EM Dipeptide Enhances Milk Protein Secretion: Evidence from Integrated Metabolomic and Transcriptomic Analysis

 , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Cell Viability
2.3. 5-Ethynyl-2′-Deoxyuridine (EdU) Incorporation Assay for Cell Proliferation
2.4. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
2.5. Immunofluorescence Assay
2.6. RNA-Seq and Transcriptomic Data Analysis
2.7. Untargeted Metabolomics Analysis
2.8. Multi-Omics Integration of Transcriptome and Metabolome
2.9. Quantitative Real-Time PCR (qRT-PCR) Validation of Selected DEGs
2.10. Statistical Analysis
3. Results
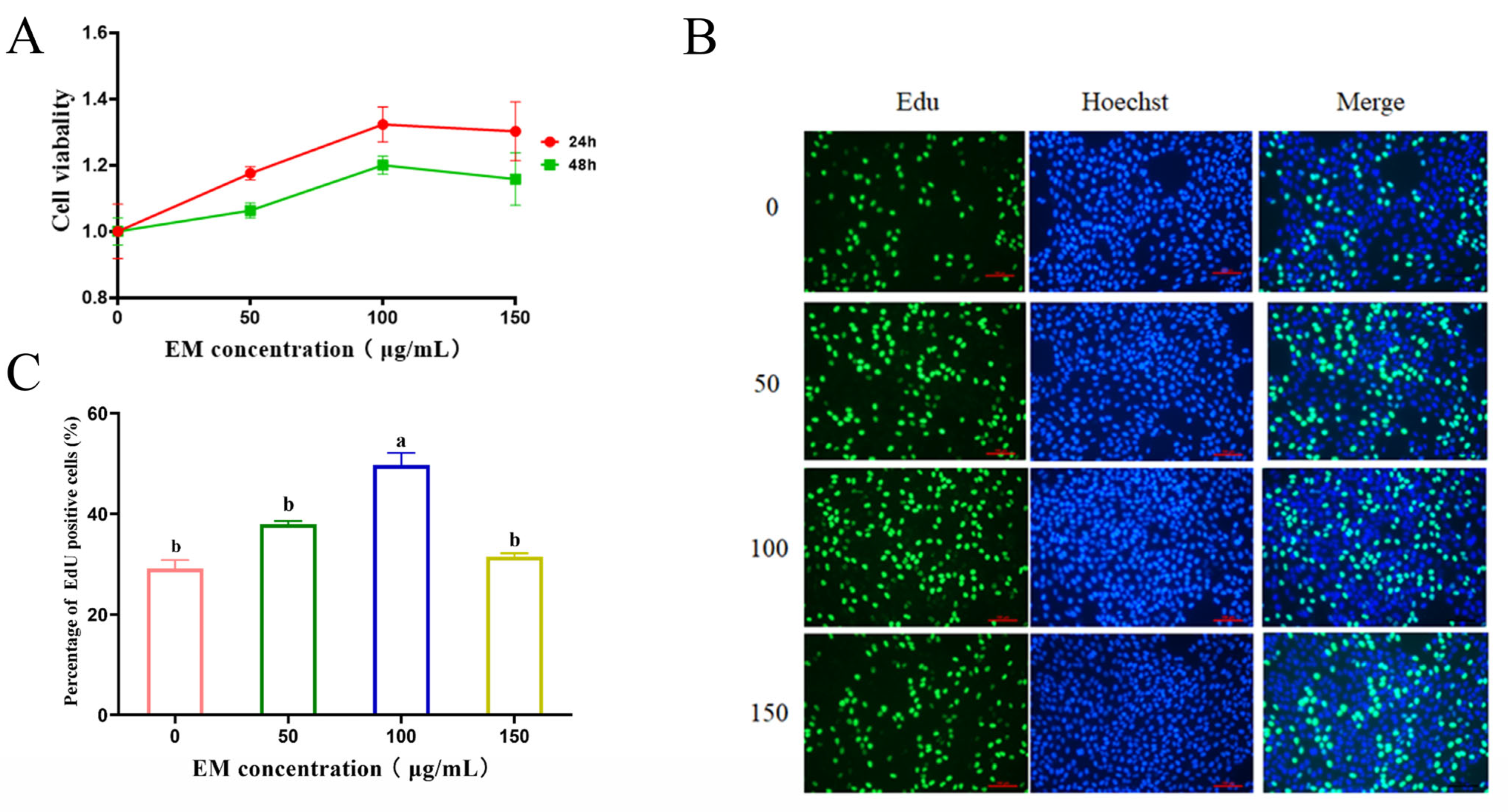
3.1. EM Promotes MCF-10A Cell Proliferation
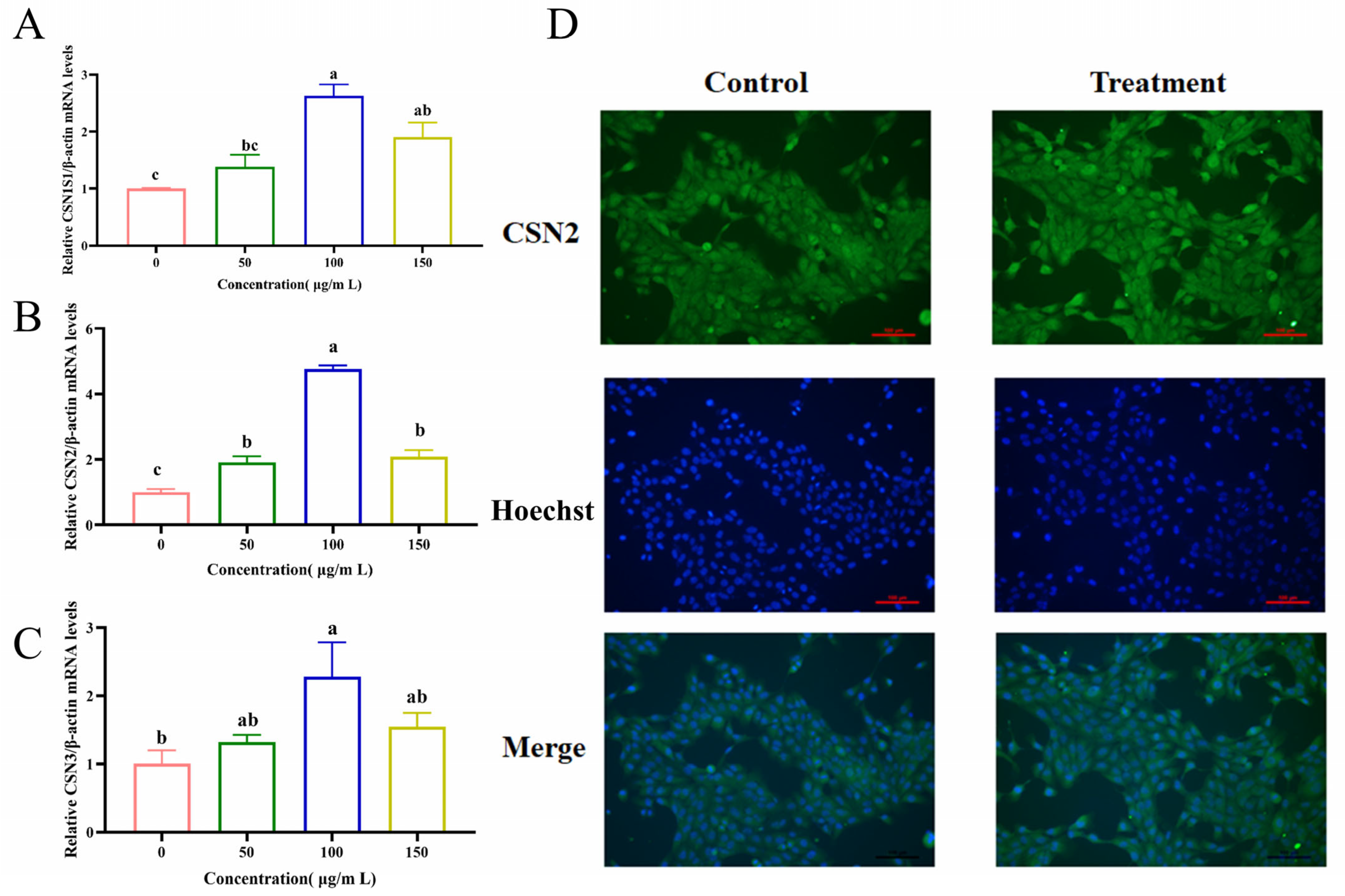
3.2. EM Enhances Milk Protein Synthesis at the Transcriptional and Protein Levels
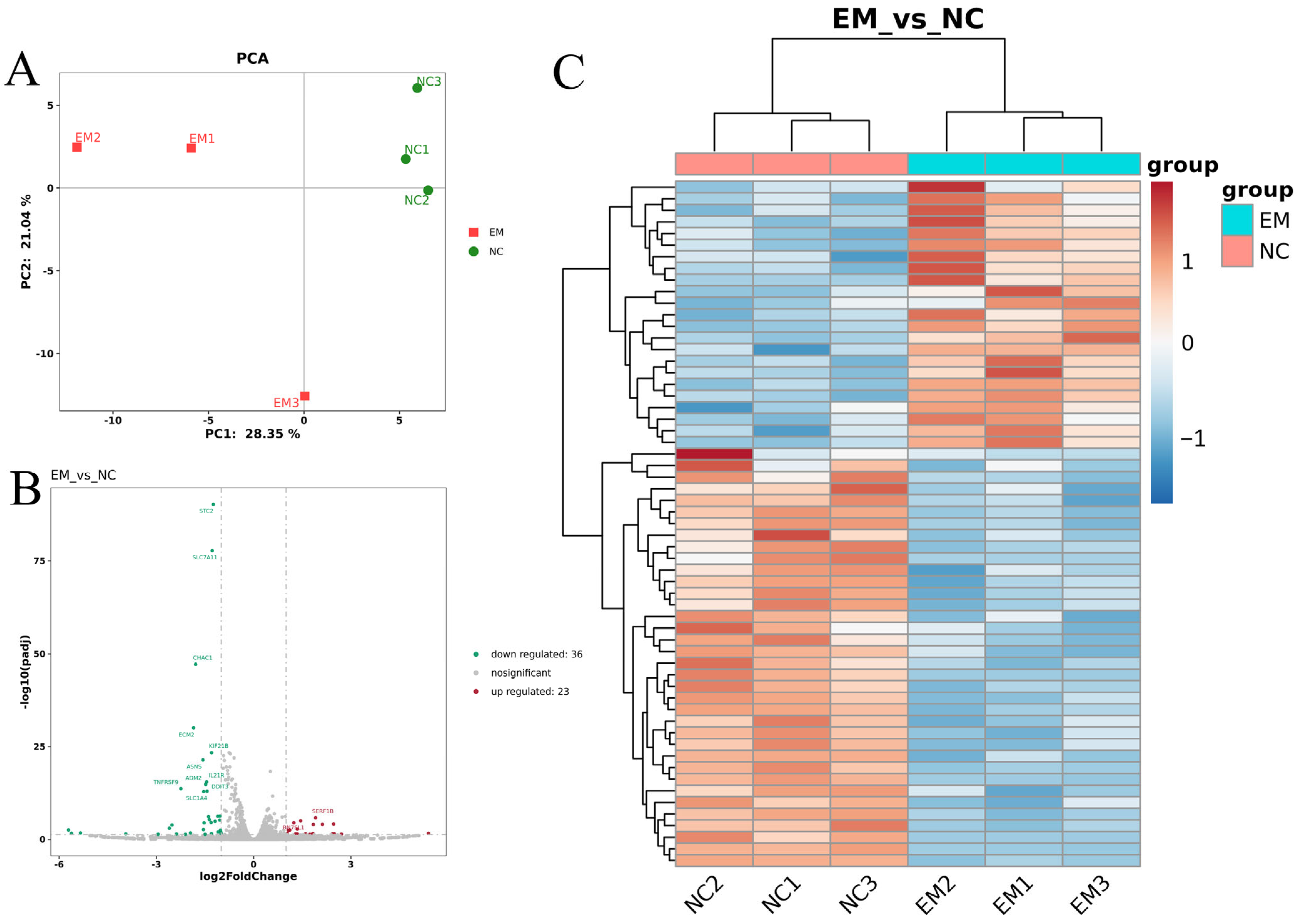
3.3. EM Reprograms Transcription to Promote Lactation
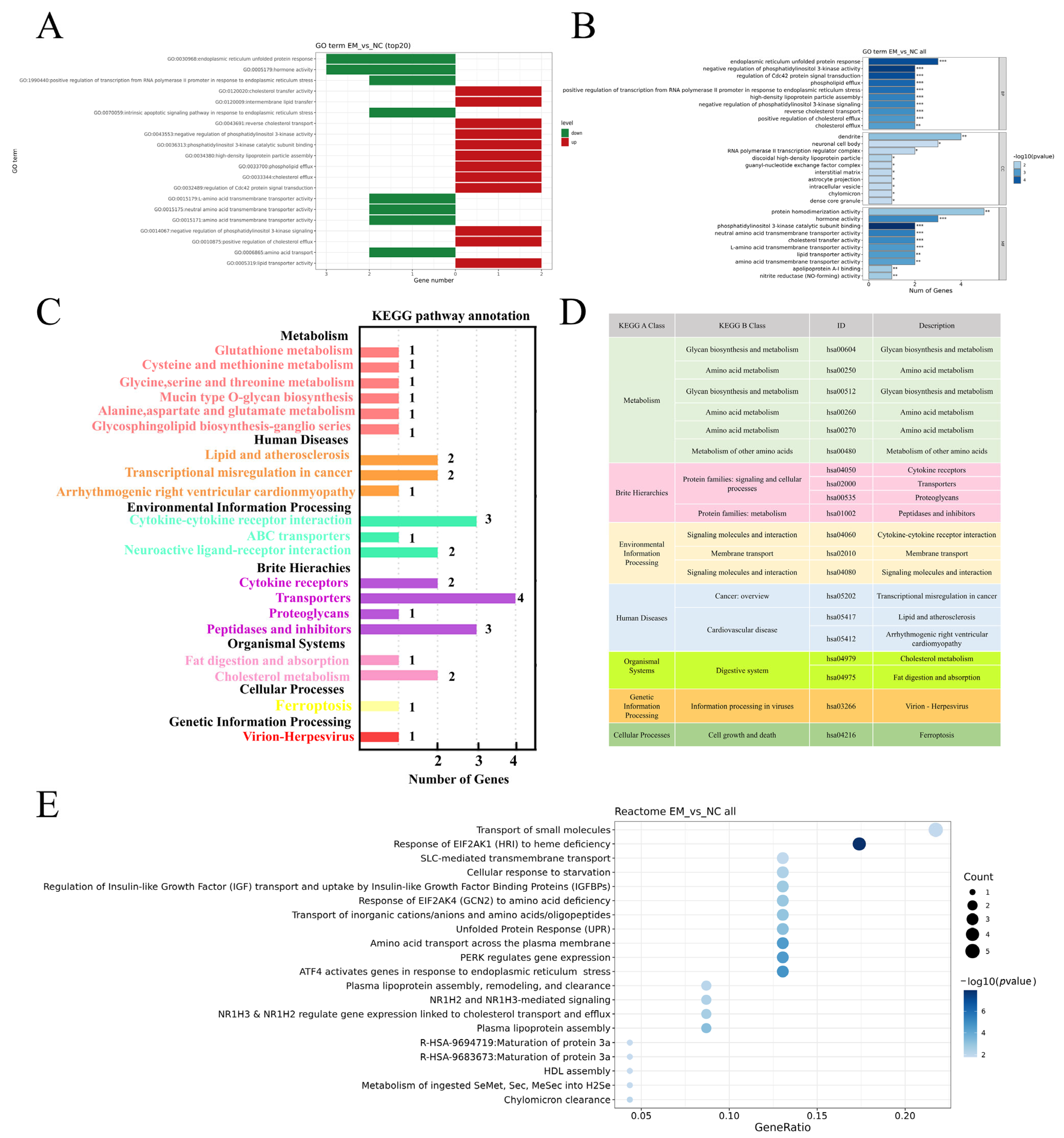
3.4. EM Reprograms Metabolism to Support Milk Component Synthesis
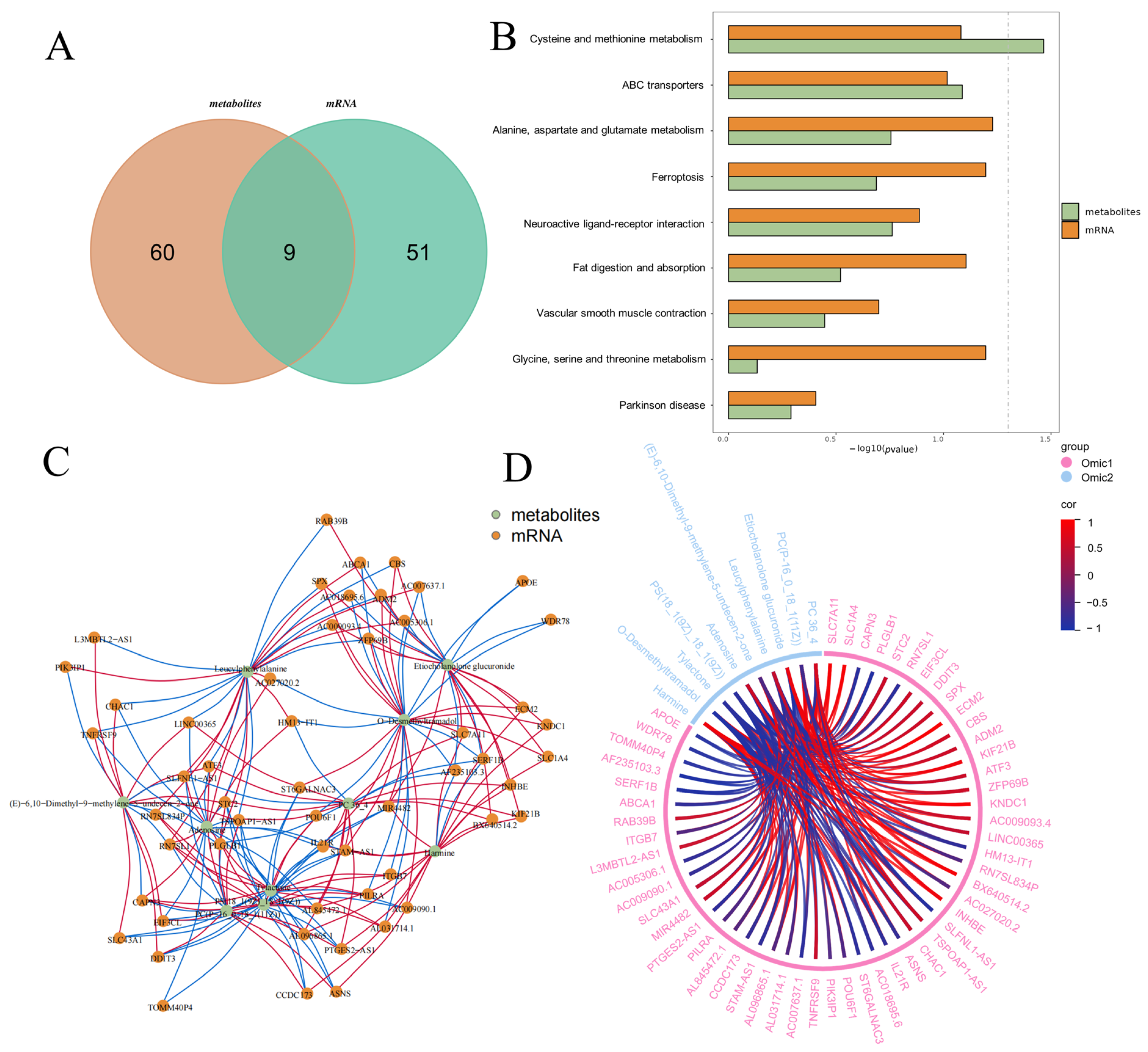
3.5. Integrated Omics Reveal Core Lactogenic Regulatory Networks Induced by EM
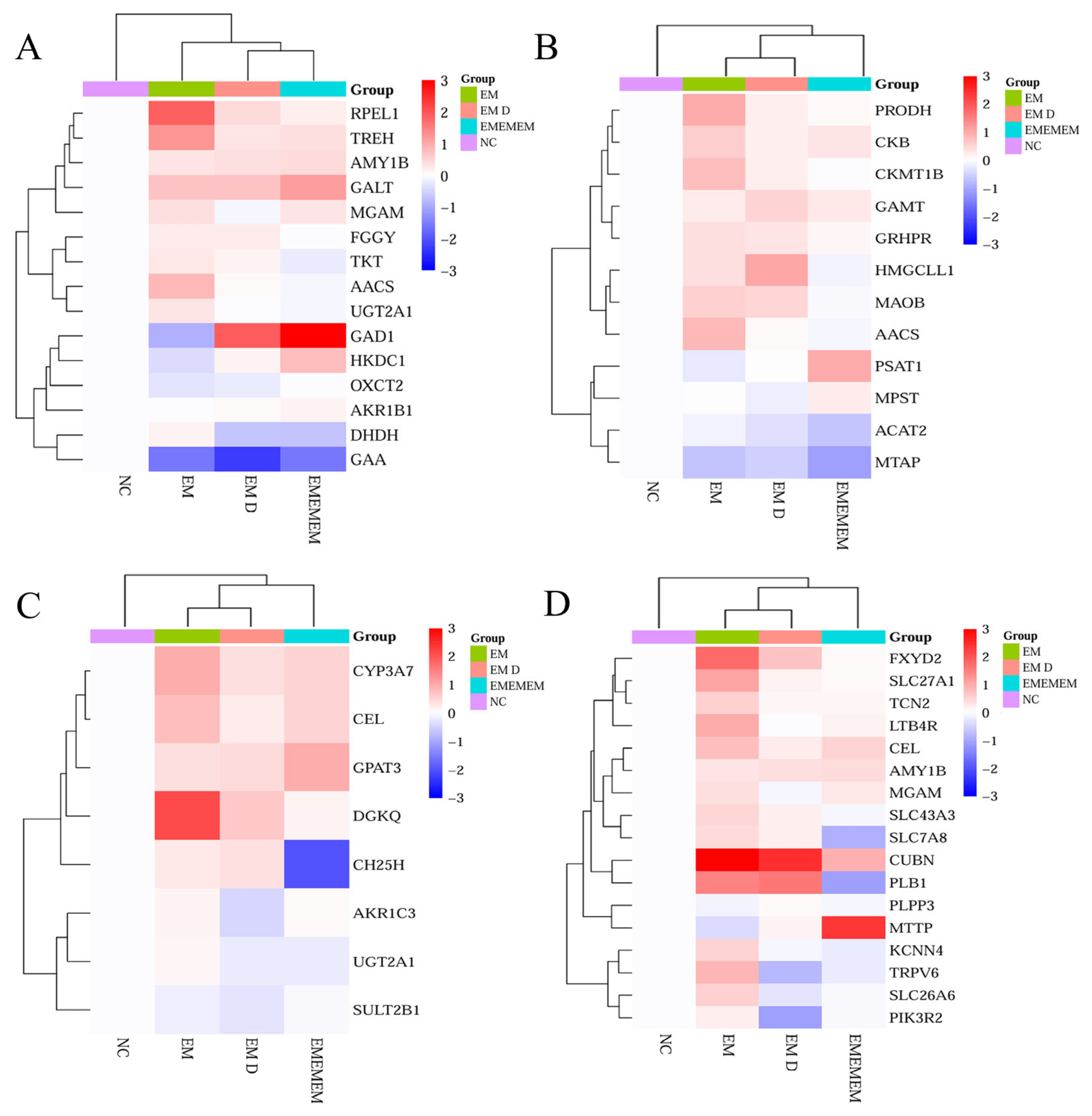
3.6. Validation of Differential Genes of Differential Metabolites
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Full Term |
| ABC | ATP-binding cassette (transporters) |
| AICAR | 5-Aminoimidazole-4-carboxamide ribonucleotide |
| AMPK | AMP-activated protein kinase |
| ATF4 | Activating transcription factor 4 |
| CCK-8 | Cell Counting Kit-8 |
| CSN1S1/CSN2/CSN3 | Casein genes (alpha-S1, beta, and kappa-casein) |
| DEG | Differentially expressed gene |
| DMEM/F-12 | Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 |
| EdU | 5-ethynyl-2′-deoxyuridine |
| EIF2AK1/EIF2AK4 | Eukaryotic translation initiation factor 2 alpha kinase 1/4 |
| EM | Methionine-containing dipeptide |
| FBS | Fetal bovine serum |
| GO | Gene ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LC-MS/MS | Liquid chromatography–tandem mass spectrometry |
| MCF-10A | Human mammary epithelial cell line MCF-10A |
| mTOR | Mammalian target of rapamycin |
| PBS | Phosphate-buffered saline |
| PCA | Principal component analysis |
| PepT1 | Peptide transporter 1 |
| PERK | Protein kinase RNA-like endoplasmic reticulum kinase |
| PI3K | Phosphoinositide 3-kinase |
| PLS-DA | Partial least squares discriminant analysis |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| RNA-seq | RNA sequencing |
| SAM | S-adenosylmethionine |
| VIP | Variable importance in projection |
References
- Garwolińska, D.; Namieśnik, J.; Kot-Wasik, A.; Hewelt-Belka, W. Chemistry of Human Breast Milk-A Comprehensive Review of the Composition and Role of Milk Metabolites in Child Development. J. Agric. Food Chem. 2018, 66, 11881–11896. [Google Scholar] [CrossRef] [PubMed]
