
Comparative Metabolomics in Single Ventricle Patients after Fontan Palliation: A Strong Case for a Targeted Metabolic Therapy


Abstract
1. Introduction
2. Cardiac Energy Metabolism
2.1. Energy Metabolism in the Healthy Heart
2.2. Energy Metabolism in Biventricular Patients with Congestive Heart Failure
2.3. Right Ventricular Failure: The Case of Pulmonary Arterial Hypertension
2.4. Energy Metabolism and the Single Ventricle after Fontan Palliation
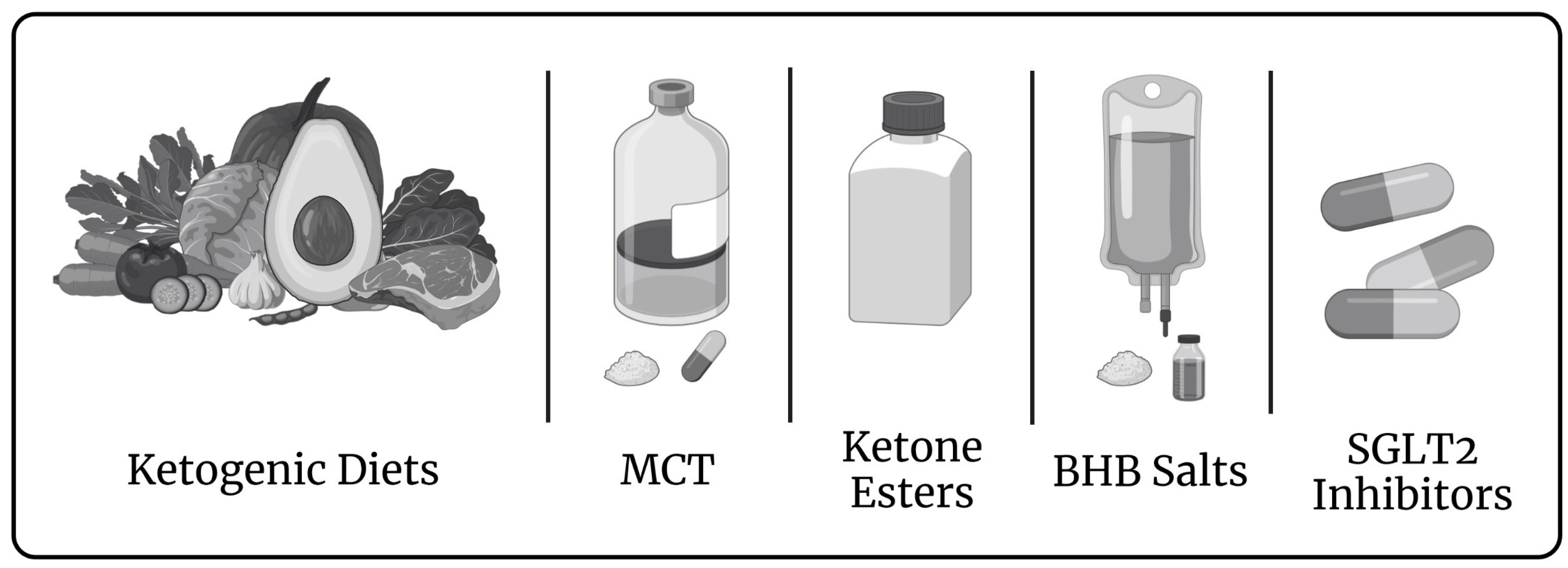
3. Induced Ketosis in Patients with a Failing Biventricular Heart
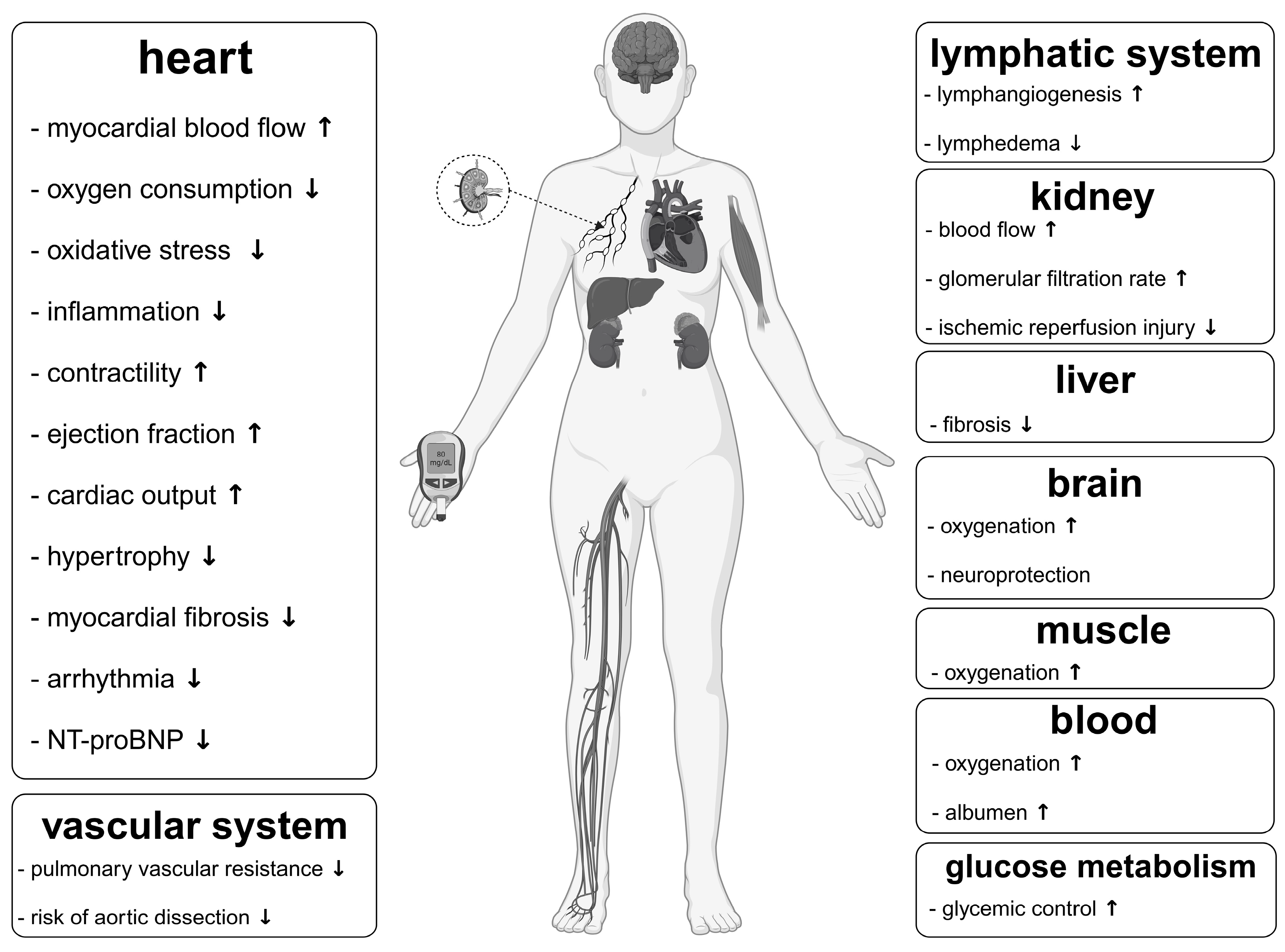
4. Impact of Ketones Apart from That on the Cardiovascular System Relevant to Fontan Circulation
5. Rationale of a Targeted Metabolic Therapy in Fontan Patients
6. Limitations
7. Methods
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rychik, J.; Atz, A.M.; Celermajer, D.S.; Deal, B.J.; Gatzoulis, M.A.; Gewillig, M.H.; Hsia, T.-Y.; Hsu, D.T.; Kovacs, A.H.; McCrindle, B.W.; et al. Evaluation and Management of the Child and Adult with Fontan Circulation: A Scientific Statement from the American Heart Association. Circulation 2019, 140, e234–e284. [Google Scholar] [CrossRef]
- Fontan, F.; Baudet, E. Surgical Repair of Tricuspid Atresia. Thorax 1971, 26, 240–248. [Google Scholar] [CrossRef]
- Kreutzer, G.; Galíndez, E.; Bono, H.; De Palma, C.; Laura, J.P. An Operation for the Correction of Tricuspid Atresia. J. Thorac. Cardiovasc. Surg. 1973, 66, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Gewillig, M.; Brown, S.C. The Fontan Circulation after 45 Years: Update in Physiology. Heart 2016, 102, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Alsoufi, B.; Gillespie, S.; Kim, D.; Shashidharan, S.; Kanter, K.; Maher, K.; Kogon, B. The Impact of Dominant Ventricle Morphology on Palliation Outcomes of Single Ventricle Anomalies. Ann. Thorac. Surg. 2016, 102, 593–601. [Google Scholar] [CrossRef]
- Kutty, S.; Jacobs, M.L.; Thompson, W.R.; Danford, D.A. Fontan Circulation of the Next Generation: Why It’s Necessary, What It Might Look Like. J. Am. Heart Assoc. 2020, 9, e013691. [Google Scholar] [CrossRef] [PubMed]
- Rychik, J. Path Taken in a Fontan Circulation: Room for Optimism in the Face of Uncertainty. Heart 2021, 107, 521–522. [Google Scholar] [CrossRef]
- Zhu, A.; Meza, J.M.; Prabhu, N.K.; McCrary, A.W.; Allareddy, V.; Turek, J.W.; Andersen, N.D. Survival After Intervention for Single-Ventricle Heart Disease Over 15 Years at a Single Institution. Ann. Thorac. Surg. 2022, 114, 2303–2312. [Google Scholar] [CrossRef]
- Lewis, M.; Rosenbaum, M. The Miracle Baby Grows Up: Hypoplastic Left Heart Syndrome in the Adult. Curr. Cardiol. Rep. 2017, 19, 74. [Google Scholar] [CrossRef]
- Julsrud, P.R.; Weigel, T.J.; Van Son, J.A.; Edwards, W.D.; Mair, D.D.; Driscoll, D.J.; Danielson, G.K.; Puga, F.J.; Offord, K.P. Influence of Ventricular Morphology on Outcome after the Fontan Procedure. Am. J. Cardiol. 2000, 86, 319–323. [Google Scholar] [CrossRef]
- McGuirk, S. The Impact of Ventricular Morphology on Midterm Outcome Following Completion Total Cavopulmonary Connection. Eur. J. Cardiothorac. Surg. 2003, 24, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.A.W.; Sleeper, L.A.; Mahony, L.; Colan, S.D.; Atz, A.M.; Breitbart, R.E.; Gersony, W.M.; Gallagher, D.; Geva, T.; Margossian, R.; et al. Contemporary Outcomes after the Fontan Procedure. J. Am. Coll. Cardiol. 2008, 52, 85–98. [Google Scholar] [CrossRef]
- Backer, C.L. The Functionally Univentricular Heart. J. Am. Coll. Cardiol. 2012, 59, 1186–1187. [Google Scholar] [CrossRef] [PubMed]
- d’Udekem, Y.; Xu, M.Y.; Galati, J.C.; Lu, S.; Iyengar, A.J.; Konstantinov, I.E.; Wheaton, G.R.; Ramsay, J.M.; Grigg, L.E.; Millar, J.; et al. Predictors of Survival After Single-Ventricle Palliation. J. Am. Coll. Cardiol. 2012, 59, 1178–1185. [Google Scholar] [CrossRef]
- King, G.; Buratto, E.; Daley, M.; Iyengar, A.; Alphonso, N.; Grigg, L.; Cordina, R.; d’Udekem, Y.; Konstantinov, I.E. Impact of Aortic Atresia After Fontan Operation in Patients With Hypoplastic Left Heart Syndrome. Ann. Thorac. Surg. 2022, 116, 95–102. [Google Scholar] [CrossRef]
- Iyengar, A.J.; Winlaw, D.S.; Galati, J.C.; Wheaton, G.R.; Gentles, T.L.; Grigg, L.E.; Justo, R.N.; Radford, D.J.; Weintraub, R.G.; Bullock, A.; et al. The Extracardiac Conduit Fontan Procedure in Australia and New Zealand: Hypoplastic Left Heart Syndrome Predicts Worse Early and Late Outcomes. Eur. J. Cardiothorac. Surg. 2014, 46, 465–473. [Google Scholar] [CrossRef]
- Book, W.M.; Gerardin, J.; Saraf, A.; Marie Valente, A.; Rodriguez, F. Clinical Phenotypes of Fontan Failure: Implications for Management: Fontan Phenotypes. Congenit. Heart Dis. 2016, 11, 296–308. [Google Scholar] [CrossRef]
- Sable, C.; Foster, E.; Uzark, K.; Bjornsen, K.; Canobbio, M.M.; Connolly, H.M.; Graham, T.P.; Gurvitz, M.Z.; Kovacs, A.; Meadows, A.K.; et al. Best Practices in Managing Transition to Adulthood for Adolescents with Congenital Heart Disease: The Transition Process and Medical and Psychosocial Issues: A Scientific Statement from the American Heart Association. Circulation 2011, 123, 1454–1485. [Google Scholar] [CrossRef]
- Michel, M.; Zlamy, M.; Entenmann, A.; Pichler, K.; Scholl-Bürgi, S.; Karall, D.; Geiger, R.; Salvador, C.; Niederwanger, C.; Ohuchi, H. Impact of the Fontan Operation on Organ Systems. Cardiovasc. Hematol. Disord. Drug Targets 2019, 19, 205–214. [Google Scholar] [CrossRef]
- Harteveld, L.M.; Blom, N.A.; Terol Espinosa de Los Monteros, C.; Kuipers, I.M.; Rammeloo, L.A.J.; Hazekamp, M.G.; van Dijk, J.G.; ten Harkel, A.D.J. 3-Month Enalapril Treatment in Pediatric Fontan Patients with Moderate to Good Systolic Ventricular Function. Am. J. Cardiol. 2022, 163, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Shaddy, R.E.; Boucek, M.M.; Hsu, D.T.; Boucek, R.J.; Canter, C.E.; Mahony, L.; Ross, R.D.; Pahl, E.; Blume, E.D.; Dodd, D.A.; et al. Carvedilol for Children and Adolescents With Heart Failure: A Randomized Controlled Trial. JAMA 2007, 298, 1171. [Google Scholar] [CrossRef] [PubMed]
