
Preliminary Study to Understand the Role of Gut Microbiota in Coronary Slow Flow Phenomenon (CSFP)

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Patient Cohort
2.2. 16S rRNA Microbiome Sequencing Analysis
2.3. Statistical Analysis
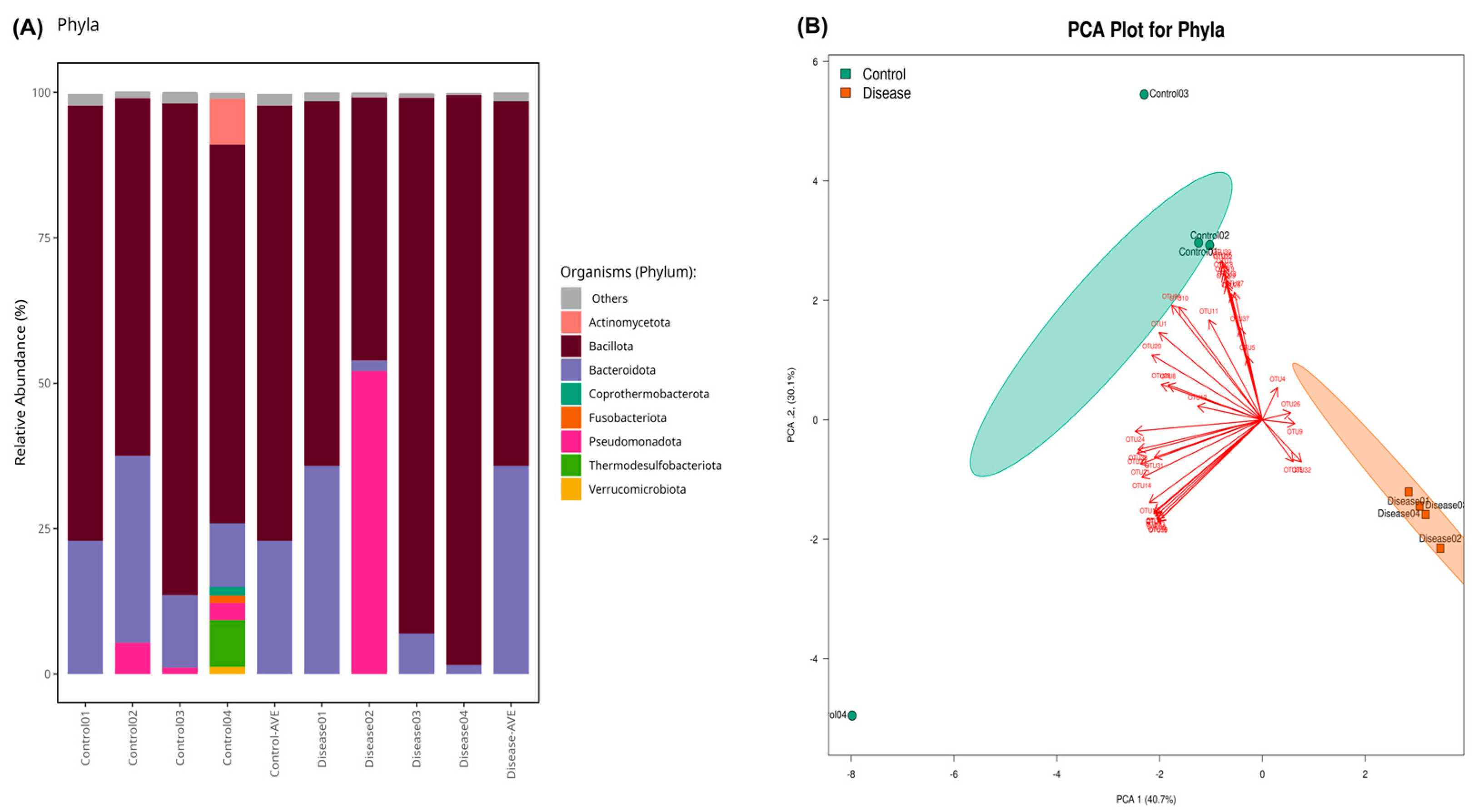
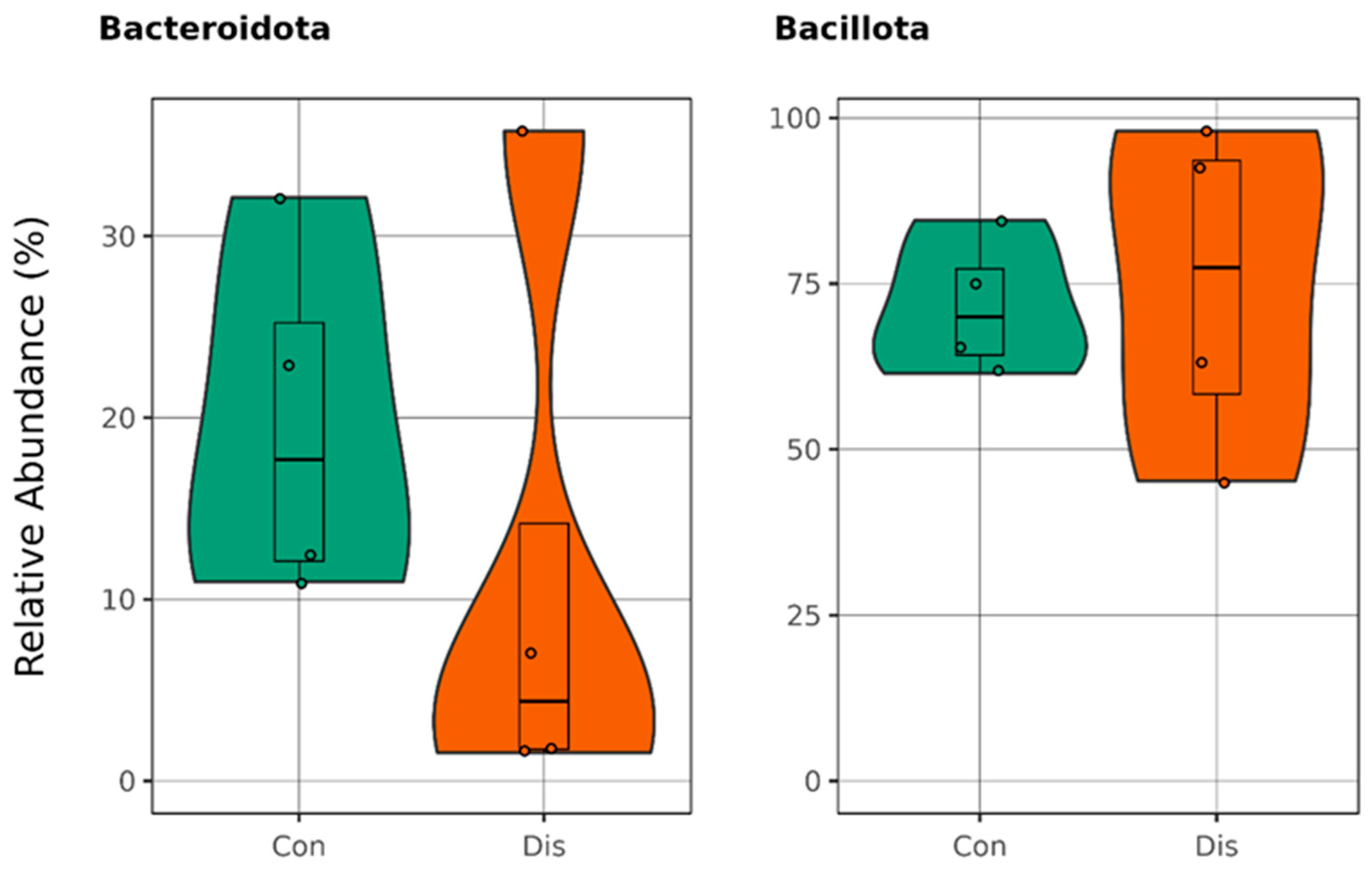
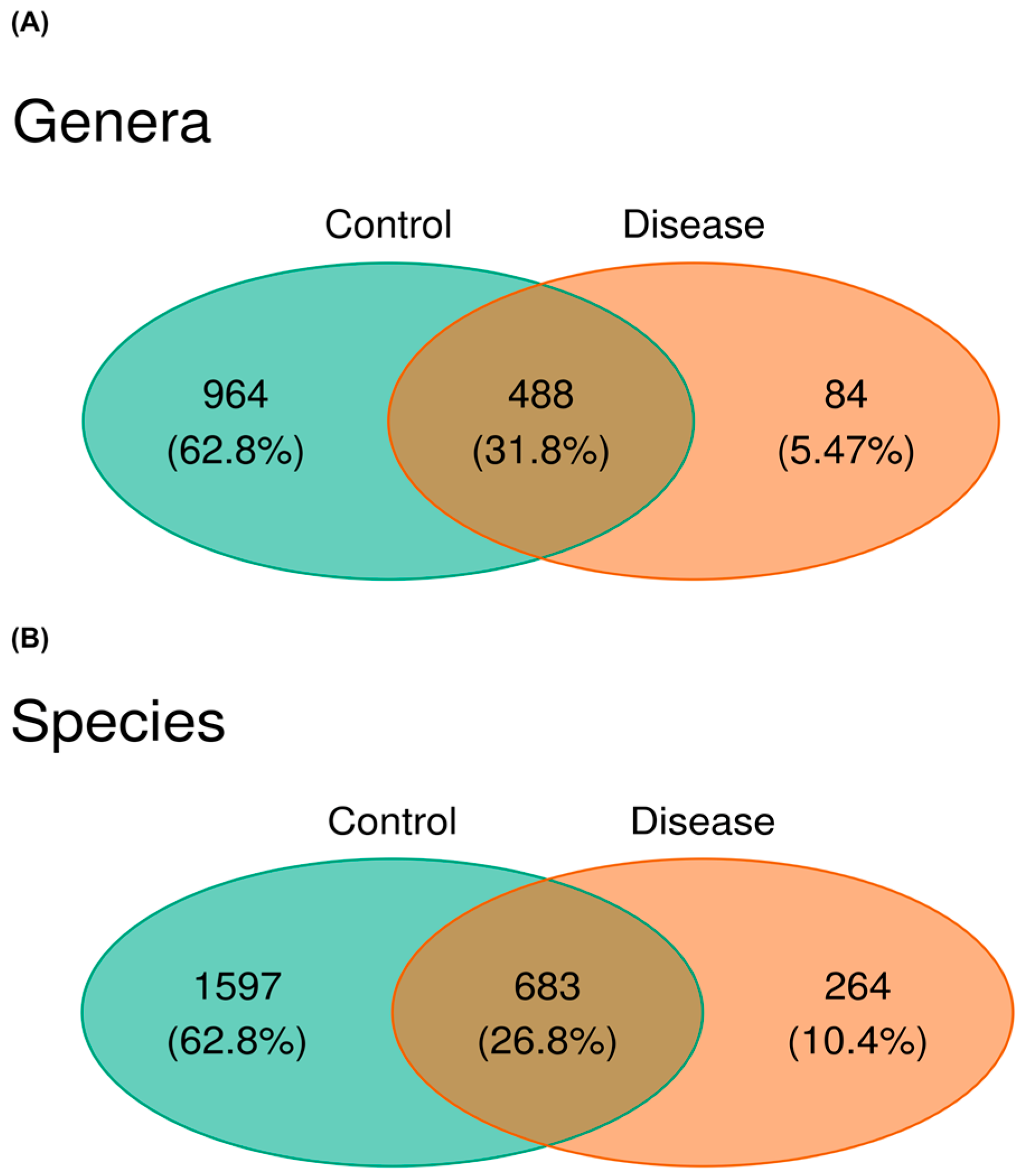
3. Results
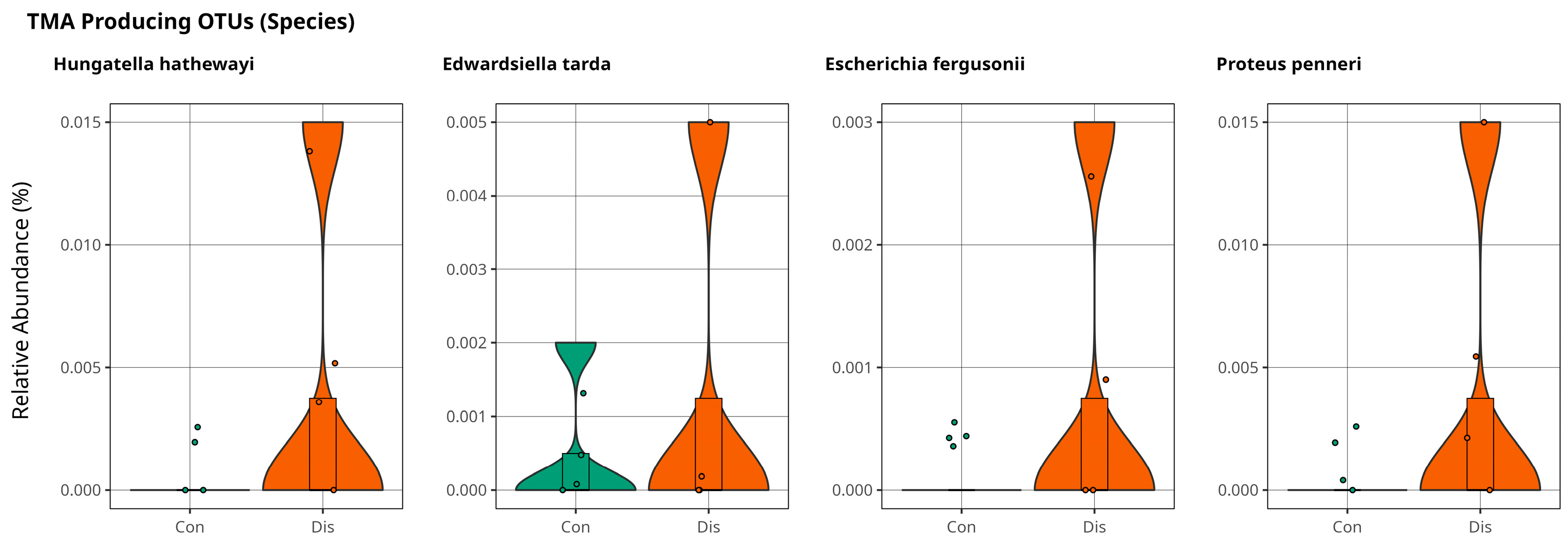
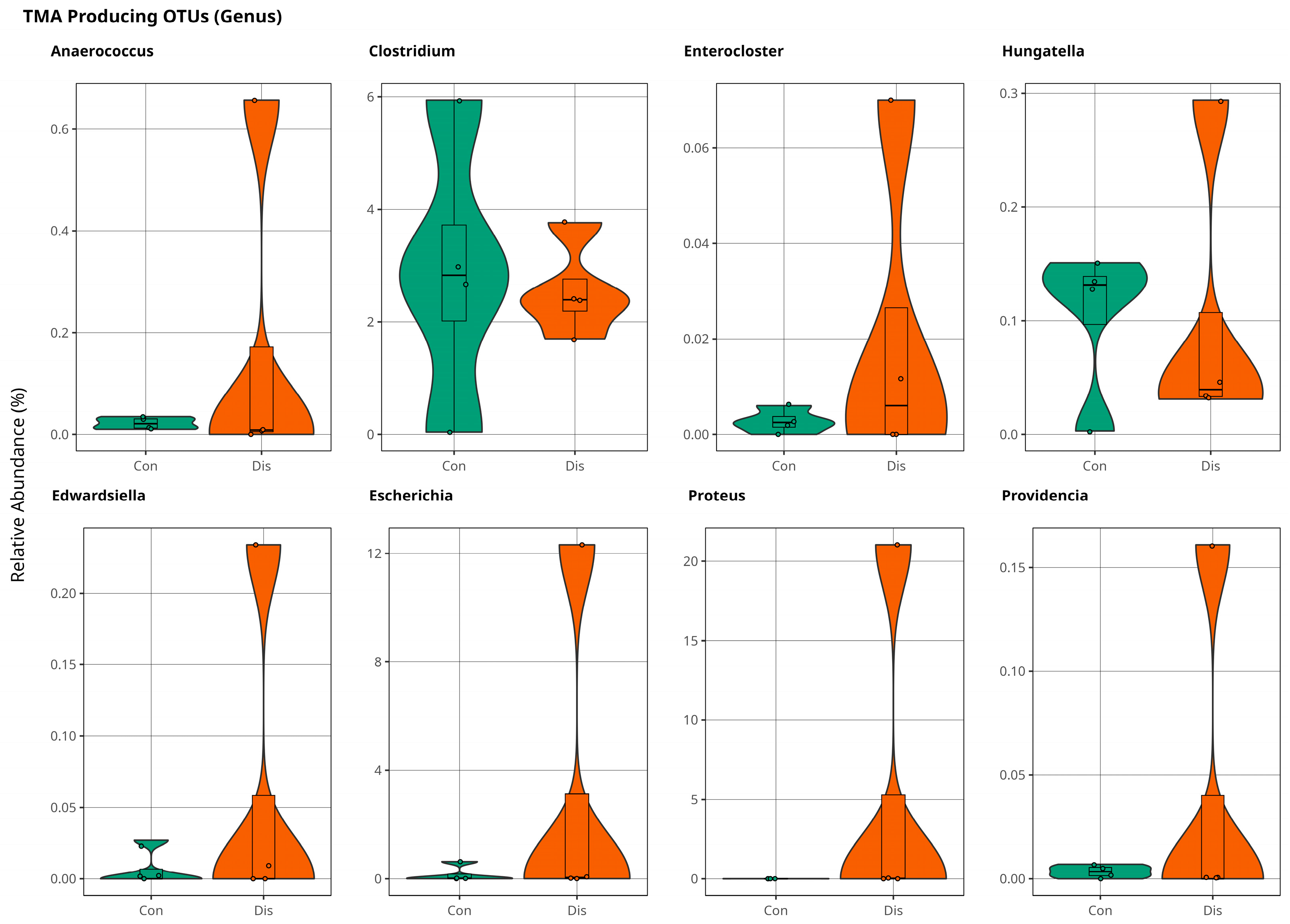
Association of Trimethylamine on CSFP Disease via Gut Microbiota
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CSFP | Coronary Slow Flow Phenomenon |
| TMA | Trimethylamine |
| SD | Standard Deviation |
| TSS | Total Sum Scaling |
| TMAO | Trimethyl-N- Oxide |
| ROS | Reactive Oxygen Species |
References
- Alvarez, C.; Siu, H. Coronary Slow-Flow Phenomenon as an Underrecognized and Treatable Source of Chest Pain: Case Series and Literature Review. J. Investig. Med. High Impact Case Rep. 2018, 6, 2324709618789194. [Google Scholar] [CrossRef]
- Hawkins, B.M.; Stavrakis, S.; Rousan, T.A.; Abu-Fadel, M.; Schechter, E. Coronary Slow Flow—Prevalence and Clinical Correlations. Circ. J. 2012, 76, 936–942. [Google Scholar] [CrossRef]
- Vane, J.R.; Botting, R.M. Secretory functions of the vascular endothelium. J. Physiol. Pharmacol. 1992, 43, 195–207. [Google Scholar] [PubMed]
- Hadi, H.A.; Carr, C.S.; Al Suwaidi, J. Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005, 1, 183–198. [Google Scholar] [PubMed] [PubMed Central]