- Ramiro-Cortijo, D.; Singh, P.; Liu, Y.; Medina-Morales, E.; Yakah, W.; Freedman, S.D.; Martin, C.R. Breast Milk Lipids and Fatty Acids in Regulating Neonatal Intestinal Development and Protecting against Intestinal Injury. Nutrients 2020, 12, 534. [Google Scholar] [CrossRef] [PubMed]
- Thai, J.D.; Gregory, K.E. Bioactive Factors in Human Breast Milk Attenuate Intestinal Inflammation during Early Life. Nutrients 2020, 12, 581. [Google Scholar] [CrossRef] [PubMed]
- Gridneva, Z.; Tie, W.J.; Rea, A.; Lai, C.T.; Ward, L.C.; Murray, K.; Hartmann, P.E.; Geddes, D.T. Human Milk Casein and Whey Protein and Infant Body Composition over the First 12 Months of Lactation. Nutrients 2018, 10, 1332. [Google Scholar] [CrossRef] [PubMed]
- Sangild, P.T.; Vonderohe, C.; Melendez Hebib, V.; Burrin, D.G. Potential Benefits of Bovine Colostrum in Pediatric Nutrition and Health. Nutrients 2021, 13, 2551. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Khan, M.Z.; Khan, I.M.; Ullah, Q.; Cisang, Z.M.; Zhang, N.; Wu, D.; Huang, B.; Ma, Y.; Khan, A.; et al. Genetics, environmental stress, and amino acid supplementation affect lactational performance via mTOR signaling pathway in bovine mammary epithelial cells. Front. Genet. 2023, 14, 1195774. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Li, P.; Hao, G.; Liu, Y.; Wang, T.; Liu, B. Enhancing Milk Production by Nutrient Supplements: Strategies and Regulatory Pathways. Animals 2023, 13, 419. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Wang, L.; Zhang, M.; Wang, Z.; Gao, X.; Li, M. Methionine and leucine induce ARID1A degradation to promote mTOR expression and milk synthesis in mammary epithelial cells. J. Nutr. Biochem. 2021, 101, 108924. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Zhang, M. The regulatory mechanism of amino acids on milk protein and fat synthesis in mammary epithelial cells: A mini review. Anim. Biotechnol. 2021, 34, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Yu, M.; Fan, X.; Zhou, Y.; Zhang, M.; Gao, X. Methionine and Leucine Promote mTOR Gene Transcription and Milk Synthesis in Mammary Epithelial Cells through the eEF1Bα-UBR5-ARID1A Signaling. J. Agric. Food Chem. 2024, 72, 11733–11745. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, R.; Wu, Z.; Hou, Y.; Bazer, F.W.; Wu, G. Amino acids and mammary gland development: Nutritional implications for milk production and neonatal growth. J. Anim. Sci. Biotechnol. 