative Stress: An Intimate Relationship. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef] [PubMed]
- Pu, F.; Chen, F.; Zhang, Z.; Shi, D.; Zhong, B.; Lv, X.; Tucker, A.B.; Fan, J.; Li, A.J.; Qin, K.; et al. Ferroptosis as a Novel Form of Regulated Cell Death: Implications in the Pathogenesis, Oncometabolism and Treatment of Human Cancer. Genes Dis. 2022, 9, 347–357. [Google Scholar] [CrossRef] [PubMed]
- El-Sehrawy, A.A.M.A.; Rashid, T.A.; Ullah, M.I.; Uthirapathy, S.; Ganesan, S.; Singh, A.; Devi, A.; Joshi, K.K.; Jasim, A.S.; Kadhim, A.J. Cutting Edge: Ferroptosis in Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD) Pathogenesis and Therapy. Funct. Integr. Genom. 2025, 25, 71. [Google Scholar] [CrossRef]
- Peleman, C.; Hellemans, S.; Veeckmans, G.; Arras, W.; Zheng, H.; Koeken, I.; Van San, E.; Hassannia, B.; Walravens, M.; Kayirangwa, E.; et al. Ferroptosis Is a Targetable Detrimental Factor in Metabolic Dysfunction-Associated Steatotic Liver Disease. Cell Death Differ. 2024, 31, 1113–1126. [Google Scholar] [CrossRef]
- Makri, E.; Orfanidou, M.; Makri, E.S.; Goulas, A.; Terpos, E.; Polyzos, S.A. Circulating Ferritin in Patients with Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. J. Clin. Exp. Hepatol. 2024, 14, 101353. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Liu, L.; Qin, T.; Luo, Y.; Song, C.; Chen, X.; Duan, H.; Jiang, Y.; Zeng, H.; Wan, H.; et al. Associations of Serum Iron Status with MAFLD and Liver Fibrosis in the USA: A Nationwide Cross-Section Study. Biol. Trace Elem. Res. 2024, 202, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Suresh, D.; Li, A.; Miller, M.J.; Wijarnpreecha, K.; Chen, V.L. Associations between Metabolic Hyperferritinaemia, Fibrosis-Promoting Alleles and Clinical Outcomes in Steatotic Liver Disease. Liver Int. 2024, 44, 389–398. [Google Scholar] [CrossRef]
- Guo, W.; Weng, T.; Song, Y. Association of Serum Iron Status with MASLD and Liver Fibrosis. PLoS ONE 2025, 20, e0319057. [Google Scholar] [CrossRef]
- Yang, H.-H.; Chen, G.-C.; Li, D.-M.; Lan, L.; Chen, L.-H.; Xu, J.-Y.; Qin, L.-Q. Serum Iron and Risk of Nonalcoholic Fatty Liver Disease and Advanced Hepatic Fibrosis in US Adults. Sci. Rep. 2021, 11, 10387. [Google Scholar] [CrossRef]
- Yu, Y.-C.; Luu, H.N.; Wang, R.; Thomas, C.E.; Glynn, N.W.; Youk, A.O.; Behari, J.; Yuan, J.-M. Serum Biomarkers of Iron Status and Risk of Hepatocellular Carcinoma Development in Patients with Nonalcoholic Fatty Liver Disease. Cancer Epidemiol. Biomark. Prev. 2022, 31, 230–235. [Google Scholar] [CrossRef]
- Yu, Y.; Jiang, L.; Wang, H.; Shen, Z.; Cheng, Q.; Zhang, P.; Wang, J.; Wu, Q.; Fang, X.; Duan, L.; et al. Hepatic Transferrin Plays a Role in Systemic Iron Homeostasis and Liver Ferroptosis. Blood 2020, 136, 726–739. [Google Scholar] [CrossRef]
- Moksnes, M.R.; Graham, S.E.; Wu, K.-H.; Hansen, A.F.; Gagliano Taliun, S.A.; Zhou, W.; Thorstensen, K.; Fritsche, L.G.; Gill, D.; Mason, A.; et al. Genome-Wide Meta-Analysis of Iron Status Biomarkers and the Effect of Iron on All-Cause Mortality in HUNT. Commun. Biol. 2022, 5, 591. [Google Scholar] [CrossRef]
- Kurki, M.I.; Karjalainen, J.; Palta, P.; Sipilä, T.P.; Kristiansson, K.; Donner, K.M.; Reeve, M.P.; Laivuori, H.; Aavikko, M.; Kaunisto, M.A.; et al. FinnGen Provides Genetic Insights from a Well-Phenotyped Isolated Population. Nature 2023, 613, 508–518. [Google Scholar] [CrossRef]