- Schranz, D.; Voelkel, N.F. “Nihilism” of Chronic Heart Failure Therapy in Children and Why Effective Therapy Is Withheld. Eur. J. Pediatr. 2016, 175, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.A.W.; Breitbart, R.E.; McCrindle, B.W.; Sleeper, L.A.; Atz, A.M.; Hsu, D.T.; Lu, M.; Margossian, R.; Williams, R.V. The Fontan Patient: Inconsistencies in Medication Therapy Across Seven Pediatric Heart Network Centers. Pediatr. Cardiol. 2010, 31, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Ghanayem, N.S.; Berger, S.; Tweddell, J.S. Medical Management of the Failing Fontan. Pediatr. Cardiol. 2007, 28, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Taegtmeyer, H. Metabolism—The Lost Child of Cardiology. J. Am. Coll. Cardiol. 2000, 36, 1386–1388. [Google Scholar] [CrossRef]
- Taegtmeyer, H. Cardiac Metabolism as a Target for the Treatment of Heart Failure. Circulation 2004, 110, 894–896. [Google Scholar] [CrossRef]
- Ashrafian, H.; Frenneaux, M.P.; Opie, L.H. Metabolic Mechanisms in Heart Failure. Circulation 2007, 116, 434–448. [Google Scholar] [CrossRef]
- Kimball, T.H.; Vondriska, T.M. Metabolism, Epigenetics, and Causal Inference in Heart Failure. Trends Endocrinol. Metab. 2020, 31, 181–191. [Google Scholar] [CrossRef]
- Selvaraj, S.; Kelly, D.P.; Margulies, K.B. Implications of Altered Ketone Metabolism and Therapeutic Ketosis in Heart Failure. Circulation 2020, 141, 1800–1812. [Google Scholar] [CrossRef]
- Taegtmeyer, H. Energy Metabolism of the Heart: From Basic Concepts to Clinical Applications Applications. Curr. Probl. Cardiol. 1994, 19, 61–86. [Google Scholar] [CrossRef]
- Christensen, K.H. Treatment with the Ketone Body 3-Hydroxybutyrate in Patients with Acute Heart Failure. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04442555 (accessed on 20 July 2022).
- Yurista, S.R.; Chong, C.-R.; Badimon, J.J.; Kelly, D.P.; de Boer, R.A.; Westenbrink, B.D. Therapeutic Potential of Ketone Bodies for Patients with Cardiovascular Disease. J. Am. Coll. Cardiol. 2021, 77, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Takahara, S.; Soni, S.; Phaterpekar, K.; Kim, T.T.; Maayah, Z.H.; Levasseur, J.L.; Silver, H.L.; Freed, D.H.; Ferdaoussi, M.; Dyck, J.R.B. Chronic Exogenous Ketone Supplementation Blunts the Decline of Cardiac Function in the Failing Heart. ESC Heart Fail. 2021, 8, 5606–5612. [Google Scholar] [CrossRef] [PubMed]
- Takahara, S.; Soni, S.; Maayah, Z.H.; Ferdaoussi, M.; Dyck, J.R.B. Ketone Therapy for Heart Failure: Current Evidence for Clinical Use. Cardiovasc. Res. 2022, 118, 977–987. [Google Scholar] [CrossRef]
- Monzo, L.; Sedlacek, K.; Hromanikova, K.; Tomanova, L.; Borlaug, B.A.; Jabor, A.; Kautzner, J.; Melenovsky, V. Myocardial Ketone Body Utilization in Patients with Heart Failure: The Impact of Oral Ketone Ester. Metabolism 2021, 115, 154452. [Google Scholar] [CrossRef] [PubMed]
- Papazafiropoulou, A.; Georgopoulos, M.; Katsilambros, N. Ketone Bodies and the Heart. Arch. Med. Sci. Atheroscler. Dis. 2021, 6, 209–214. [Google Scholar] [CrossRef]
- Schulze, P.C.; Wu, J.M.F. Ketone Bodies for the Starving Heart. Nat. Metab. 2020, 2, 1183–1185. [Google Scholar] [CrossRef]
- Horton, J.L.; Davidson, M.T.; Kurishima, C.; Vega, R.B.; Powers, J.C.; Matsuura, T.R.; Petucci, C.; Lewandowski, E.D.; Crawford, P.A.; Muoio, D.M.; et al. The Failing Heart Utilizes 3-Hydroxybutyrate as a Metabolic Stress Defense. JCI Insight 2019, 4, e124079. [Google Scholar] [CrossRef]
- Uchihashi, M.; Hoshino, A.; Okawa, Y.; Ariyoshi, M.; Kaimoto, S.; Tateishi, S.; Ono, K.; Yamanaka, R.; Hato, D.; Fushimura, Y.; et al. Cardiac-Specific Bdh1 Overexpression Ameliorates Oxidative Stress and Cardiac Remodeling in Pressure Overload–Induced Heart Failure. Circ. Heart Fail. 2017, 10, e004417. [Google Scholar] [CrossRef]
- Schugar, R.C.; Moll, A.R.; André d’Avignon, D.; Weinheimer, C.J.; Kovacs, A.; Crawford, P.A. Cardiomyocyte-Specific Deficiency of Ketone Body Metabolism Promotes Accelerated Pathological Remodeling. Mol. Metab. 2014, 3, 754–769. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Röhling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone Bodies: From Enemy to Friend and Guardian Angel. BMC Med. 2021, 19, 313. [Google Scholar] [CrossRef]
- Michel, M.; Dubowy, K.-O.; Entenmann, A.; Karall, D.; Adam, M.G.; Zlamy, M.; Odri Komazec, I.; Geiger, R.; Niederwanger, C.; Salvador, C.; et al. Targeted Metabolomic Analysis of Serum Amino Acids in the Adult Fontan Patient with a Dominant Left Ventricle. Sci. Rep. 2020, 10, 8930. [Google Scholar] [CrossRef]
- Michel, M.; Dubowy, K.-O.; Zlamy, M.; Karall, D.; Adam, M.G.; Entenmann, A.; Keller, M.A.; Koch, J.; Odri Komazec, I.; Geiger, R.; et al. Targeted Metabolomic Analysis of Serum Phospholipid and Acylcarnitine in the Adult Fontan Patient with a Dominant Left Ventricle. Ther. Adv. Chronic Dis. 2020, 11, 204062232091603. [Google Scholar] [CrossRef] [PubMed]
- Opie, L.H. Heart Physiology: From Cell to Circulation, 4th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2004. [Google Scholar]
- Barth, E. Ultrastructural Quantitation of Mitochondria and Myofilaments in Cardiac Muscle from 10 Different Animal Species Including Man. J. Mol. Cell. Cardiol. 1992, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Ingwall, J.S.; Weiss, R.G. Is the Failing Heart Energy Starved?: On Using Chemical Energy to Support Cardiac Function. Circ. Res. 2004, 95, 135–145. [Google Scholar] [CrossRef]
- Herrmann, G.; Decherd, G. The chemical nature of heart failure. Ann. Intern. Med. 1939, 12, 1233. [Google Scholar] [CrossRef]
- Neubauer, S. The Failing Heart—An Engine Out of Fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef]
- Sack, M.N.; Rader, T.A.; Park, S.; Bastin, J.; McCune, S.A.; Kelly, D.P. Fatty Acid Oxidation Enzyme Gene Expression Is Downregulated in the Failing Heart. Circulation 1996, 94, 2837–2842. [Google Scholar] [CrossRef]
- Katz, A.M. Energetics and the Failing Heart. Hosp. Pract. 1991, 26, 78–90. [Google Scholar] [CrossRef]
- Taegtmeyer, H. Failing Heart and Starving Brain. Circulation 2016, 134, 265–266. [Google Scholar] [CrossRef] [PubMed]
- Giussani, D.A.; Bennet, L.; Sferruzzi-Perri, A.N.; Vaughan, O.R.; Fowden, A.L. Hypoxia, fetal and neonatal physiology: 100 years on from Sir Joseph Barcroft. J. Physiol. 2016, 594, 1105–1111. [Google Scholar] [CrossRef]
- Girard, J.; Ferre, P.; Pegorier, J.P.; Duee, P.H. Adaptations of Glucose and Fatty Acid Metabolism during Perinatal Period and Suckling-Weaning Transition. Physiol. Rev. 1992, 72, 507–562. [Google Scholar] [CrossRef]