2016, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.C.; Zhao, S.G.; Wang, S.S.; Luo, C.C.; Gao, H.N.; Zheng, N.; Wang, J.Q. d-Glucose and amino acid deficiency inhibits casein synthesis through JAK2/STAT5 and AMPK/mTOR signaling pathways in mammary epithelial cells of dairy cows. J. Dairy Sci. 2018, 101, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, Y.; Yu, X.Y.; Zeng, T.Y. The rates and factors of perceived insufficient milk supply: A systematic review. Matern. Child Nutr. 2022, 18, e13255. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Han, N.; Liu, H. Supplementation of Methionine Dipeptide Enhances the Milking Performance of Lactating Dairy Cows. Animals 2024, 14, 1339. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xu, L.; Zhao, F.; Liu, H. Regulation of Milk Protein Synthesis by Free and Peptide-Bound Amino Acids in Dairy Cows. Biology 2021, 10, 1044. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.X.; Wang, C.H.; Xu, Q.B.; Zhao, F.Q.; Liu, J.X.; Liu, H.Y. Methionyl-Methionine Promotes α-s1 Casein Synthesis in Bovine Mammary Gland Explants by Enhancing Intracellular Substrate Availability and Activating JAK2-STAT5 and mTOR-Mediated Signaling Pathways. J. Nutr. 2015, 145, 1748–1753. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Wei, X.S.; Dai, W.T.; Xu, L.B.; Wang, C.; Liu, J.X. The functional and regulatory entities underlying free and peptide-bound amino acid transporters in the bovine mammary gland. JDS Commun. 2023, 4, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Xu, Y. The role and molecular mechanisms of amino acids in enhancing mammary gland milk synthesis - a comprehensive review. Food Res. Int. 2025, 211, 116355. [Google Scholar] [CrossRef] [PubMed]
- Gilbreath, K.R.; Bazer, F.W.; Satterfield, M.C.; Wu, G. Amino Acid Nutrition and Reproductive Performance in Ruminants. Adv. Exp. Med. Biol. 2021, 1285, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Neville, M.C.; McFadden, T.B.; Forsyth, I. Hormonal Regulation of Mammary Differentiation and Milk Secretion. J. Mammary Gland. Biol. Neoplasia 2002, 7, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Neville, M.C.; Morton, J. Physiology and Endocrine Changes Underlying Human Lactogenesis II. J. Nutr. 2001, 131, 3005S–3008S. [Google Scholar] [CrossRef] [PubMed]
- Macias, H.; Hinck, L. Mammary gland development. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 533–557. [Google Scholar] [CrossRef] [PubMed]
- Brandsch, M. Drug transport via the intestinal peptide transporter PepT1. Curr. Opin. Pharmacol. 2013, 13, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Daniel, H. Molecular and integrative physiology of intestinal peptide transport. Annu. Rev. Physiol. 2004, 66, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Fang, X.; Hao, G.; Li, X.; Cai, Y.; Yan, Y.; Zan, L.; Yang, R.; Liu, B. Methionine Promotes Milk Protein Synthesis via the PI3K-mTOR Signaling Pathway in Human Mammary Epithelial Cells. Metabolites 2023, 13, 1149. [Google Scholar] [CrossRef] [PubMed]