- Bell, S.; Rigas, A.S.; Magnusson, M.K.; Ferkingstad, E.; Allara, E.; Bjornsdottir, G.; Ramond, A.; Sørensen, E.; Halldorsson, G.H.; Paul, D.S.; et al. A Genome-Wide Meta-Analysis Yields 46 New Loci Associating with Biomarkers of Iron Homeostasis. Commun. Biol. 2021, 4, 156. [Google Scholar] [CrossRef] [PubMed]
- Staley, J.R.; Blackshaw, J.; Kamat, M.A.; Ellis, S.; Surendran, P.; Sun, B.B.; Paul, D.S.; Freitag, D.; Burgess, S.; Danesh, J.; et al. PhenoScanner: A Database of Human Genotype–Phenotype Associations. Bioinformatics 2016, 32, 3207–3209. [Google Scholar] [CrossRef]
- Trowsdale, J.; Knight, J.C. Major Histocompatibility Complex Genomics and Human Disease. Annu. Rev. Genom. Hum. Genet. 2013, 14, 301–323. [Google Scholar] [CrossRef]
- EPIC-InterAct Consortium; Burgess, S.; Scott, R.A.; Timpson, N.J.; Davey Smith, G.; Thompson, S.G. Using Published Data in Mendelian Randomization: A Blueprint for Efficient Identification of Causal Risk Factors. Eur. J. Epidemiol. 2015, 30, 543–552. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Thompson, S.G. Interpreting Findings from Mendelian Randomization Using the MR-Egger Method. Eur. J. Epidemiol. 2017, 32, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Chatterjee, N. Mendelian Randomization Analysis Using Mixture Models for Robust and Efficient Estimation of Causal Effects. Nat. Commun. 2019, 10, 1941. [Google Scholar] [CrossRef] [PubMed]
- Zuber, V.; Colijn, J.M.; Klaver, C.; Burgess, S. Selecting Likely Causal Risk Factors from High-Throughput Experiments Using Multivariable Mendelian Randomization. Nat. Commun. 2020, 11, 29. [Google Scholar] [CrossRef]
- Palmer, T.M.; Lawlor, D.A.; Harbord, R.M.; Sheehan, N.A.; Tobias, J.H.; Timpson, N.J.; Smith, G.D.; Sterne, J.A. Using Multiple Genetic Variants as Instrumental Variables for Modifiable Risk Factors. Stat. Methods Med. Res. 2012, 21, 223–242. [Google Scholar] [CrossRef]
- Higgins, J.P.T.; Thompson, S.G.; Deeks, J.J.; Altman, D.G. Measuring Inconsistency in Meta-Analyses. BMJ 2003, 327, 557–560. [Google Scholar] [CrossRef]
- Bowden, J.; Del Greco, M.F.; Minelli, C.; Davey Smith, G.; Sheehan, N.; Thompson, J. A Framework for the Investigation of Pleiotropy in Two-Sample Summary Data Mendelian Randomization. Stat. Med. 2017, 36, 1783–1802. [Google Scholar] [CrossRef]
- Del Greco, M.F.; Foco, L.; Pichler, I.; Eller, P.; Eller, K.; Benyamin, B.; Whitfield, J.B.; Genetics of Iron Status Consortium; CKDGen Consortium; Pramstaller, P.P.; et al. Serum Iron Level and Kidney Function: A Mendelian Randomization Study. Nephrol. Dial. Transplant. 2017, 32, 273–278. [Google Scholar] [CrossRef]
- Gill, D.; Monori, G.; Tzoulaki, I.; Dehghan, A. Iron Status and Risk of Stroke: A Mendelian Randomization Study. Stroke 2018, 49, 2815–2821. [Google Scholar] [CrossRef]
- Barad, A.; Clark, A.G.; Pressman, E.K.; O’Brien, K.O. Associations Between Genetically Predicted Iron Status and Cardiovascular Disease Risk: A Mendelian Randomization Study. J. Am. Heart Assoc. 2024, 13, e034991. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.; Del Greco, M.F.; Walker, A.P.; Srai, S.K.S.; Laffan, M.A.; Minelli, C. The Effect of Iron Status on Risk of Coronary Artery Disease: A Mendelian Randomization Study—Brief Report. Arterioscler. Thromb. Vasc. Biol 2017, 37, 1788–1792. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liu, Y.; Xu, S.; Wang, Q.; Xu, F.; Liu, Y. Mendelian Randomization Study Reveals a Causal Relationship between Serum Iron Status and Coronary Heart Disease and Related Cardiovascular Diseases. Front. Cardiovasc. Med. 2023, 10, 1152201. [Google Scholar] [CrossRef]