- Fosse, J.H.; Haraldsen, G.; Falk, K.; Edelmann, R. Endothelial Cells in Emerging Viral Infections. Front. Cardiovasc. Med. 2021, 8, 619690. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Z. Gut Microbiome and Cardiovascular Disease. Curr. Opin. Cardiol. 2020, 35, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Ye, L.; Li, J.; Jin, L.; Wang, W.; Li, S.; Bao, M.; Wu, S.; Li, L.; Geng, B.; et al. Metagenomic and Metabolomic Analyses Unveil Dysbiosis of Gut Microbiota in Chronic Heart Failure Patients. Sci. Rep. 2018, 8, 635. [Google Scholar] [CrossRef]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut Microbiota Dysbiosis Contributes to the Development of Hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef]
- Jie, Z.; Xia, H.; Zhong, S.-L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The Gut Microbiome in Atherosclerotic Cardiovascular Disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef]
- Wang, X.; Nie, S.-P. The Coronary Slow Flow Phenomenon: Characteristics, Mechanisms and Implications. Cardiovasc. Diagn. Ther. 2011, 1, 37–43. [Google Scholar]
- Mangieri, E.; Macchiarelli, G.; Ciavolella, M.; Barillà, F.; Avella, A.; Martinotti, A.; Dell’Italia, L.J.; Scibilia, G.; Motta, P.; Campa, P.P. Slow Coronary Flow: Clinical and Histopathological Features in Patients with Otherwise Normal Epicardial Coronary Arteries. Cathet. Cardiovasc. Diagn. 1996, 37, 375–381. [Google Scholar] [CrossRef]
- Beltrame, J.F.; Limaye, S.B.; Horowitz, J.D. The Coronary Slow Flow Phenomenon—A New Coronary Microvascular Disorder. Cardiology 2002, 97, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Goel, P.K.; Gupta, S.K.; Agarwal, A.; Kapoor, A. Slow Coronary Flow: A Distinct Angiographic Subgroup in Syndrome X. Angiology 2001, 52, 507–514. [Google Scholar] [CrossRef]