- Pirkmajer, S.; Chibalin, A.V. Serum starvation: Caveat emptor. Am. J. Physiol. Cell Physiol. 2011, 301, C272–C279. [Google Scholar] [CrossRef] [PubMed]
- Hennighausen, L.; Robinson, G.W. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008, 22, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Salic, A.; Mitchison, T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 2415–2420. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Jamur, M.C.; Oliver, C. Permeabilization of cell membranes. Methods Mol. Biol. 2010, 588, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic. Acids. Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome. Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K. Culture Models to Investigate Mechanisms of Milk Production and Blood-Milk Barrier in Mammary Epithelial Cells: A Review and a Protocol. J. Mammary Gland. Biol. Neoplasia 2023, 28, 8. [Google Scholar] [CrossRef] [PubMed]
- Bionaz, M.; Loor, J.J. Gene networks driving bovine milk fat synthesis during the lactation cycle. BMC Genom. 2008, 9, 366. [Google Scholar] [CrossRef] [PubMed]
- Bionaz, M.; Loor, J.J. Gene networks driving bovine mammary protein synthesis during the lactation cycle. Bioinform. Biol. Insights 2011, 5, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Mauerer, R.; Ebert, S.; Langmann, T. High glucose, unsaturated and saturated fatty acids differentially regulate expression of ATP-binding cassette transporters ABCA1 and ABCG1 in human macrophages. Exp. Mol. Med. 2009, 41, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; La, S.K.; Zhang, L.Y.; Li, S.; Yu, Z.B.; Ao, L.M.; Gao, T.Y.; Huang, H.T. Metabolomics and amino acid profiling of plasma reveals the metabolite profiles associated with nitrogen utilisation efficiency in primiparous dairy cows. Animal 2024, 18, 101202. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Hou, G.; Zeng, C.; Ren, Y.; Chen, X.; Peng, C. Metabolomic profiling reveals amino acid and carnitine alterations as metabolic signatures in psoriasis. Theranostics 2021, 11, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Bu, D.; Ma, L. Integration of Multiplied Omics, a Step Forward in Systematic Dairy Research. Metabolites 2022, 12, 225. [Google Scholar] [CrossRef] [PubMed]
- Shennan, D.B.; Peaker, M. Transport of milk constituents by the mammary gland. Physiol. Rev. 2000, 80, 925–951. [Google Scholar] [CrossRef] [PubMed]