- Ascuitto, R.J.; Ross-Ascuitto, N.T. Substrate Metabolism in the Developing Heart. Semin. Perinatol. 1996, 20, 542–563. [Google Scholar] [CrossRef]
- Itoi, T.; Lopaschuk, G.D. The Contribution of Glycolysis, Glucose Oxidation, Lactate Oxidation, and Fatty Acid Oxidation to ATP Production in Isolated Biventricular Working Hearts from 2-Week-Old Rabbits. Pediatr. Res. 1993, 34, 735–741. [Google Scholar] [CrossRef]
- Dimasi, C.G.; Darby, J.R.T.; Morrison, J.L. A change of heart: Understanding the mechanisms regulating cardiac proliferation and metabolism before and after birth. J. Physiol. 2023, 601, 1319–1341. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Jaswal, J.S. Energy Metabolic Phenotype of the Cardiomyocyte During Development, Differentiation, and Postnatal Maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Ritterhoff, J.; Tian, R. Metabolism in Cardiomyopathy: Every Substrate Matters. Cardiovasc. Res. 2017, 113, 411–421. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; Sen, S.; Vela, D. Return to the Fetal Gene Program: A Suggested Metabolic Link to Gene Expression in the Heart. Ann. N. Y. Acad. Sci. 2010, 1188, 191–198. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Shannon, R.P. Insulin Resistance in Dilated Cardiomyopathy. Rev. Cardiovasc. Med. 2003, 4, S50–S57. [Google Scholar] [PubMed]
- Nikolaidis, L. The Development of Myocardial Insulin Resistance in Conscious Dogs with Advanced Dilated Cardiomyopathy. Cardiovasc. Res. 2004, 61, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Taegtmeyer, H.; Golfman, L.; Sharma, S.; Razeghi, P.; Arsdall, M. Linking Gene Expression to Function: Metabolic Flexibility in the Normal and Diseased Heart. Ann. N. Y. Acad. Sci. 2004, 1015, 202–213. [Google Scholar] [CrossRef]
- Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid Use and Misuse by the Heart. Circ. Res. 2016, 118, 1736–1751. [Google Scholar] [CrossRef]
- Kolwicz, S.C.; Airhart, S.; Tian, R. Ketones Step to the Plate: A Game Changer for Metabolic Remodeling in Heart Failure? Circulation 2016, 133, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Bedi, K.C.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B.; et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 2016, 133, 706–716. [Google Scholar] [CrossRef]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Krüger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016, 133, 698–705. [Google Scholar] [CrossRef]
- Huynh, K. Ketone Bodies as Fuel in Heart Failure. Nat. Rev. Cardiol. 2016, 13, 123. [Google Scholar] [CrossRef]
- Manolis, A.S.; Manolis, T.A.; Manolis, A.A. Ketone Bodies and Cardiovascular Disease: An Alternate Fuel Source to the Rescue. Int. J. Mol. Sci. 2023, 24, 3534. [Google Scholar] [CrossRef]
- Voros, G.; Ector, J.; Garweg, C.; Droogne, W.; Van Cleemput, J.; Peersman, N.; Vermeersch, P.; Janssens, S. Increased Cardiac Uptake of Ketone Bodies and Free Fatty Acids in Human Heart Failure and Hypertrophic Left Ventricular Remodeling. Circ. Heart Fail. 2018, 11, e004953. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Ketone Body Can Be a Fuel Substrate for Failing Heart. Cardiovasc. Res. 2019, 115, 1567–1569. [Google Scholar] [CrossRef]
- Liao, S.; Tang, Y.; Yue, X.; Gao, R.; Yao, W.; Zhou, Y.; Zhang, H. β-Hydroxybutyrate Mitigated Heart Failure with Preserved Ejection Fraction by Increasing Treg Cells via Nox2/GSK-3β. J. Inflamm. Res. 2021, 14, 4697–4706. [Google Scholar] [CrossRef]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circ. Res. 2021, 128, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative Stress and Heart Failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef]
- Beer, M.; Seyfarth, T.; Sandstede, J.; Landschütz, W.; Lipke, C.; Köstler, H.; von Kienlin, M.; Harre, K.; Hahn, D.; Neubauer, S. Absolute Concentrations of High-Energy Phosphate Metabolites in Normal, Hypertrophied, and Failing Human Myocardium Measured Noninvasively with 31P-SLOOP Magnetic Resonance Spectroscopy. J. Am. Coll. Cardiol. 2002, 40, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.G.; Gerstenblith, G.; Bottomley, P.A. ATP Flux through Creatine Kinase in the Normal, Stressed, and Failing Human Heart. Proc. Natl. Acad. Sci. USA 2005, 102, 808–813. [Google Scholar] [CrossRef]
- Smith, C.S.; Bottomley, P.A.; Schulman, S.P.; Gerstenblith, G.; Weiss, R.G. Altered Creatine Kinase Adenosine Triphosphate Kinetics in Failing Hypertrophied Human Myocardium. Circulation 2006, 114, 1151–1158. [Google Scholar] [CrossRef]
- Nielsen, R.; Møller, N.; Gormsen, L.C.; Tolbod, L.P.; Hansson, N.H.; Sorensen, J.; Harms, H.J.; Frøkiær, J.; Eiskjaer, H.; Jespersen, N.R.; et al. Cardiovascular Effects of Treatment with the Ketone Body 3-Hydroxybutyrate in Chronic Heart Failure Patients. Circulation 2019, 139, 2129–2141. [Google Scholar] [CrossRef]
- Brittain, E.L.; Talati, M.; Fessel, J.P.; Zhu, H.; Penner, N.; Calcutt, M.W.; West, J.D.; Funke, M.; Lewis, G.D.; Gerszten, R.E.; et al. Fatty Acid Metabolic Defects and Right Ventricular Lipotoxicity in Human Pulmonary Arterial Hypertension. Circulation 2016, 133, 1936–1944. [Google Scholar] [CrossRef] [PubMed]
- Pugh, M.E.; Robbins, I.M.; Rice, T.W.; West, J.; Newman, J.H.; Hemnes, A.R. Unrecognized Glucose Intolerance Is Common in Pulmonary Arterial Hypertension. J. Heart Lung Transplant. 2011, 30, 904–911. [Google Scholar] [CrossRef]
- West, J.; Niswender, K.D.; Johnson, J.A.; Pugh, M.E.; Gleaves, L.; Fessel, J.P.; Hemnes, A.R. A Potential Role for Insulin Resistance in Experimental Pulmonary Hypertension. Eur. Respir. J. 2013, 41, 861–871. [Google Scholar] [CrossRef]
- Zamanian, R.T.; Hansmann, G.; Snook, S.; Lilienfeld, D.; Rappaport, K.M.; Reaven, G.M.; Rabinovitch, M.; Doyle, R.L. Insulin Resistance in Pulmonary Arterial Hypertension. Eur. Respir. J. 2008, 33, 318–324. [Google Scholar] [CrossRef]
- Zare, E.; Kafshbani, P.; Chenaghlou, M.; Noori, M.; Ghaemmaghami, Z.; Amin, A.; Taghavi, S.; Naderi, N. Prognostic Significance of Insulin Resistance in Pulmonary Hypertension. ESC Heart Fail. 2022, 9, 318–326. [Google Scholar] [CrossRef]
- University of Aarhus. Ketones for Pulmonary Hypertension—Effects on Hemodynamics (KEPAH). 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04615754 (accessed on 22 March 2023).