- Tambe, A.A.; Demany, M.A.; Zimmerman, H.A.; Mascarenhas, E. Angina Pectoris and Slow Flow Velocity of Dye in Coronary Arteries—A New Angiographic Finding. Am. Heart J. 1972, 84, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Saya, S.; Hennebry, T.A.; Lozano, P.; Lazzara, R.; Schechter, E. Coronary Slow Flow Phenomenon and Risk for Sudden Cardiac Death Due to Ventricular Arrhythmias: A Case Report and Review of Literature. Clin. Cardiol. 2008, 31, 352–355. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Shi, X.-R.; Chen, B.-Y.; Lin, W.-Z.; Li, Y.-L.; Wang, Y.-L.; Liu, Y.; Huang, J.-J.; Zhang, W.-W.; Ma, X.-X.; Shao, S.; et al. Microbiota in Gut, Oral Cavity, and Mitral Valves Are Associated With Rheumatic Heart Disease. Front. Cell. Infect. Microbiol. 2021, 11, 643092. [Google Scholar] [CrossRef]
- Amiri, P.; Hosseini, S.A.; Ghaffari, S.; Tutunchi, H.; Ghaffari, S.; Mosharkesh, E.; Asghari, S.; Roshanravan, N. Role of Butyrate, a Gut Microbiota Derived Metabolite, in Cardiovascular Diseases: A Comprehensive Narrative Review. Front. Pharmacol. 2022, 12, 837509. [Google Scholar] [CrossRef]
- Thomas, M.S.; Fernandez, M.L. Trimethylamine N-Oxide (TMAO), Diet and Cardiovascular Disease. Curr. Atheroscler. Rep. 2021, 23, 12. [Google Scholar] [CrossRef]
- Rath, S.; Rud, T.; Pieper, D.H.; Vital, M. Potential TMA-Producing Bacteria Are Ubiquitously Found in Mammalia. Front. Microbiol. 2020, 10, 2966. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the Trimethylamine-Producing Bacteria of the Human Gut Microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, M. Trimethylamine N-Oxide Generated by the Gut Microbiota Is Associated with Vascular Inflammation: New Insights into Atherosclerosis. Mediat. Inflamm. 2020, 2020, 4634172. [Google Scholar] [CrossRef]