- Baumgard, L.H.; Collier, R.J.; Bauman, D.E. A 100-Year Review: Regulation of nutrient partitioning to support lactation. J. Dairy Sci. 2017, 100, 10353–10366. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhao, F.-Q.; Ren, Y.; Han, J.; Liu, J.; Li, Y.; Liu, H. Parenterally Delivered Methionyl-Methionine Dipeptide During Pregnancy Enhances Mammogenesis and Lactation Performance Over Free Methionine by Activating PI3K-AKT Signaling in Methionine-Deficient Mice. J. Nutr. 2020, 150, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- van Sadelhoff, J.H.J.; Wiertsema, S.P.; Garssen, J.; Hogenkamp, A. Free Amino Acids in Human Milk: A Potential Role for Glutamine and Glutamate in the Protection Against Neonatal Allergies and Infections. Front. Immunol. 2020, 11, 1007. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Adelman, A.S.; Rai, D.; Boettcher, J.; Lőnnerdal, B. Amino Acid Profiles in Term and Preterm Human Milk through Lactation: A Systematic Review. Nutrients 2013, 5, 4800–4821. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yan, X.; Lu, R.; Meng, X.; Nie, G. Peptide transporter 1 (PepT1) in fish: A review. Aquac. Fish. 2017, 2, 193–206. [Google Scholar] [CrossRef]
- Spanier, B. Transcriptional and functional regulation of the intestinal peptide transporter PEPT1. J. Physiol. 2014, 592, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Han, B.; Yu, Y.; Yao, W.; Bose, S.; Karlan, B.Y.; Giuliano, A.E.; Cui, X. Evaluation of MCF10A as a Reliable Model for Normal Human Mammary Epithelial Cells. PLoS ONE 2015, 10, e0131285. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Lee-Pullen, T.; Rigby, P.; Hartmann, P.; Xu, J.; Zeps, N. Receptor activator of NF-κB ligand promotes proliferation of a putative mammary stem cell unique to the lactating epithelium. Stem. Cells 2012, 30, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Pinu, F.R.; Beale, D.J.; Paten, A.M.; Kouremenos, K.; Swarup, S.; Schirra, H.J.; Wishart, D. Systems Biology and Multi-Omics Integration: Viewpoints from the Metabolomics Research Community. Metabolites 2019, 9, 76. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequence (5′-3′) |
|---|---|
| CSN1S1-F | AGGGCACCTAATCAGAGGGT |
| CSN1S1-R | AATTGATGGCACTTACAGAACTGG |
| CSN2-F | GGAGAGGATGAACACCAGGATA |
| CSN2-R | GCTGAGCAAGAGGCAGAATG |
| CSN3-F | GCAGGTCCCTCAGCCTATTC |
| CSN3-R | ACAGCTCTCTGAGGGTAGGG |
| β-actin-F | AGACCTGTACGCCAACACAG |
| β-actin-R | CGCTCAGGAGGAGCAATGAT |
| MPST-F | GCCGTGTCACTGCTTGATG |
| MPST-R | GGTTCTCCTTGATGTCCTCGTA |
| PSAT1-F | GTGTGCTGACTATGTGGTGAC |
| PSAT1-R | GTTGAGGTTCCAGGTGCTTG |
| MTAP-F | TCAACTACCAGGCGAACATCT |
| MTAP-R | GGCACAAGAATGACTTCCATCA |
| GAMT-F | GCCCATTGATGAGCATTGGAT |
| GAMT-R | GGCGTGGTTCTTGATGAAGTT |
| GRHPR-F | CCAGATGTCCTGACAGATACCA |
| GRHPR-R | AGCCACCATTCTTCACTTCCT |
| MAOB-F | GGCAGGACTTACACTCTTAGGA |
| MAOB-R | AGACGCTCAACCTCATTCACT |
| ACAT2-F | GGCTCCTCACTTGGCTTACT |
| ACAT2-R | GCAACCTTGTCCTGATCTTCTC |
| HMGCLL1-F | GGTATGTGTCTTGTGCTCTGG |