- Wang, X.; Fang, X.; Zheng, W.; Zhou, J.; Song, Z.; Xu, M.; Min, J.; Wang, F. Genetic Support of A Causal Relationship Between Iron Status and Type 2 Diabetes: A Mendelian Randomization Study. J. Clin. Endocrinol. Metab. 2021, 106, e4641–e4651. [Google Scholar] [CrossRef]
- Zhou, J.; Shi, W.; Wu, D.; Wang, S.; Wang, X.; Min, J.; Wang, F. Mendelian Randomization Analysis of Systemic Iron Status and Risk of Different Types of Kidney Disease. Nutrients 2024, 16, 1978. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Lian, F.; Fang, X. Iron Status and Mental Disorders: A Mendelian Randomization Study. Front. Nutr. 2022, 9, 1084860. [Google Scholar] [CrossRef]
- Wang, Z.; He, Y.; Wang, Y.; Shu, X.; Long, B.; Zhou, W.; Sui, J. Iron Status and Cancer Risk across 14 Cancers: Evidence from Cross-Sectional Analyses and a Three-Stage Mendelian Randomization. Int. J. Surg. 2026, 112, 8120–8135. [Google Scholar] [CrossRef]
- Yu, Z.; Xu, C.; Fang, C.; Zhang, F. Causal Effect of Iron Status on Lung Function: A Mendelian Randomization Study. Front. Nutr. 2022, 9, 1025212. [Google Scholar] [CrossRef]
- Sun, K.; Zhao, J.V.; Nelson, E.A.S.; Wong, V.W.S.; Lam, H.S.H.S.; Hui, L.L. Iron Status and Non-Alcoholic Fatty Liver Disease: A Mendelian Randomization Study. Nutrition 2024, 118, 112295. [Google Scholar] [CrossRef]
- Liu, C.; Chen, Y.; Zhang, Z.; Xie, J.; Yu, C.; Xu, L.; Li, Y. Iron Status and NAFLD among European Populations: A Bidirectional Two-Sample Mendelian Randomization Study. Nutrients 2022, 14, 5237. [Google Scholar] [CrossRef]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on Iron and Its Importance for Human Health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar]
- Sudarev, V.V.; Dolotova, S.M.; Bukhalovich, S.M.; Bazhenov, S.V.; Ryzhykau, Y.L.; Uversky, V.N.; Bondarev, N.A.; Osipov, S.D.; Mikhailov, A.E.; Kuklina, D.D.; et al. Ferritin Self-Assembly, Structure, Function, and Biotechnological Applications. Int. J. Biol. Macromol. 2023, 224, 319–343. [Google Scholar] [CrossRef]
- Faruk, M.O.; Ahmed, K.U.; Ishrat, N.; Mrittika, S.A.; Alam, S.; Hasan, M.; Tanjum, F.A.; Mohammad, N.; Mahmud, A.; Amin, M.N. Liver Cirrhosis: Epidemiology, Risk Factors, Potential Complications, and Possible Treatment Strategies. Gastroenterol. Endosc. 2026, 4, 117–129. [Google Scholar] [CrossRef]
- Li, J.; Xiang, Z.; Xu, Y.; Luo, X.; Jiang, Y.; Huang, Y.; Yang, Z.; Chen, R.; Xu, X. Targeting Cellular Senescence: A New Therapeutic Axis in Chronic Liver Disease. Engineering 2026. [Google Scholar] [CrossRef]
- Tran, V.V.; Kanwal, F.; Cholankeril, G. Beyond the Baseline: Longitudinal Surveillance with Noninvasive Tests for Fibrosis in MASLD. Hepatology 2025, 81, 1397–1399. [Google Scholar] [CrossRef] [PubMed]
- St Aubin, C.R.; Fisher, A.L.; Hernandez, J.A.; Broderick, T.L.; Al-Nakkash, L. Mitigation of MAFLD in High Fat-High Sucrose-Fructose Fed Mice by a Combination of Genistein Consumption and Exercise Training. Diabetes Metab. Syndr. Obes. 2022, 15, 2157–2172. [Google Scholar] [CrossRef]