- Zheng, Y.; He, J.-Q. Pathogenic Mechanisms of Trimethylamine N-Oxide-Induced Atherosclerosis and Cardiomyopathy. Curr. Vasc. Pharmacol. 2022, 20, 29–36. [Google Scholar] [CrossRef]
- Rahman, M.M.; Islam, F.; Or-Rashid, M.H.; Mamun, A.A.; Rahaman, M.S.; Islam, M.M.; Meem, A.F.K.; Sutradhar, P.R.; Mitra, S.; Mimi, A.A.; et al. The Gut Microbiota (Microbiome) in Cardiovascular Disease and Its Therapeutic Regulation. Front. Cell. Infect. Microbiol. 2022, 12, 903570. [Google Scholar] [CrossRef] [PubMed]
- Sawicka-Smiarowska, E.; Bondarczuk, K.; Bauer, W.; Niemira, M.; Szalkowska, A.; Raczkowska, J.; Kwasniewski, M.; Tarasiuk, E.; Dubatowka, M.; Lapinska, M.; et al. Gut Microbiome in Chronic Coronary Syndrome Patients. J. Clin. Med. 2021, 10, 5074. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, J.; Liu, H.; Tang, Y.; Zhan, Q.; Lai, W.; Ao, L.; Meng, X.; Ren, H.; Xu, D.; et al. The Intestinal Microbiota Associated with Cardiac Valve Calcification Differs from That of Coronary Artery Disease. Atherosclerosis 2019, 284, 121–128. [Google Scholar] [CrossRef]
- Ahmad, A.F.; Dwivedi, G.; O’Gara, F.; Caparros-Martin, J.; Ward, N.C. The Gut Microbiome and Cardiovascular Disease: Current Knowledge and Clinical Potential. Am. J. Physiol.-Heart Circ. Physiol. 2019, 317, H923–H938. [Google Scholar] [CrossRef]
- Astudillo, A.A.; Mayrovitz, H.N. The Gut Microbiome and Cardiovascular Disease. Cureus 2021, 13, e14519. [Google Scholar] [CrossRef]