| HMGCLL1-R | GCTAAGGCTTGTCCGTATGTG |
| AACS-F | TGGCTGTCAGTGCTGGAAG |
| AACS-R | CAGGAGGATGCTGCTCTTGA |
| PRODH-F | AGAGCACAAGGAGATGGAGTC |
| PRODH-R | CTCATTGGCGTAGAAGTAGGTG |
| CKMT1B-F | GTGGCTGGAGATGAGGAGAC |
| CKMT1B-R | TAGATCCGTGGTGTGCTTCAT |
| CKB-F | CTCATCGACGACCACTTCCT |
| CKB-R | ATTGTCATTGTGCCAGATACCG |
| GAD1-F | ACAGCCTGGAAGAGAAGAGTC |
| GAD1-R | TTGCGGACATAGTTGAGGAGTA |
| OXCT2-F | GTTCAACGGCGACCACTTC |
| OXCT2-R | CCACCTCCACGATCTCTTCC |
| RPEL1-F | TGCCATCAAACCAGGAACCT |
| RPEL1-R | TGTCAGAACCTACTCCACCATC |
| GAA-F | CGTTCATGCCGCCATACTG |
| GAA-R | TGTAGTCCAGGTCGTTCCAC |
| AKR1B1-F | GATCGCAGCCAAGCACAATA |
| AKR1B1-R | CAGCTCAACAAGGCACAGAC |
| GALT-F | GCGTGATGATCTAGCCTCCA |
| GALT-R | TCTGAGCCTGAGCAAGCATT |
| UGT2A1-F | CCATAGACCAACTCCTCTCACA |
| UGT2A1-R | GAGCAACAAGATCACCACAGAT |
| TKT-F | AGTGATGGCGTTGCTACAGA |
| TKT-R | GACCTGGAAGTCCTCATTGTTG |
| DHDH-F | TCGGGCAGAATTTGGGAAGA |
| DHDH-R | TGGAGGTGAACTGGACACAG |
| FGGY-F | GCTGGCTGTCATCTGTGGA |
| FGGY-R | ACTCTGGCATCTGGCTGTG |
| AMY1B-F | CGGCACAGTTATTCGCAAGT |
| AMY1B-R | GTCCTCGTTGATTGTCATGGTT |
| MGAM-F | TGACATACCGCACCACAGG |
| MGAM-R | GCAGCCACCATCTCATCATAC |
| TREH-F | TGCTACTTGACTCACACCAATG |
| TREH-R | AGCGATTCAGGAGGTAGTTCTT |
| HKDC1-F | GATGTGGTGAGCCGTCTGA |
| HKDC1-R | TGCCAGTTCCGATGATGACA |
| CH25H-F | GCTACAAGATCCACCCTGACT |
| CH25H-R | TGCCACACGAAGAACTCCAT |
| CYP3A7-F | TGCTCTAGTCAGAGTCCTTCAG |
| CYP3A7-R | GGCTCCACTTACGGTCTCAT |
| AKR1C3-F | GATGGTCACTTCATGCCTGTC |
| AKR1C3-R | CAGTCCAACCTGCTCCTCAT |
| SULT2B1-F | TCGGATGAAGGGCAAAGACA |
| SULT2B1-R | AGCAGCGTGTAGTTGGACAT |
| GPAT3-F | CTGGTTCTCGGCTTCATCCT |
| GPAT3-R | CTCAAAGTCCCTTCCTCGTAGA |
| DGKQ-F | CGTTCTCCGTACTGCTGTCT |
| DGKQ-R | GTCTGCCGTGTCGTTCTCT |
| CEL-F | CACATCTTCGCCAGCATCG |
| CEL-R | AGTCCACCACAGTCTTCTTCTT |
| TRPV6-F | GCCGAGATGAGCAGAACCT |
| TRPV6-R | CAGCCTCCATCAGCACCAT |
| FXYD2-F | GTGGACCCGTTCTACTATGACT |
| FXYD2-R | ATCTTCTGCTGAGGAGGATGAG |
| SLC43A3-F | CTGGTGTGGCTGTCTGTGA |
| SLC43A3-R | TGCTTCCTTCTGGTACTTCTGT |
| SLC26A6-F | ATCTTGCTGAACCTGGACCTT |
| SLC26A6-R | CCACACCACACCTCTGCTT |
| PLB1-F | TTGTGGATTCAGGCTCAAGAAC |
| PLB1-R | GAGAGGAGAACCGTGCTAAGT |
| TCN2-F | CTCTACCTGCTCGCTCTCAG |
| TCN2-R | TTGTCCACCACGCTGTCAT |
| CUBN-F | TCAGCAAGGATGTGGTGGTT |
| CUBN-R | GTTCCAGCCAAGTTCGCATT |
| PLPP3-F | CTTCGTGTCTGACCTCTTCAAG |
| PLPP3-R | TGTTGTGGTGATTGTTCCTGTC |
| MTTP-F | TGACAGCAGCATTATCCTCCA |
| MTTP-R | CAGCCTTCATTCTGACACAACT |
| SLC27A1-F | ACCACAGGCACCTTCAAGAT |
| SLC27A1-R | CCGAGCAGATGCGAGTGTA |
| LTB4R-F | TCACTATGTCTGCGGAGTCAG |
| LTB4R-R | AGCACAGGCTCATGTTCGT |
| KCNN4-F | ATGCTGCTGCGTCTCTACC |
| KCNN4-R | GCGGAAGCGGACTTGATTG |
| SLC7A8-F | GCCTTCCTGCTGGTCTTCA |
| SLC7A8-R | ATGTCCTCATTAGCCTCCTCTG |
| PIK3R2-F | CACTTGGAAGAGCAGGAGGT |