- Shao, T.; Chung, R.T. Ironing out MAFLD: Therapeutic Targeting of Liver Ferroptosis. Cell Metab. 2024, 36, 2167–2169. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Yu, Y.; Xie, E.; Wu, Q.; Yin, X.; Zhao, B.; Min, J.; Wang, F. Hepatic HDAC3 Regulates Systemic Iron Homeostasis and Ferroptosis via the Hippo Signaling Pathway. Research 2023, 6, 0281. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Belt, P.; Wilson, L.A.; Yeh, M.M.; Neuschwander-Tetri, B.A.; Chalasani, N.; Sanyal, A.J.; Nelson, J.E. NASH Clinical Research Network. Serum Ferritin Is an Independent Predictor of Histologic Severity and Advanced Fibrosis in Patients with Nonalcoholic Fatty Liver Disease. Hepatology 2012, 55, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.-W.; Chang, Y.; Sung, E.; Shin, H.; Ryu, S. Serum Ferritin Levels Predict Incident Non-Alcoholic Fatty Liver Disease in Healthy Korean Men. Metabolism 2012, 61, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of Hepatic Fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exposure | Hepatic Steatosis—MRMix | Hepatic Fibrosis/Cirrhosis—MRMix | ||||
|---|---|---|---|---|---|---|
| θ | π0 | σ2 | θ | π0 | σ2 | |
| Iron | 0.45 | 0.999 | 1.47 × 10−3 | 0.46 | 0.999 | 5.48 × 10−3 |
| Ferritin | 0.20 | 0.999 | 1.59 × 10−2 | 0.60 | 0.999 | 4.24 × 10−2 |
| TfSat | 0.02 | 0.923 | 8.26 × 10−4 | 0.26 | 0.999 | 2.80 × 10−3 |
| TIBC | −0.15 | 0.999 | 1.92 × 10−2 | −0.31 | 0.999 | 4.07 × 10−2 |
| (A) Model averaging for risk factors | |||
| Ranking by MIP | Risk factor | MIP | MACE |
| 1 | Iron | 0.850 | 0.295 |
| 2 | TfSat | 0.187 | 0.035 |
| 3 | TIBC | 0.067 | 0.005 |
| 4 | Ferritin | 0.057 | 0.005 |
| (B) The best 10 individual models | |||
| Ranking by PP | Model | PP | λ |
| 1 | Iron | 0.725 | 0.344 |
| 3 | TfSat | 0.117 | 0.217 |
| 1, 2 | Iron, Ferritin | 0.042 | 0.313, 0.069 |
| 1, 3 | Iron, TfSat | 0.038 | 0.338, 0.004 |
| 1, 4 | Iron, TIBC | 0.036 | 0.448, 0.007 |
| 3, 4 | TfSat, TIBC | 0.016 | 0.393, 0.182 |
| 2, 3 | Ferritin, TfSat | 0.008 | 0.104, 0.186 |
| 4 | TIBC | 0.006 | −0.215 |
| 1, 3, 4 | Iron, TfSat, TIBC | 0.004 | 0.309, 0.175, 0.159 |
| 1, 2, 4 | Iron, Ferritin, TIBC | 0.002 | 0.421, 0.079, 0.076 |
| (A) Model averaging for risk factors | |||
| Ranking by MIP | Risk factor | MIP | MACE |
| 1 | TIBC | 0.604 | −0.240 |
| 2 | TfSat | 0.235 | 0.051 |
| 3 | Iron | 0.212 | 0.049 |
| 4 | Ferritin | 0.125 | 0.057 |
| (B) The best 10 individual models | |||
| Ranking by PP | Model | PP | λ |
| 4 | TIBC | 0.476 | −0.358 |
| 3 | TfSat | 0.162 | 0.330 |
| 1 | Iron | 0.132 | 0.493 |
| 2 | Ferritin | 0.066 | 0.825 |
| 1, 4 | Iron, TIBC | 0.047 | −0.331, −0.569 |
| 3, 4 | TfSat, TIBC | 0.040 | −0.271, −0.632 |
| 2, 4 | Ferritin, TIBC | 0.031 | −0.018, −0.363 |
| 1, 3 | Iron, TfSat | 0.015 | −0.137, 0.416 |
| 2, 3 | Ferritin, TfSat | 0.011 | 0.103, 0.300 |
| 1, 2 | Iron, Ferritin | 0.01 | 0.400, 0.205 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Yue, W.; Yang, Y.; Ma, J.; Zhang, J.; Wang, X.; Min, J.; Wang, F. Mendelian Randomization Analysis of Systemic Iron Status and Risk of Metabolic Dysfunction-Associated Steatotic Liver Disease. Metabolites 2026, 16, 356. https://doi.org/10.3390/metabo16060356
Yue W, Yang Y, Ma J, Zhang J, Wang X, Min J, Wang F. Mendelian Randomization Analysis of Systemic Iron Status and Risk of Metabolic Dysfunction-Associated Steatotic Liver Disease. Metabolites. 2026; 16(6):356. https://doi.org/10.3390/metabo16060356
Chicago/Turabian StyleYue, Wuyang, Yi Yang, Jinling Ma, Jiale Zhang, Xinhui Wang, Junxia Min, and Fudi Wang. 2026. "Mendelian Randomization Analysis of Systemic Iron Status and Risk of Metabolic Dysfunction-Associated Steatotic Liver Disease" Metabolites 16, no. 6: 356. https://doi.org/10.3390/metabo16060356
APA StyleYue, W., Yang, Y., Ma, J., Zhang, J., Wang, X., Min, J., & Wang, F. (2026). Mendelian Randomization Analysis of Systemic Iron Status and Risk of Metabolic Dysfunction-Associated Steatotic Liver Disease. Metabolites, 16(6), 356. https://doi.org/10.3390/metabo16060356