- Toya, T.; Corban, M.T.; Marrietta, E.; Horwath, I.E.; Lerman, L.O.; Murray, J.A.; Lerman, A. Coronary Artery Disease Is Associated with an Altered Gut Microbiome Composition. PLoS ONE 2020, 15, e0227147. [Google Scholar] [CrossRef]
- Yoshida, N.; Yamashita, T.; Hirata, K. Gut Microbiome and Cardiovascular Diseases. Diseases 2018, 6, 56. [Google Scholar] [CrossRef]
- Liu, H.; Chen, X.; Hu, X.; Niu, H.; Tian, R.; Wang, H.; Pang, H.; Jiang, L.; Qiu, B.; Chen, X.; et al. Alterations in the Gut Microbiome and Metabolism with Coronary Artery Disease Severity. Microbiome 2019, 7, 68. [Google Scholar] [CrossRef]
- Ferreira, R.L.U.; Sena-Evangelista, K.C.M.; De Azevedo, E.P.; Pinheiro, F.I.; Cobucci, R.N.; Pedrosa, L.F.C. Selenium in Human Health and Gut Microflora: Bioavailability of Selenocompounds and Relationship With Diseases. Front. Nutr. 2021, 8, 685317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Meng, S.; Yu, Y.; Bi, L.; Tian, J.; Zhang, L. Associations of Dietary Selenium Intake with the Risk of Chronic Diseases and Mortality in US Adults. Front. Nutr. 2024, 11, 1363299. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Su, W.; Chen, X.; Zheng, H. Advances in the Study of Selenium and Human Intestinal Bacteria. Front. Nutr. 2022, 9, 1059358. [Google Scholar] [CrossRef]
- Weng, Y.J.; Gan, H.Y.; Li, X.; Huang, Y.; Li, Z.C.; Deng, H.M.; Chen, S.Z.; Zhou, Y.; Wang, L.S.; Han, Y.P.; et al. Correlation of Diet, Microbiota and Metabolite Networks in Inflammatory Bowel Disease. J. Dig. Dis. 2019, 20, 447–459. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- Flores-Mateo, G.; Navas-Acien, A.; Pastor-Barriuso, R.; Guallar, E. Selenium and Coronary Heart Disease: A Meta-Analysis. Am. J. Clin. Nutr. 2006, 84, 762–773. [Google Scholar] [CrossRef]