| PIK3R2-R | AAGCATCTCGGACTAGGAAGG |
| Sample_Name | Raw_Reads | Clean_Reads | Q20 (%) | Q30 (%) |
|---|---|---|---|---|
| EM1 | 48,596,854 | 47,342,498 | 98.7 | 95.67 |
| EM2 | 43,440,298 | 42,454,230 | 98.69 | 95.65 |
| EM3 | 57,191,982 | 55,852,078 | 98.83 | 96.09 |
| NC1 | 67,290,316 | 66,527,492 | 98.83 | 96.06 |
| NC2 | 61,570,630 | 60,938,366 | 98.72 | 95.68 |
| NC3 | 42,081,762 | 40,672,606 | 98.71 | 95.66 |
| Sample Name | Total Reads | Total Mapped | Multiple Mapped | Uniquely Mapped |
|---|---|---|---|---|
| EM1 | 47,342,498 | 46,430,209 (98.07%) | 1,632,481 (3.45%) | 44,797,728 (94.62%) |
| EM2 | 42,454,230 | 41,558,282 (97.89%) | 1,555,859 (3.66%) | 40,002,423 (94.22%) |
| EM3 | 55,852,078 | 54,868,973 (98.24%) | 1,797,530 (3.22%) | 53,071,443 (95.02%) |
| NC1 | 66,527,492 | 65,366,885 (98.26%) | 2,191,223 (3.29%) | 63,175,662 (94.96%) |
| NC2 | 60,938,366 | 59,887,762 (98.28%) | 2,008,530 (3.3%) | 57,879,232 (94.98%) |
| NC3 | 40,672,606 | 39,918,832 (98.15%) | 1,277,923 (3.14%) | 38,640,909 (95%) |
| Category | KEGG Pathway | DEGs |
|---|---|---|
| Amino acid metabolism | Cysteine and methionine metabolism | PSAT1, MPST, MTAP |
| Glycine, serine, and threonine metabolism | GAMT, MAOB, GRHPR | |
| Valine, leucine, and isoleucine degradation | AACS, ACAT2, HMGCLL1 | |
| Arginine and proline metabolism | PRODH, CKB, CKMT1B | |
| Carbohydrate metabolism | Butanoate metabolism | GAD1, OXCT2, AACS |
| Pentose phosphate pathway | TKT, RPEL1 | |
| Galactose metabolism | AKR1B15, AKR1B1, GALT | |
| Pentose and glucuronate interconversions | FGGY, UGT2A1, DHDH | |
| Starch and sucrose metabolism | AMY1B, MGAM, TREH, HKDC1 | |
| Lipid metabolism | Primary bile acid biosynthesis | CH25H, CYP27A1 |
| Steroid hormone biosynthesis | CYP3A7, UGT2A1, AKR1C3, SULT2B1 | |
| Glycerolipid metabolism | CEL, DGKQ | |
| Material absorption | Mineral absorption | TRPV6, FXYD2, SLC26A6 |
| Vitamin digestion and absorption | PLB1, TCN2, CUBN | |
| Fat digestion and absorption | CEL, SLC27A1, PLPP3, MTTP | |
| Protein digestion and absorption | SLC7A8, KCNN4, LTB4R | |
| Carbohydrate digestion and absorption | PIK3R2, MGAM, HKDC1, AMY1B |
| Pathway ID | Pathway Name | Hits | Compound Name | p-Value |
|---|---|---|---|---|
| hsa04978 | Mineral absorption | 4 | L-Alanine; L-Methionine; L-Asparagine; L-Valine | 0.007 |
| hsa04974 | Protein digestion and absorption | 5 | L-Alanine; L-Methionine; L-Asparagine; L-Valine; Tyramine | 0.008 |
| hsa00290 | Valine, leucine and isoleucine biosynthesis | 3 | 2-Ketobutyric acid; L-Valine; D-erythro-3-Methylmalate | 0.023 |
| hsa00270 | Cysteine and methionine metabolism | 5 | L-Alanine; L-Methionine; 2-Ketobutyric acid; S-Adenosylmethioninamine; Methionine sulfoxide | 0.034 |