- Nielsen, R.; Christensen, K.H.; Gopalasingam, N.; Berg-Hansen, K.; Seefeldt, J.; Homilius, C.; Boedtkjer, E.; Andersen, M.J.; Wiggers, H.; Møller, N.; et al. Hemodynamic Effects of Ketone Bodies in Patients with Pulmonary Hypertension. J. Am. Heart Assoc. 2023, 12, e028232. [Google Scholar] [CrossRef]
- Blake, M.; Puchalska, P.; Kazmirczak, F.; Thenappan, T.; Crawford, P.A.; Prins, K.W. Ketone Bodies in Right Ventricular Failure: A Unique Therapeutic Opportunity. bioRxiv 2023. [Google Scholar] [CrossRef]
- McCullough, D.J.; Kue, N.; Mancini, T.; Vang, A.; Clements, R.T.; Choudhary, G. Endurance Exercise Training in Pulmonary Hypertension Increases Skeletal Muscle Electron Transport Chain Supercomplex Assembly. Pulm. Circ. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Cawthon, D.; Beers, K.; Bottje, W.G. Electron Transport Chain Defect and Inefficient Respiration May Underlie Pulmonary Hypertension Syndrome (Ascites)-Associated Mitochondrial Dysfunction in Broilers. Poult. Sci. 2001, 80, 474–484. [Google Scholar] [CrossRef]
- Xu, W.; Comhair, S.A.A.; Chen, R.; Hu, B.; Hou, Y.; Zhou, Y.; Mavrakis, L.A.; Janocha, A.J.; Li, L.; Zhang, D.; et al. Integrative Proteomics and Phosphoproteomics in Pulmonary Arterial Hypertension. Sci. Rep. 2019, 9, 18623. [Google Scholar] [CrossRef]
- Huertas, A.; Tu, L.; Humbert, M.; Guignabert, C. Chronic Inflammation within the Vascular Wall in Pulmonary Arterial Hypertension: More than a Spectator. Cardiovasc. Res. 2020, 116, 885–893. [Google Scholar] [CrossRef]
- Fowler, E.D.; Hauton, D.; Boyle, J.; Egginton, S.; Steele, D.S.; White, E. Energy Metabolism in the Failing Right Ventricle: Limitations of Oxygen Delivery and the Creatine Kinase System. Int. J. Mol. Sci. 2019, 20, 1805. [Google Scholar] [CrossRef]
- Garcia, A.M.; Toni, L.S.; Miyano, C.A.; Sparagna, G.C.; Jonscher, R.; Phillips, E.K.; Karimpour-Fard, A.; Chapman, H.L.; Baybayon-Grandgeorge, A.N.; Pietra, A.E.; et al. Cardiac Transcriptome Remodeling and Impaired Bioenergetics in Single-Ventricle Congenital Heart Disease. JACC Basic Transl. Sci. 2023, 8, 258–279. [Google Scholar] [CrossRef]
- Pires da Silva, J.; Pietra, A.E.; Baybayon-Grandgeorge, A.N.; Garcia, A.M. Serum Metabolic Profiling Identifies Key Differences between Patients with Single-Ventricle Heart Disease and Healthy Controls. Int. J. Transl. Med. 2022, 2, 78–96. [Google Scholar] [CrossRef]
- Xu, X.; Lin, J.-H.I.; Bais, A.S.; Reynolds, M.J.; Tan, T.; Gabriel, G.C.; Kondos, Z.; Liu, X.; Shiva, S.S.; Lo, C.W. Mitochondrial Respiration Defects in Single-Ventricle Congenital Heart Disease. Front. Cardiovasc. Med. 2021, 8, 734388. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA Damage and Dysfunction Associated with Oxidative Stress in Failing Hearts After Myocardial Infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef]
- Tsutsui, H. Mitochondrial Oxidative Stress and Heart Failure. Intern. Med. 2006, 45, 809–813. [Google Scholar] [CrossRef]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 281, 785–789. [Google Scholar] [CrossRef]
- Koeslag, J.H.; Noakes, T.D.; Sloan, A.W. Post-Exercise Ketosis. J. Physiol. 1980, 301, 79–90. [Google Scholar] [CrossRef]
- Robinson, A.M.; Williamson, D.H. Physiological Roles of Ketone Bodies as Substrates and Signals in Mammalian Tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Ho, K.L.; Pherwani, S.; Ketema, E.B. Ketone Metabolism in the Failing Heart. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2020, 1865, 158813. [Google Scholar] [CrossRef]
- Hue, L.; Taegtmeyer, H. The Randle Cycle Revisited: A New Head for an Old Hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E578–E591. [Google Scholar] [CrossRef]
- Barger, P.M.; Kelly, D.P. PPAR Signaling in the Control of Cardiac Energy Metabolism. Trends Cardiovasc. Med. 2000, 10, 238–245. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; Wilson, C.R.; Razeghi, P.; Sharma, S. Metabolic Energetics and Genetics in the Heart. Ann. N. Y. Acad. Sci. 2005, 1047, 208–218. [Google Scholar] [CrossRef]
- Kelly, D.P. PPARs of the Heart: Three Is a Crowd. Circ. Res. 2003, 92, 482–484. [Google Scholar] [CrossRef]
- Lehman, J.J.; Kelly, D.P. Gene Regulatory Mechanisms Governing Energy Metabolism during Cardiac Hypertrophic Growth. Heart Fail. Rev. 2002, 7, 175–185. [Google Scholar] [CrossRef]
- Razeghi, P.; Young, M.E.; Abbasi, S.; Taegtmeyer, H. Hypoxia in Vivo Decreases Peroxisome Proliferator-Activated Receptor α-Regulated Gene Expression in Rat Heart. Biochem. Biophys. Res. Commun. 2001, 287, 5–10. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, Y.; Ren, J. Acetylation in Cardiovascular Diseases: Molecular Mechanisms and Clinical Implications. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165836. [Google Scholar] [CrossRef]
- Shogren-Knaak, M.; Ishii, H.; Sun, J.-M.; Pazin, M.J.; Davie, J.R.; Peterson, C.L. Histone H4-K16 Acetylation Controls Chromatin Structure and Protein Interactions. Science 2006, 311, 844–847. [Google Scholar] [CrossRef]
- Peleg, S.; Feller, C.; Ladurner, A.G.; Imhof, A. The Metabolic Impact on Histone Acetylation and Transcription in Ageing. Trends Biochem. Sci. 2016, 41, 700–711. [Google Scholar] [CrossRef]
- Verdin, E.; Dequiedt, F.; Kasler, H.G. Class II Histone Deacetylases: Versatile Regulators. Trends Genet. 2003, 19, 286–293. [Google Scholar] [CrossRef]
- McKinsey, T.A.; Olson, E.N. Cardiac Histone Acetylation—Therapeutic Opportunities Abound. Trends Genet. 2004, 20, 206–213. [Google Scholar] [CrossRef]
- Backs, J.; Olson, E.N. Control of Cardiac Growth by Histone Acetylation/Deacetylation. Circ. Res. 2006, 98, 15–24. [Google Scholar] [CrossRef]
- Allard, M.F.; Schonekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of Oxidative Metabolism and Glycolysis to ATP Production in Hypertrophied Hearts. Am. J. Physiol. Heart Circ. Physiol. 1994, 267, H742–H750. [Google Scholar] [CrossRef]
- Diakos, N.A.; Navankasattusas, S.; Abel, E.D.; Rutter, J.; McCreath, L.; Ferrin, P.; McKellar, S.H.; Miller, D.V.; Park, S.Y.; Richardson, R.S.; et al. Evidence of Glycolysis Up-Regulation and Pyruvate Mitochondrial Oxidation Mismatch During Mechanical Unloading of the Failing Human Heart. JACC Basic Transl. Sci. 2016, 1, 432–444. [Google Scholar] [CrossRef]
- Bottomley, P.A.; Panjrath, G.S.; Lai, S.; Hirsch, G.A.; Wu, K.; Najjar, S.S.; Steinberg, A.; Gerstenblith, G.; Weiss, R.G. Metabolic Rates of ATP Transfer Through Creatine Kinase (CK Flux) Predict Clinical Heart Failure Events and Death. Sci. Transl. Med. 2013, 5, 215re3. [Google Scholar] [CrossRef]
- Mey, J.T.; Hari, A.; Axelrod, C.L.; Fealy, C.E.; Erickson, M.L.; Kirwan, J.P.; Dweik, R.A.; Heresi, G.A. Lipids and Ketones Dominate Metabolism at the Expense of Glucose Control in Pulmonary Arterial Hypertension: A Hyperglycaemic Clamp and Metabolomics Study. Eur. Respir. J. 2020, 55, 1901700. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial Dysfunction in Pathophysiology of Heart Failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- Takimoto, E.; Kass, D.A. Role of Oxidative Stress in Cardiac Hypertrophy and Remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating Oxidative Stress in Heart Failure: Past, Present and Future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Nascimben, L.; Ingwall, J.S.; Pauletto, P.; Friedrich, J.; Gwathmey, J.K.; Saks, V.; Pessina, A.C.; Allen, P.D. Creatine Kinase System in Failing and Nonfailing Human Myocardium. Circulation 1996, 94, 1894–1901. [Google Scholar] [CrossRef]
- Keceli, G.; Gupta, A.; Sourdon, J.; Gabr, R.; Schär, M.; Dey, S.; Tocchetti, C.G.; Stuber, A.; Agrimi, J.; Zhang, Y.; et al. Mitochondrial Creatine Kinase Attenuates Pathologic Remodeling in Heart Failure. Circ. Res. 2022, 130, 741–759. [Google Scholar] [CrossRef]
- Olson, E.N.; Backs, J.; McKinsey, T.A. Control of Cardiac Hypertrophy and Heart Failure by Histone Acetylation/Deacetylation. In Novartis Foundation Symposia; Bock, G., Goode, J., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2008; pp. 3–19. [Google Scholar] [CrossRef]
- Fukushima, A.; Zhang, L.; Huqi, A.; Lam, V.H.; Rawat, S.; Altamimi, T.; Wagg, C.S.; Dhaliwal, K.K.; Hornberger, L.K.; Kantor, P.F.; et al. Acetylation Contributes to Hypertrophy-Caused Maturational Delay of Cardiac Energy Metabolism. JCI Insight 2018, 3, e99239. [Google Scholar] [CrossRef]