- Hu, T.; Wu, Q.; Yao, Q.; Jiang, K.; Yu, J.; Tang, Q. Short-Chain Fatty Acid Metabolism and Multiple Effects on Cardiovascular Diseases. Ageing Res. Rev. 2022, 81, 101706. [Google Scholar] [CrossRef]
- Modrego, J.; Ortega-Hernández, A.; Goirigolzarri, J.; Restrepo-Córdoba, M.A.; Bäuerl, C.; Cortés-Macías, E.; Sánchez-González, S.; Esteban-Fernández, A.; Pérez-Villacastín, J.; Collado, M.C.; et al. Gut Microbiota and Derived Short-Chain Fatty Acids Are Linked to Evolution of Heart Failure Patients. Int. J. Mol. Sci. 2023, 24, 13892. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Kaur, H.; Mande, S.S. Comparative In Silico Analysis of Butyrate Production Pathways in Gut Commensals and Pathogens. Front. Microbiol. 2016, 7, 1945. [Google Scholar] [CrossRef]
- Singh, V.; Lee, G.; Son, H.; Koh, H.; Kim, E.S.; Unno, T.; Shin, J.-H. Butyrate Producers, “The Sentinel of Gut”: Their Intestinal Significance with and beyond Butyrate, and Prospective Use as Microbial Therapeutics. Front. Microbiol. 2023, 13, 1103836. [Google Scholar] [CrossRef]
- Tian, Q.; Leung, F.P.; Chen, F.M.; Tian, X.Y.; Chen, Z.; Tse, G.; Ma, S.; Wong, W.T. Butyrate Protects Endothelial Function through PPARδ/miR-181b Signaling. Pharmacol. Res. 2021, 169, 105681. [Google Scholar] [CrossRef] [PubMed]
- Canyelles, M.; Borràs, C.; Rotllan, N.; Tondo, M.; Escolà-Gil, J.C.; Blanco-Vaca, F. Gut Microbiota-Derived TMAO: A Causal Factor Promoting Atherosclerotic Cardiovascular Disease? Int. J. Mol. Sci. 2023, 24, 1940. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Ke, B.; Du, J. TMAO: How Gut Microbiota Contributes to Heart Failure. Transl. Res. 2021, 228, 109–125. [Google Scholar] [CrossRef]