| hsa00970 | Aminoacyl-tRNA biosynthesis | 4 | L-Alanine; L-Methionine; L-Asparagine; L-Valine | 0.051 |
| hsa00051 | Fructose and mannose metabolism | 4 | Fructose 1,6-bisphosphate; D-Tagatose 1-phosphate; 2-dehydro-3-deoxy-L-rhamnonate; L-Fucono-1,5-lactone | 0.060 |
| hsa02010 | ABC transporters | 7 | L-Alanine; L-Valine; Adenosine; Uridine; Phthalic acid; Xanthosine; Nickel cation | 0.081 |
| hsa04152 | AMPK signaling pathway | 2 | Fructose 1,6-bisphosphate; AICAR | 0.127 |
| hsa00250 | Alanine, aspartate and glutamate metabolism | 2 | L-Alanine; L-Asparagine | 0.175 |
| hsa00750 | Vitamin B6 metabolism | 2 | D-Ribose 5-phosphate; 4-Pyridoxic acid | 0.185 |
| hsa00330 | Arginine and proline metabolism | 3 | S-Adenosylmethioninamine; N(omega)-Hydroxyarginine; cis-3-Hydroxy-DL-proline | 0.288 |
| hsa04975 | Fat digestion and absorption | 1 | Coenzyme A | 0.301 |
| hsa00564 | Glycerophospholipid metabolism | 2 | Citicoline; Demanyl phosphate | 0.453 |
| hsa00450 | Selenocompound metabolism | 1 | L-Alanine | 0.526 |
| hsa00470 | D-Amino acid metabolism | 2 | L-Alanine; L-Methionine | 0.564 |
| hsa00400 | Phenylalanine, tyrosine and tryptophan biosynthesis | 1 | Fructose 1,6-bisphosphate | 0.620 |
| hsa00500 | Starch and sucrose metabolism | 1 | 3-Ketosucrose | 0.641 |
| hsa04977 | Vitamin digestion and absorption | 1 | Coenzyme A | 0.660 |
| hsa00340 | Histidine metabolism | 1 | AICAR | 0.728 |
| hsa00260 | Glycine, serine and threonine metabolism | 1 | 2-Ketobutyric acid | 0.735 |
| hsa00360 | Phenylalanine metabolism | 1 | 2-Phenylethanol | 0.743 |
| hsa00350 | Tyrosine metabolism | 1 | Tyramine | 0.886 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Yan, Y.; Yang, R.; Li, X.; Zhai, C.; Wu, X.; Fang, X.; Liu, B. EM Dipeptide Enhances Milk Protein Secretion: Evidence from Integrated Metabolomic and Transcriptomic Analysis. Metabolites 2025, 15, 476. https://doi.org/10.3390/metabo15070476
Liu Y, Yan Y, Yang R, Li X, Zhai C, Wu X, Fang X, Liu B. EM Dipeptide Enhances Milk Protein Secretion: Evidence from Integrated Metabolomic and Transcriptomic Analysis. Metabolites. 2025; 15(7):476. https://doi.org/10.3390/metabo15070476
Chicago/Turabian StyleLiu, Yuqing, Yuhao Yan, Runjun Yang, Xiaohui Li, Chuang Zhai, Xuan Wu, Xibi Fang, and Boqun Liu. 2025. "EM Dipeptide Enhances Milk Protein Secretion: Evidence from Integrated Metabolomic and Transcriptomic Analysis" Metabolites 15, no. 7: 476. https://doi.org/10.3390/metabo15070476
APA StyleLiu, Y., Yan, Y., Yang, R., Li, X., Zhai, C., Wu, X., Fang, X., & Liu, B. (2025). EM Dipeptide Enhances Milk Protein Secretion: Evidence from Integrated Metabolomic and Transcriptomic Analysis. Metabolites, 15(7), 476. https://doi.org/10.3390/metabo15070476