- Castillo, E.C.; Morales, J.A.; Chapoy-Villanueva, H.; Silva-Platas, C.; Treviño-Saldaña, N.; Guerrero-Beltrán, C.E.; Bernal-Ramírez, J.; Torres-Quintanilla, A.; García, N.; Youker, K.; et al. Mitochondrial Hyperacetylation in the Failing Hearts of Obese Patients Mediated Partly by a Reduction in SIRT3: The Involvement of the Mitochondrial Permeability Transition Pore. Cell Physiol. Biochem. 2019, 53, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Ooi, J.Y.Y.; Tuano, N.K.; Rafehi, H.; Gao, X.-M.; Ziemann, M.; Du, X.-J.; El-Osta, A. HDAC Inhibition Attenuates Cardiac Hypertrophy by Acetylation and Deacetylation of Target Genes. Epigenetics 2015, 10, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-H.; Seok, Y.M.; Song, M.; Lee, H.-A.; Kurz, T.; Kim, I. Histone Deacetylase Inhibition Attenuates Cardiac Hypertrophy and Fibrosis through Acetylation of Mineralocorticoid Receptor in Spontaneously Hypertensive Rats. Mol. Pharmacol. 2015, 87, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Scholz, B.; Schulte, J.S.; Hamer, S.; Himmler, K.; Pluteanu, F.; Seidl, M.D.; Stein, J.; Wardelmann, E.; Hammer, E.; Völker, U.; et al. HDAC (Histone Deacetylase) Inhibitor Valproic Acid Attenuates Atrial Remodeling and Delays the Onset of Atrial Fibrillation in Mice. Circ. Arrhythm. Electrophysiol. 2019, 12, e007071. [Google Scholar] [CrossRef]
- Colussi, C.; Berni, R.; Rosati, J.; Straino, S.; Vitale, S.; Spallotta, F.; Baruffi, S.; Bocchi, L.; Delucchi, F.; Rossi, S.; et al. The Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid Reduces Cardiac Arrhythmias in Dystrophic Mice. Cardiovasc. Res. 2010, 87, 73–82. [Google Scholar] [CrossRef]
- Müller, J.; Bertsch, T.; Volke, J.; Schmid, A.; Klingbeil, R.; Metodiev, Y.; Karaca, B.; Kim, S.-H.; Lindner, S.; Schupp, T.; et al. Narrative Review of Metabolomics in Cardiovascular Disease. J. Thorac. Dis. 2021, 13, 2532–2550. [Google Scholar] [CrossRef]
- Bassareo, P.P.; McMahon, C.J. Metabolomics: A New Tool in Our Understanding of Congenital Heart Disease. Children 2022, 9, 1803. [Google Scholar] [CrossRef]
- Michel, M.; Laser, K.T.; Dubowy, K.-O.; Scholl-Bürgi, S.; Michel, E. Metabolomics and Random Forests in Patients with Complex Congenital Heart Disease. Front. Cardiovasc. Med. 2022, 9, 994068. [Google Scholar] [CrossRef]
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive Quantification of Fuel Use by the Failing and Nonfailing Human Heart. Science 2020, 370, 364–368. [Google Scholar] [CrossRef]
- Smith, E.; Fernandez, C.; Melander, O.; Ottosson, F. Altered Acylcarnitine Metabolism Is Associated With an Increased Risk of Atrial Fibrillation. J. Am. Heart Assoc. 2020, 9, e016737. [Google Scholar] [CrossRef]
- Ruiz, M.; Labarthe, F.; Fortier, A.; Bouchard, B.; Thompson Legault, J.; Bolduc, V.; Rigal, O.; Chen, J.; Ducharme, A.; Crawford, P.A.; et al. Circulating Acylcarnitine Profile in Human Heart Failure: A Surrogate of Fatty Acid Metabolic Dysregulation in Mitochondria and Beyond. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H768–H781. [Google Scholar] [CrossRef] [PubMed]
- Penny, D.J.; Redington, A.N. Function of the Left and Right Ventricles and the Interactions Between Them. Pediatr. Crit. Care Med. 2016, 17, S112–S118. [Google Scholar] [CrossRef] [PubMed]
- Sanz, J.; Sánchez-Quintana, D.; Bossone, E.; Bogaard, H.J.; Naeije, R. Anatomy, Function, and Dysfunction of the Right Ventricle. J. Am. Coll. Cardiol. 2019, 73, 1463–1482. [Google Scholar] [CrossRef] [PubMed]
- Kondo, R.P.; Dederko, D.A.; Teutsch, C.; Chrast, J.; Catalucci, D.; Chien, K.R.; Giles, W.R. Comparison of Contraction and Calcium Handling between Right and Left Ventricular Myocytes from Adult Mouse Heart: A Role for Repolarization Waveform: Interventricular Heterogeneity of Cardiac Myocyte Contractions. J. Physiol. 2006, 571, 131–146. [Google Scholar] [CrossRef]
- Sedmera, D. Form Follows Function: Developmental and Physiological View on Ventricular Myocardial Architecture. Eur. J. Cardiothorac. Surg. 2005, 28, 526–528. [Google Scholar] [CrossRef]
- Garcia, A.M.; Beatty, J.-T.; Nakano, S.J. Heart Failure in Single Right Ventricle Congenital Heart Disease: Physiological and Molecular Considerations. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H947–H965. [Google Scholar] [CrossRef] [PubMed]
- Friehs, I.; Cowan, D.B.; Choi, Y.-H.; Black, K.M.; Barnett, R.; Bhasin, M.K.; Daly, C.; Dillon, S.J.; Libermann, T.A.; McGowan, F.X.; et al. Pressure-Overload Hypertrophy of the Developing Heart Reveals Activation of Divergent Gene and Protein Pathways in the Left and Right Ventricular Myocardium. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H697–H708. [Google Scholar] [CrossRef]
- Schreckenberg, R.; Rebelo, M.; Deten, A.; Weber, M.; Rohrbach, S.; Pipicz, M.; Csonka, C.; Ferdinandy, P.; Schulz, R.; Schlüter, K.-D. Specific Mechanisms Underlying Right Heart Failure: The Missing Upregulation of Superoxide Dismutase-2 and Its Decisive Role in Antioxidative Defense. Antioxid. Redox Signal. 2015, 23, 1220–1232. [Google Scholar] [CrossRef]
- Nagendran, J.; Gurtu, V.; Fu, D.Z.; Dyck, J.R.B.; Haromy, A.; Ross, D.B.; Rebeyka, I.M.; Michelakis, E.D. A Dynamic and Chamber-Specific Mitochondrial Remodeling in Right Ventricular Hypertrophy Can Be Therapeutically Targeted. J. Thorac. Cardiovasc. Surg. 2008, 136, 168–178.e3. [Google Scholar] [CrossRef] [PubMed]
- Bokhari, S.; Raina, A.; Berman Rosenweig, E.; Schulze, P.C.; Bokhari, J.; Einstein, A.J.; Barst, R.J.; Johnson, L.L. PET Imaging May Provide a Novel Biomarker and Understanding of Right Ventricular Dysfunction in Patients With Idiopathic Pulmonary Arterial Hypertension. Circ. Cardiovasc. Imaging 2011, 4, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arroyo, J.; Mizuno, S.; Szczepanek, K.; Van Tassell, B.; Natarajan, R.; dos Remedios, C.G.; Drake, J.I.; Farkas, L.; Kraskauskas, D.; Wijesinghe, D.S.; et al. Metabolic Gene Remodeling and Mitochondrial Dysfunction in Failing Right Ventricular Hypertrophy Secondary to Pulmonary Arterial Hypertension. Circ. Heart Fail. 2013, 6, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Marsboom, G.; Archer, S.L. Mitochondrial Metabolic Adaptation in Right Ventricular Hypertrophy and Failure. J. Mol. Med. 2010, 88, 1011–1020. [Google Scholar] [CrossRef]
- Fessel, J.P.; Hamid, R.; Wittmann, B.M.; Robinson, L.J.; Blackwell, T.; Tada, Y.; Tanabe, N.; Tatsumi, K.; Hemnes, A.R.; West, J.D. Metabolomic Analysis of Bone Morphogenetic Protein Receptor Type 2 Mutations in Human Pulmonary Endothelium Reveals Widespread Metabolic Reprogramming. Pulm. Circ. 2012, 2, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Schooneman, M.G.; Vaz, F.M.; Houten, S.M.; Soeters, M.R. Acylcarnitines. Diabetes 2013, 62, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kalim, S.; Clish, C.B.; Wenger, J.; Elmariah, S.; Yeh, R.W.; Deferio, J.J.; Pierce, K.; Deik, A.; Gerszten, R.E.; Thadhani, R.; et al. A Plasma Long-Chain Acylcarnitine Predicts Cardiovascular Mortality in Incident Dialysis Patients. J. Am. Heart Assoc. 2013, 2, e000542. [Google Scholar] [CrossRef]
- Aitken-Buck, H.M.; Krause, J.; Zeller, T.; Jones, P.P.; Lamberts, R.R. Long-Chain Acylcarnitines and Cardiac Excitation-Contraction Coupling: Links to Arrhythmias. Front. Physiol. 2020, 11, 577856. [Google Scholar] [CrossRef]
- Brunner, N.W.; Skhiri, M.; Fortenko, O.; Hsi, A.; Haddad, F.; Khazeni, N.; Zamanian, R.T. Impact of Insulin Resistance on Ventricular Function in Pulmonary Arterial Hypertension. J. Heart Lung Transplant. 2014, 33, 721–726. [Google Scholar] [CrossRef]
- Assad, T.R.; Hemnes, A.R. Metabolic Dysfunction in Pulmonary Arterial Hypertension. Curr. Hypertens. Rep. 2015, 17, 20. [Google Scholar] [CrossRef]
- Graham, B.B.; Kumar, R.; Mickael, C.; Sanders, L.; Gebreab, L.; Huber, K.M.; Perez, M.; Smith-Jones, P.; Serkova, N.J.; Tuder, R.M. Severe Pulmonary Hypertension Is Associated with Altered Right Ventricle Metabolic Substrate Uptake. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L435–L440. [Google Scholar] [CrossRef]
- Ohuchi, H.; Negishi, J.; Hayama, Y.; Miike, H.; Suzuki, D.; Nakajima, K.; Konagai, N.; Iwasa, T.; Sakaguchi, H.; Kurosaki, K.; et al. Abnormal Glucose Metabolism in Patients with Fontan Circulation: Unique Characteristics and Associations with Fontan Pathophysiology. Am. Heart J. 2019, 216, 125–135. [Google Scholar] [CrossRef]
- Sharma, S.; Sud, N.; Wiseman, D.A.; Carter, A.L.; Kumar, S.; Hou, Y.; Rau, T.; Wilham, J.; Harmon, C.; Oishi, P.; et al. Altered Carnitine Homeostasis Is Associated with Decreased Mitochondrial Function and Altered Nitric Oxide Signaling in Lambs with Pulmonary Hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L46–L56. [Google Scholar] [CrossRef]
- Fowler, E.D.; Benoist, D.; Drinkhill, M.J.; Stones, R.; Helmes, M.; Wüst, R.C.I.; Stienen, G.J.M.; Steele, D.S.; White, E. Decreased Creatine Kinase Is Linked to Diastolic Dysfunction in Rats with Right Heart Failure Induced by Pulmonary Artery Hypertension. J. Mol. Cell. Cardiol. 2015, 86, 1–8. [Google Scholar] [CrossRef]
- Bernal-Ramirez, J.; Díaz-Vesga, M.C.; Talamilla, M.; Méndez, A.; Quiroga, C.; Garza-Cervantes, J.A.; Lázaro-Alfaro, A.; Jerjes-Sanchez, C.; Henríquez, M.; García-Rivas, G.; et al. Exploring Functional Differences between the Right and Left Ventricles to Better Understand Right Ventricular Dysfunction. Oxidative Med. Cell. Longev. 2021, 2021, 9993060. [Google Scholar] [CrossRef]