- Chen, H.-C.; Liu, Y.-W.; Chang, K.-C.; Wu, Y.-W.; Chen, Y.-M.; Chao, Y.-K.; You, M.-Y.; Lundy, D.J.; Lin, C.-J.; Hsieh, M.L.; et al. Gut Butyrate-Producers Confer Post-Infarction Cardiac Protection. Nat. Commun. 2023, 14, 7249. [Google Scholar] [CrossRef]
- Shimada, B.K.; Alfulaij, N.; Seale, L.A. The Impact of Selenium Deficiency on Cardiovascular Function. Int. J. Mol. Sci. 2021, 22, 10713. [Google Scholar] [CrossRef]
- Zhang, C.; Zeng, Q.; Liu, X.; He, Q.; Zhang, J.; Zhao, S.; Hu, H. Association of Blood Selenium Levels with Diabetes and Heart Failure in American General Adults: A Cross-Sectional Study of NHANES 2011–2020 Pre. Biol. Trace Elem. Res. 2024, 202, 3413–3424. [Google Scholar] [CrossRef]
- Leszto, K.; Biskup, L.; Korona, K.; Marcinkowska, W.; Możdżan, M.; Węgiel, A.; Młynarska, E.; Rysz, J.; Franczyk, B. Selenium as a Modulator of Redox Reactions in the Prevention and Treatment of Cardiovascular Diseases. Antioxidants 2024, 13, 688. [Google Scholar] [CrossRef]
- Jaworska, K.; Hering, D.; Mosieniak, G.; Bielak-Zmijewska, A.; Pilz, M.; Konwerski, M.; Gasecka, A.; Kapłon-Cieślicka, A.; Filipiak, K.; Sikora, E.; et al. TMA, A Forgotten Uremic Toxin, but Not TMAO, Is Involved in Cardiovascular Pathology. Toxins 2019, 11, 490. [Google Scholar] [CrossRef]