- Bogaard, H.J.; Mizuno, S.; Hussaini, A.A.A.; Toldo, S.; Abbate, A.; Kraskauskas, D.; Kasper, M.; Natarajan, R.; Voelkel, N.F. Suppression of Histone Deacetylases Worsens Right Ventricular Dysfunction after Pulmonary Artery Banding in Rats. Am. J. Respir. Crit. Care Med. 2011, 183, 1402–1410. [Google Scholar] [CrossRef]
- Chelladurai, P.; Boucherat, O.; Stenmark, K.; Kracht, M.; Seeger, W.; Bauer, U.; Bonnet, S.; Pullamsetti, S.S. Targeting Histone Acetylation in Pulmonary Hypertension and Right Ventricular Hypertrophy. Br. J. Pharmacol. 2021, 178, 54–71. [Google Scholar] [CrossRef]
- Chen, C.; Luo, F.; Wu, P.; Huang, Y.; Das, A.; Chen, S.; Chen, J.; Hu, X.; Li, F.; Fang, Z.; et al. Metabolomics Reveals Metabolite Changes of Patients with Pulmonary Arterial Hypertension in China. J. Cell. Mol. Med. 2020, 24, 2484–2496. [Google Scholar] [CrossRef]
- Rawat, S.; Fukushima, A.; Zhang, L.; Hugi, A.; Lam, V.; Altamimi, T.; Wagg, C.; Petinelli, R.; Dhaliwal, K.; Hornberger, L.; et al. Control of cardiac fatty acid metabolism in infants with hypoplastic left heart syndrome. J. Mol. Cell. Cardiol. 2018, 124, 91–92. [Google Scholar] [CrossRef]
- Motoki, N.; Motoki, H.; Utsumi, M.; Yamazaki, S.; Obinata, H.; Takei, K.; Yasukochi, S. Identification of metabolomic profile related to adult Fontan pathophysiology. Int. J. Cardiol. Heart Vasc. 2021, 37, 100921. [Google Scholar] [CrossRef]
- Li, M.; Zhou, S.; Chen, C.; Ma, L.; Luo, D.; Tian, X.; Dong, X.; Zhou, Y.; Yang, Y.; Cui, Y. Therapeutic Potential of Pyruvate Therapy for Patients with Mitochondrial Diseases: A Systematic Review. Ther. Adv. Endocrinol. Metab. 2020, 11, 204201882093824. [Google Scholar] [CrossRef]
- Des Rosiers, C.; Labarthe, F.; Lloyd, S.G.; Chatham, J.C. Cardiac Anaplerosis in Health and Disease: Food for Thought. Cardiovasc. Res. 2011, 90, 210–219. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Verma, S. Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors. JACC Basic Transl. Sci. 2020, 5, 632–644. [Google Scholar] [CrossRef]
- Maejima, Y. SGLT2 Inhibitors Play a Salutary Role in Heart Failure via Modulation of the Mitochondrial Function. Front. Cardiovasc. Med. 2020, 6, 186. [Google Scholar] [CrossRef]
- Pietschner, R.; Kolwelter, J.; Bosch, A.; Striepe, K.; Jung, S.; Kannenkeril, D.; Ott, C.; Schiffer, M.; Achenbach, S.; Schmieder, R.E. Effect of Empagliflozin on Ketone Bodies in Patients with Stable Chronic Heart Failure. Cardiovasc. Diabetol. 2021, 20, 219. [Google Scholar] [CrossRef]
- Takada, S.; Sabe, H.; Kinugawa, S. Treatments for Skeletal Muscle Abnormalities in Heart Failure: Sodium-Glucose Transporter 2 and Ketone Bodies. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H117–H128. [Google Scholar] [CrossRef]
- Muneuchi, J.; Sugitani, Y.; Kobayashi, M.; Ezaki, H.; Yamada, H.; Watanabe, M. Feasibility and Safety of Sodium Glucose Cotransporter-2 Inhibitors in Adults with Heart Failure after the Fontan Procedure. Case Rep. Cardiol. 2022, 2022, 5243594. [Google Scholar] [CrossRef]
- Ghelani, S.J.; Opotowsky, A.R.; Harrild, D.M.; Powell, A.J.; Azcue, N.; Ahmad, S.; Clair, N.S.; Bradwin, G.; Rathod, R.H. Characterization of Circulating and Urinary Biomarkers in the Fontan Circulation and Their Correlation with Cardiac Imaging. Am. J. Cardiol. 2022, 162, 177–183. [Google Scholar] [CrossRef]
- van den Bosch, E.; Bossers, S.S.M.; Kamphuis, V.P.; Boersma, E.; Roos-Hesselink, J.W.; Breur, J.M.P.J.; Ten Harkel, A.D.J.; Kapusta, L.; Bartelds, B.; Roest, A.A.W.; et al. Associations Between Blood Biomarkers, Cardiac Function, and Adverse Outcome in a Young Fontan Cohort. J. Am. Heart Assoc. 2021, 10, e015022. [Google Scholar] [CrossRef]
- Gom, R.C.; Bhatt, D.; Villa, B.R.; George, A.G.; Lohman, A.W.; Mychasiuk, R.; Rho, J.M.; Teskey, G.C. The Ketogenic Diet Raises Brain Oxygen Levels, Attenuates Postictal Hypoxia, and Protects against Learning Impairments. Neurobiol. Dis. 2021, 154, 105335. [Google Scholar] [CrossRef]
- Trevisan, R.; Nosadini, R.; Fioretto, P.; Avogaro, A.; Duner, E.; Jori, E.; Valerio, A.; Doria, A.; Crepaldi, G. Ketone Bodies Increase Glomerular Filtration Rate in Normal Man and in Patients with Type 1 (Insulin-Dependent) Diabetes Mellitus. Diabetologia 1987, 30, 214–221. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, D.D.-H.; Qiu, Y.; Airhart, S.; Liu, Y.; Stempien-Otero, A.; O’Brien, K.D.; Tian, R. Boosting NAD Level Suppresses Inflammatory Activation of PBMCs in Heart Failure. J. Clin. Investig. 2020, 130, 6054–6063. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Persad, K.L. Failure to Launch. JACC Basic Transl. Sci. 2023, 8, 280–282. [Google Scholar] [CrossRef]
- Frank, B.S.; Khailova, L.; Silveira, L.; Mitchell, M.B.; Morgan, G.J.; Hsieh, E.W.Y.; DiMaria, M.V.; Twite, M.; Klawitter, J.; Davidson, J.A. Proteomic profiling identifies key differences between inter-stage infants with single ventricle heart disease and healthy controls. Transl. Res. 2021, 229, 24–37. [Google Scholar] [CrossRef]
- Ohuchi, H.; Miyamoto, Y.; Yamamoto, M.; Ishihara, H.; Takata, H.; Miyazaki, A.; Yamada, O.; Yagihara, T. High Prevalence of Abnormal Glucose Metabolism in Young Adult Patients with Complex Congenital Heart Disease. Am. Heart J. 2009, 158, 30–39. [Google Scholar] [CrossRef]
- Whiteside, W.; Tan, M.; Ostlund, R.E.; Yu, S.; Ma, L.; Rocchini, A. Altered Cholesterol Metabolism and Hypocholesterolemia in Patients with Single Ventricle Following Fontan Palliation. J. Pediatr. 2016, 171, 73–77. [Google Scholar] [CrossRef]
- Whiteside, W.; Tan, M.; Yu, S.; Rocchini, A. Low Total, Low-Density Lipoprotein, High-Density Lipoprotein, and Non–High-Density Lipoprotein Cholesterol Levels in Patients with Complex Congenital Heart Disease after Fontan Palliation. J. Pediatr. 2013, 162, 1199–1204. [Google Scholar] [CrossRef]
- Lubert, A.M.; Alsaied, T.; Palermo, J.J.; Anwar, N.; Urbina, E.M.; Brown, N.M.; Alexander, C.; Almeneisi, H.; Wu, F.; Leventhal, A.R.; et al. Fontan-Associated Dyslipidemia. J. Am. Heart Assoc. 2021, 10, e019578. [Google Scholar] [CrossRef]
- Zyblewski, S.C.; Argraves, W.S.; Graham, E.M.; Slate, E.H.; Atz, A.M.; Bradley, S.M.; McQuinn, T.C.; Wilkerson, B.A.; Wing, S.B.; Argraves, K.M. Reduction in postoperative high-density lipoprotein cholesterol levels in children undergoing the Fontan operation. Pediatr. Cardiol. 2012, 33, 1154–1159. [Google Scholar] [CrossRef]
- Saraf, A.; De Staercke, C.; Everitt, I.; Haouzi, A.; Ko, Y.A.; Jennings, S.; Kim, J.H.; Rodriguez, F.H.; Kalogeropoulos, A.P.; Quyyumi, A.; et al. Biomarker profile in stable Fontan patients. Int. J. Cardiol. 2020, 305, 56–62. [Google Scholar] [CrossRef]
- Fillmore, N.; Lopaschuk, G.D. Targeting Mitochondrial Oxidative Metabolism as an Approach to Treat Heart Failure. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 857–865. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Biswas, D.; Pulinilkunnil, T.; Lopaschuk, G.D. Myocardial Ketones Metabolism in Heart Failure. J. Card. Fail. 2020, 26, 998–1005. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Dyck, J.R.B. Ketones and the Cardiovascular System. Nat. Cardiovasc. Res. 2023, 2, 425–437. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Sato, K.; Tsuchiya, N.; Thomas, S.; Fell, D.A.; Veech, R.L.; Passonneau, J.V. Control of Glucose Utilization in Working Perfused Rat Heart. J. Biol. Chem. 1994, 269, 25502–25514. [Google Scholar] [CrossRef]
- Cahill, G.F.; Veech, R.L. Ketoacids? Good Medicine? Trans. Am. Clin. Climatol. Assoc. 2003, 114, 149–163. [Google Scholar]
- Veech, R.L.; Chance, B.; Kashiwaya, Y.; Lardy, H.A.; Cahill, G.F., Jr. Ketone Bodies, Potential Therapeutic Uses. IUBMB Life Int. Union Biochem. Mol. Biol. Life 2001, 51, 241–247. [Google Scholar] [CrossRef]
- Veech, R.L. The Therapeutic Implications of Ketone Bodies: The Effects of Ketone Bodies in Pathological Conditions: Ketosis, Ketogenic Diet, Redox States, Insulin Resistance, and Mitochondrial Metabolism. Prostaglandins Leukot. Essent. Fatty Acids 2004, 70, 309–319. [Google Scholar] [CrossRef]
- Sato, K.; Kashiwaya, Y.; Keon, C.A.; Tsuchiya, N.; King, M.T.; Radda, G.K.; Chance, B.; Clarke, K.; Veech, R.L. Insulin, Ketone Bodies, and Mitochondrial Energy Transduction. FASEB J. 1995, 9, 651–658. [Google Scholar] [CrossRef]
- Mudaliar, S.; Alloju, S.; Henry, R.R. Can a Shift in Fuel Energetics Explain the Beneficial Cardiorenal Outcomes in the EMPA-REG OUTCOME Study? A Unifying Hypothesis. Diabetes Care 2016, 39, 1115–1122. [Google Scholar] [CrossRef]
- Ho, K.L.; Karwi, Q.G.; Wagg, C.; Zhang, L.; Vo, K.; Altamimi, T.; Uddin, G.M.; Ussher, J.R.; Lopaschuk, G.D. Ketones Can Become the Major Fuel Source for the Heart but Do Not Increase Cardiac Efficiency. Cardiovasc. Res. 2021, 117, 1178–1187. [Google Scholar] [CrossRef]