- He, S.; Jiang, H.; Zhuo, C.; Jiang, W. Trimethylamine/Trimethylamine-N-Oxide as a Key Between Diet and Cardiovascular Diseases. Cardiovasc. Toxicol. 2021, 21, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Roncal, C.; Martínez-Aguilar, E.; Orbe, J.; Ravassa, S.; Fernandez-Montero, A.; De Pipaon, G.S.; Ugarte, A.; Estella-Hermoso De Mendoza, A.; Rodriguez, J.A.; Fernández-Alonso, S.; et al. Trimethylamine (Tma) And Trimethylamine-N-Oxide (Tmao) As Predictors Of Cardiovascular Mortality In Peripheral Artery Disease. Atherosclerosis 2019, 287, e233. [Google Scholar] [CrossRef]
- He, Y.; Chen, S.; Xue, Y.; Lu, H.; Li, Z.; Jia, X.; Ning, Y.; Yuan, Q.; Wang, S. Analysis of Alterations in Intestinal Flora in Chinese Elderly with Cardiovascular Disease and Its Association with Trimethylamine. Nutrients 2024, 16, 1864. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurol, T.; Karaman, T.; Gurol, Y.; Sezerman, O.U.; Oktem Okullu, S. Preliminary Study to Understand the Role of Gut Microbiota in Coronary Slow Flow Phenomenon (CSFP). Metabolites 2025, 15, 475. https://doi.org/10.3390/metabo15070475
Gurol T, Karaman T, Gurol Y, Sezerman OU, Oktem Okullu S. Preliminary Study to Understand the Role of Gut Microbiota in Coronary Slow Flow Phenomenon (CSFP). Metabolites. 2025; 15(7):475. https://doi.org/10.3390/metabo15070475
Chicago/Turabian StyleGurol, Tayfun, Tayyip Karaman, Yesim Gurol, Osman Ugur Sezerman, and Sinem Oktem Okullu. 2025. "Preliminary Study to Understand the Role of Gut Microbiota in Coronary Slow Flow Phenomenon (CSFP)" Metabolites 15, no. 7: 475. https://doi.org/10.3390/metabo15070475
APA StyleGurol, T., Karaman, T., Gurol, Y., Sezerman, O. U., & Oktem Okullu, S. (2025). Preliminary Study to Understand the Role of Gut Microbiota in Coronary Slow Flow Phenomenon (CSFP). Metabolites, 15(7), 475. https://doi.org/10.3390/metabo15070475