- Berg-Hansen, K.; Christensen, K.H.; Gopalasingam, N.; Nielsen, R.; Eiskjær, H.; Møller, N.; Birkelund, T.; Christensen, S.; Wiggers, H. Beneficial Effects of Ketone Ester in Patients with Cardiogenic Shock: A Randomized, Controlled, Double-Blind Trial. JACC Heart Fail. 2023; ahead of print. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G. Jump Starting the Heart: Ketone Esters Improve Cardiac Function in Patients with Cardiogenic Shock. JACC Heart Fail. 2023, in press. [CrossRef]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef]
- Grieve, D. Oxidative Stress in Heart Failure More than Just Damage. Eur. Heart J. 2003, 24, 2161–2163. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of Oxidative Stress by β-Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, C.; Shang, F.-F.; Luo, M.; You, Y.; Zhai, Q.; Xia, Y.; Suxin, L. Ketogenic Diet Ameliorates Cardiac Dysfunction via Balancing Mitochondrial Dynamics and Inhibiting Apoptosis in Type 2 Diabetic Mice. Aging Dis. 2020, 11, 229. [Google Scholar] [CrossRef]
- Murphy, S.P.; Kakkar, R.; McCarthy, C.P.; Januzzi, J.L. Inflammation in Heart Failure. J. Am. Coll. Cardiol. 2020, 75, 1324–1340. [Google Scholar] [CrossRef]
- Shirazi, L.F.; Bissett, J.; Romeo, F.; Mehta, J.L. Role of Inflammation in Heart Failure. Curr. Atheroscler. Rep. 2017, 19, 27. [Google Scholar] [CrossRef]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the Role of Inflammation in Heart Failure. Nat. Rev. Cardiol. 2020, 17, 269–285. [Google Scholar] [CrossRef]
- Suetomi, T.; Willeford, A.; Brand, C.S.; Cho, Y.; Ross, R.S.; Miyamoto, S.; Brown, J.H. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca2+/Calmodulin-Dependent Protein Kinase II δ Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation 2018, 138, 2530–2544. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The Ketone Metabolite β-Hydroxybutyrate Blocks NLRP3 Inflammasome–Mediated Inflammatory Disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones Inhibit Mitochondrial Production of Reactive Oxygen Species Production Following Glutamate Excitotoxicity by Increasing NADH Oxidation. Neuroscience 2007, 145, 256–264. [Google Scholar] [CrossRef]
- Forsythe, C.E.; Phinney, S.D.; Fernandez, M.L.; Quann, E.E.; Wood, R.J.; Bibus, D.M.; Kraemer, W.J.; Feinman, R.D.; Volek, J.S. Comparison of Low Fat and Low Carbohydrate Diets on Circulating Fatty Acid Composition and Markers of Inflammation. Lipids 2008, 43, 65–77. [Google Scholar] [CrossRef]
- Greco, T.; Glenn, T.C.; Hovda, D.A.; Prins, M.L. Ketogenic Diet Decreases Oxidative Stress and Improves Mitochondrial Respiratory Complex Activity. J. Cereb. Blood Flow Metab. 2016, 36, 1603–1613. [Google Scholar] [CrossRef]
- Yang, D.; Liu, H.-Q.; Liu, F.-Y.; Guo, Z.; An, P.; Wang, M.-Y.; Yang, Z.; Fan, D.; Tang, Q.-Z. Mitochondria in Pathological Cardiac Hypertrophy Research and Therapy. Front. Cardiovasc. Med. 2022, 8, 822969. [Google Scholar] [CrossRef]
- Abel, E.D.; Doenst, T. Mitochondrial Adaptations to Physiological vs. Pathological Cardiac Hypertrophy. Cardiovasc. Res. 2011, 90, 234–242. [Google Scholar] [CrossRef]
- Kolwicz, S.C.; Tian, R. Glucose Metabolism and Cardiac Hypertrophy. Cardiovasc. Res. 2011, 90, 194–201. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial Fatty Acid Metabolism in Health and Disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Kee, H.J.; Kook, H. Roles and Targets of Class I and IIa Histone Deacetylases in Cardiac Hypertrophy. J. Biomed. Biotechnol. 2011, 2011, 928326. [Google Scholar] [CrossRef]
- Hewitson, R.; Dargan, J.; Collis, D.; Green, A.; Moorjani, N.; Ohri, S.; Townsend, P.A. Heart Failure: The Pivotal Role of Histone Deacetylases. Int. J. Biochem. Cell Biol. 2013, 45, 448–453. [Google Scholar] [CrossRef]
- Yurista, S.R.; Matsuura, T.R.; Silljé, H.H.W.; Nijholt, K.T.; McDaid, K.S.; Shewale, S.V.; Leone, T.C.; Newman, J.C.; Verdin, E.; van Veldhuisen, D.J.; et al. Ketone Ester Treatment Improves Cardiac Function and Reduces Pathologic Remodeling in Preclinical Models of Heart Failure. Circ. Heart Fail. 2021, 14, e007684. [Google Scholar] [CrossRef]
- Nakamura, M.; Odanovic, N.; Nakada, Y.; Dohi, S.; Zhai, P.; Ivessa, A.; Yang, Z.; Abdellatif, M.; Sadoshima, J. Dietary Carbohydrates Restriction Inhibits the Development of Cardiac Hypertrophy and Heart Failure. Cardiovasc. Res. 2021, 117, 2365–2376. [Google Scholar] [CrossRef]
- Okere, I.C.; Young, M.E.; McElfresh, T.A.; Chess, D.J.; Sharov, V.G.; Sabbah, H.N.; Hoit, B.D.; Ernsberger, P.; Chandler, M.P.; Stanley, W.C. Low Carbohydrate/High-Fat Diet Attenuates Cardiac Hypertrophy, Remodeling, and Altered Gene Expression in Hypertension. Hypertension 2006, 48, 1116–1123. [Google Scholar] [CrossRef]
- Egbe, A.C.; Connolly, H.M.; Miranda, W.R.; Ammash, N.M.; Hagler, D.J.; Veldtman, G.R.; Borlaug, B.A. Hemodynamics of Fontan Failure: The Role of Pulmonary Vascular Disease. Circ. Heart Fail. 2017, 10, e004515. [Google Scholar] [CrossRef]
- Castaldi, B.; Bordin, G.; Padalino, M.; Cuppini, E.; Vida, V.; Milanesi, O. Hemodynamic Impact of Pulmonary Vasodilators on Single Ventricle Physiology. Cardiovasc. Ther. 2018, 36, e12314. [Google Scholar] [CrossRef]
- Zuchi, C.; Tritto, I.; Carluccio, E.; Mattei, C.; Cattadori, G.; Ambrosio, G. Role of Endothelial Dysfunction in Heart Failure. Heart Fail. Rev. 2020, 25, 21–30. [Google Scholar] [CrossRef]
- Giannitsi, S.; Maria, B.; Bechlioulis, A.; Naka, K. Endothelial Dysfunction and Heart Failure: A Review of the Existing Bibliography with Emphasis on Flow Mediated Dilation. JRSM Cardiovasc. Dis. 2019, 8, 204800401984304. [Google Scholar] [CrossRef]
- McCarthy, C.G.; Chakraborty, S.; Singh, G.; Yeoh, B.S.; Schreckenberger, Z.J.; Singh, A.; Mell, B.; Bearss, N.R.; Yang, T.; Cheng, X.; et al. Ketone Body β-Hydroxybutyrate Is an Autophagy-Dependent Vasodilator. JCI Insight 2021, 6, e149037. [Google Scholar] [CrossRef]
- Gormsen, L.C.; Svart, M.; Thomsen, H.H.; Søndergaard, E.; Vendelbo, M.H.; Christensen, N.; Tolbod, L.P.; Harms, H.J.; Nielsen, R.; Wiggers, H.; et al. Ketone Body Infusion With 3-Hydroxybutyrate Reduces Myocardial Glucose Uptake and Increases Blood Flow in Humans: A Positron Emission Tomography Study. J. Am. Heart Assoc. 2017, 6, e005066. [Google Scholar] [CrossRef]
- Ibrahim, A. The Effect of Ketone on β-Aminopropionitrile-Induced Vascular Remodeling. Master’s Thesis, Georgia State University, Atlanta, GA, USA, 2022. [Google Scholar] [CrossRef]
- Coleman, K.; Phillips, J.; Sciarini, M.; Stubbs, B.; Jackson, O.; Kernagis, D. A Metabolic Intervention for Improving Human Cognitive Performance During Hypoxia. Aerosp. Med. Hum. Perform. 2021, 92, 556–562. [Google Scholar] [CrossRef]
- Prins, P.J.; Buxton, J.D.; McClure, T.S.; D’Agostino, D.P.; Ault, D.L.; Welton, G.L.; Jones, D.W.; Atwell, A.D.; Slack, M.A.; Slack, M.L.; et al. Ketone Bodies Impact on Hypoxic CO2 Retention Protocol During Exercise. Front. Physiol. 2021, 12, 780755. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Pawlosky, R.; Markis, W.; King, M.T.; Bergman, C.; Srivastava, S.; Murray, A.; Clarke, K.; Veech, R.L. A Ketone Ester Diet Increases Brain Malonyl-CoA and Uncoupling Proteins 4 and 5 While Decreasing Food Intake in the Normal Wistar Rat. J. Biol. Chem. 2010, 285, 25950–25956. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-Dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Kim, D.; Roberts, C.; McKenzie, A.; George, M.P. Nutritional Ketosis to Treat Pulmonary Hypertension Associated with Obesity and Metabolic Syndrome: A Case Report. Pulm. Circ. 2021, 11, 2045894021991426. [Google Scholar] [CrossRef]
- Chowdhury, B.; Luu, A.Z.; Luu, V.Z.; Kabir, M.G.; Pan, Y.; Teoh, H.; Quan, A.; Sabongui, S.; Al-Omran, M.; Bhatt, D.L.; et al. The SGLT2 Inhibitor Empagliflozin Reduces Mortality and Prevents Progression in Experimental Pulmonary Hypertension. Biochem. Biophys. Res. Commun. 2020, 524, 50–56. [Google Scholar] [CrossRef]
- Rychik, J. The Relentless Effects of the Fontan Paradox. Semin. Thorac. Cardiovasc. Surg. Pediatr. Card. Surg. Annu. 2016, 19, 37–43. [Google Scholar] [CrossRef]
- Mooli, R.G.R.; Ramakrishnan, S.K. Emerging Role of Hepatic Ketogenesis in Fatty Liver Disease. Front. Physiol. 2022, 13, 946474. [Google Scholar] [CrossRef]
- Liao, Y.-J.; Wang, Y.-H.; Wu, C.-Y.; Hsu, F.-Y.; Chien, C.-Y.; Lee, Y.-C. Ketogenic Diet Enhances the Cholesterol Accumulation in Liver and Augments the Severity of CCl4 and TAA-Induced Liver Fibrosis in Mice. Int. J. Mol. Sci. 2021, 22, 2934. [Google Scholar] [CrossRef]
- Moore, M.P.; Cunningham, R.P.; Davis, R.A.H.; Deemer, S.E.; Roberts, B.M.; Plaisance, E.P.; Rector, R.S. A Dietary Ketone Ester Mitigates Histological Outcomes of NAFLD and Markers of Fibrosis in High-Fat Diet Fed Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G564–G572. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Dufour, S.; Lyu, K.; Zhang, X.-M.; Hakkarainen, A.; Lehtimäki, T.E.; Cline, G.W.; Petersen, K.F.; Shulman, G.I.; Yki-Järvinen, H. Effect of a Ketogenic Diet on Hepatic Steatosis and Hepatic Mitochondrial Metabolism in Nonalcoholic Fatty Liver Disease. Proc. Natl. Acad. Sci. USA 2020, 117, 7347–7354. [Google Scholar] [CrossRef]
- Sripongpun, P.; Churuangsuk, C.; Bunchorntavakul, C. Current Evidence Concerning Effects of Ketogenic Diet and Intermittent Fasting in Patients with Nonalcoholic Fatty Liver. J. Clin. Transl. Hepatol. 2022, 10, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Khuong, J.N.; Wilson, T.G.; Grigg, L.E.; Bullock, A.; Celermajer, D.; Disney, P.; Wijesekera, V.A.; Hornung, T.; Zannino, D.; Iyengar, A.J.; et al. Fontan-Associated Nephropathy: Predictors and Outcomes. Int. J. Cardiol. 2020, 306, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Levin, A.; Kiess, M.; Sexsmith, G.; Chakrabarti, S.; Barlow, A.; Human, D.; Grewal, J. Chronic Kidney Damage in the Adult Fontan Population. Int. J. Cardiol. 2018, 257, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Binotto, M.A. Renal Function and Fontan Patients: What Is the Real Impact in the Long-Term Outcomes? Int. J. Cardiol. 2020, 306, 86–87. [Google Scholar] [CrossRef]
- Niaz, T.; Stephens, E.H.; Gleich, S.J.; Dearani, J.A.; Johnson, J.N.; Sas, D.J.; Bly, S.; Driscoll, D.J.; Cetta, F. Acute Kidney Injury and Renal Replacement Therapy After Fontan Operation. Am. J. Cardiol. 2021, 161, 84–94. [Google Scholar] [CrossRef]
- Zafar, F.; Lubert, A.M.; Katz, D.A.; Hill, G.D.; Opotowsky, A.R.; Alten, J.A.; Goldstein, S.L.; Alsaied, T. Long-Term Kidney Function After the Fontan Operation. J. Am. Coll. Cardiol. 2020, 76, 334–341. [Google Scholar] [CrossRef]
- Hems, D.A.; Brosnan, J.T. Effects of Ischaemia on Content of Metabolites in Rat Liver and Kidney in Vivo. Biochem. J. 1970, 120, 105–111. [Google Scholar] [CrossRef][Green Version]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1α Drives NAD Biosynthesis Linking Oxidative Metabolism to Renal Protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef]
- Ritmeester, E.; Veger, V.A.; van der Ven, J.P.G.; van Tussenbroek, G.M.J.W.; van Capelle, C.I.; Udink ten Cate, F.E.A.; Helbing, W.A. Fontan Circulation Associated Organ Abnormalities Beyond the Heart, Lungs, Liver, and Gut: A Systematic Review. Front. Cardiovasc. Med. 2022, 9, 826096. [Google Scholar] [CrossRef]
- Puchowicz, M.A.; Emancipator, D.S.; Xu, K.; Magness, D.L.; Ndubuizu, O.I.; Lust, W.D.; LaManna, J.C. Adaptation to Chronic Hypoxia During Diet-Induced Ketosis. In Oxygen Transport to Tissue XXVI; Okunieff, P., Williams, J., Chen, Y., Eds.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2005; Volume 566, pp. 51–57. [Google Scholar] [CrossRef]
- Poffé, C.; Robberechts, R.; Podlogar, T.; Kusters, M.; Debevec, T.; Hespel, P. Exogenous Ketosis Increases Blood and Muscle Oxygenation but Not Performance during Exercise in Hypoxia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021, 321, R844–R857. [Google Scholar] [CrossRef]
- García-Caballero, M.; Zecchin, A.; Souffreau, J.; Truong, A.-C.K.; Teuwen, L.-A.; Vermaelen, W.; Martín-Pérez, R.; de Zeeuw, P.; Bouché, A.; Vinckier, S.; et al. Role and Therapeutic Potential of Dietary Ketone Bodies in Lymph Vessel Growth. Nat. Metab. 2019, 1, 666–675. [Google Scholar] [CrossRef]
- Universitaire Ziekenhuizen KU Leuven. Ketogenic Diet: A Novel Metabolic Strategy to Treat Lymphedema Patients? 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03991897 (accessed on 4 April 2023).
- Puchalska, P.; Crawford, P.A. Ketogenic Therapies for Lymphedema? Nat. Metab. 2019, 1, 656–657. [Google Scholar] [CrossRef]
- Dodeja, A.; Urbina, F.; Moore-Padilla, M.; Mah, M.L.; Bradley, D.; Bradley, E. Fontan-Associated Liver Disease: Is Insulin Sensitivity Important? J. Am. Coll. Cardiol. 2020, 75, 549. [Google Scholar] [CrossRef]
- Emamaullee, J.; Zaidi, A.N.; Schiano, T.; Kahn, J.; Valentino, P.L.; Hofer, R.E.; Taner, T.; Wald, J.W.; Olthoff, K.M.; Bucuvalas, J.; et al. Fontan-Associated Liver Disease: Screening, Management, and Transplant Considerations. Circulation 2020, 142, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, J.Z.; Day, A.; Brinkworth, G.D.; Sato, J.; Yamada, S.; Jönsson, T.; Beardsley, J.; Johnson, J.A.; Thabane, L.; Johnston, B.C. Efficacy and Safety of Low and Very Low Carbohydrate Diets for Type 2 Diabetes Remission: Systematic Review and Meta-Analysis of Published and Unpublished Randomized Trial Data. BMJ 2021, 372, m4743. [Google Scholar] [CrossRef]
- Tommerdahl, K.L.; Nelson, R.G.; Bjornstad, P. Dapagliflozin in young people with type 2 diabetes. Lancet Diabetes Endocrinol. 2022, 10, 303–304. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Liao, J.; Zhou, D.; Mu, J. Ketogenic Diet Therapy for Epilepsy: Past 100 Years of Practice. Acta Epileptol. 2022, 4, 15. [Google Scholar] [CrossRef]
- Suo, C.; Liao, J.; Lu, X.; Fang, K.; Hu, Y.; Chen, L.; Cao, D.; Huang, T.; Li, B.; Li, C. Efficacy and Safety of the Ketogenic Diet in Chinese Children. Seizure 2013, 22, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.; Swaminathan, A.; Paseka, J.; Hanson, C. Efficacy and Safety of a Ketogenic Diet in Children and Adolescents with Refractory Epilepsy—A Review. Nutrients 2020, 12, 1809. [Google Scholar] [CrossRef]
- Dressler, A.; Trimmel-Schwahofer, P. The Ketogenic Diet for Infants: How Long Can You Go? Epilepsy Res. 2020, 164, 106339. [Google Scholar] [CrossRef]
- Mady, M.A.; Kossoff, E.H.; McGregor, A.L.; Wheless, J.W.; Pyzik, P.L.; Freeman, J.M. The Ketogenic Diet: Adolescents Can Do It, Too. Epilepsia 2003, 44, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Scholl-Bürgi, S.; Höller, A.; Pichler, K.; Michel, M.; Haberlandt, E.; Karall, D. Ketogenic Diets in Patients with Inherited Metabolic Disorders. J. Inherit. Metab. Dis. 2015, 38, 765–773. [Google Scholar] [CrossRef]
- Lin, K.-L.; Lin, J.-J.; Wang, H.-S. Application of Ketogenic Diets for Pediatric Neurocritical Care. Biomed. J. 2020, 43, 218–225. [Google Scholar] [CrossRef]
- Li, J.; Zhang, H.; Dai, Z. Cancer Treatment with the Ketogenic Diet: A Systematic Review and Meta-Analysis of Animal Studies. Front. Nutr. 2021, 8, 594408. [Google Scholar] [CrossRef] [PubMed]
- Lauzier, B.; Vaillant, F.; Merlen, C.; Gélinas, R.; Bouchard, B.; Rivard, M.-E.; Labarthe, F.; Dolinsky, V.W.; Dyck, J.R.B.; Allen, B.G.; et al. Metabolic Effects of Glutamine on the Heart: Anaplerosis versus the Hexosamine Biosynthetic Pathway. J. Mol. Cell. Cardiol. 2013, 55, 92–100. [Google Scholar] [CrossRef]
- Hernandez-Saavedra, D.; Sanders, L.; Freeman, S.; Reisz, J.A.; Lee, M.H.; Mickael, C.; Kumar, R.; Kassa, B.; Gu, S.; D’ Alessandro, A.; et al. Stable Isotope Metabolomics of Pulmonary Artery Smooth Muscle and Endothelial Cells in Pulmonary Hypertension and with TGF-Beta Treatment. Sci. Rep. 2020, 10, 413. [Google Scholar] [CrossRef]
- Sun, H.; Olson, K.C.; Gao, C.; Prosdocimo, D.A.; Zhou, M.; Wang, Z.; Jeyaraj, D.; Youn, J.-Y.; Ren, S.; Liu, Y.; et al. Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation 2016, 133, 2038–2049. [Google Scholar] [CrossRef]
- Vockley, J.; Charrow, J.; Ganesh, J.; Eswara, M.; Diaz, G.A.; McCracken, E.; Conway, R.; Enns, G.M.; Starr, J.; Wang, R.; et al. Triheptanoin Treatment in Patients with Pediatric Cardiomyopathy Associated with Long Chain-Fatty Acid Oxidation Disorders. Mol. Genet. Metab. 2016, 119, 223–231. [Google Scholar] [CrossRef]
- Zöggeler, T.; Stock, K.; Jörg-Streller, M.; Spenger, J.; Konstantopoulou, V.; Hufgard-Leitner, M.; Scholl-Bürgi, S.; Karall, D. Long-Term Experience with Triheptanoin in 12 Austrian Patients with Long-Chain Fatty Acid Oxidation Disorders. Orphanet J. Rare Dis. 2021, 16, 28. [Google Scholar] [CrossRef]
- Lei, I.; Tian, S.; Gao, W.; Liu, L.; Guo, Y.; Tang, P.; Chen, E.; Wang, Z. Acetyl-CoA Production by Specific Metabolites Promotes Cardiac Repair after Myocardial Infarction via Histone Acetylation. eLife 2021, 10, e60311. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.N.; Driscoll, D.J.; O’Leary, P.W. Protein-Losing Enteropathy and the Fontan Operation. Nutr. Clin. Pract. 2012, 27, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Violante, S.; IJlst, L.; te Brinke, H.; Almeida, I.T.; Wanders, R.J.A.; Ventura, F.V.; Houten, S.M. Carnitine Palmitoyltransferase 2 and Carnitine/Acylcarnitine Translocase Are Involved in the Mitochondrial Synthesis and Export of Acylcarnitines. FASEB J. 2013, 27, 2039–2044. [Google Scholar] [CrossRef] [PubMed]
- Pereyra, A.S.; Harris, K.L.; Soepriatna, A.H.; Waterbury, Q.A.; Bharathi, S.S.; Zhang, Y.; Fisher-Wellman, K.H.; Goergen, C.J.; Goetzman, E.S.; Ellis, J.M. Octanoate Is Differentially Metabolized in Liver and Muscle and Fails to Rescue Cardiomyopathy in CPT2 Deficiency. J. Lipid Res. 2021, 62, 100069. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Metabolic State | ||||||
|---|---|---|---|---|---|---|
| Substrate Consumption | ETC | CK | Therapeutic Ketosis | |||
| Fatty acids | Glucose | Ketone bodies | ||||
| Healthy heart | 60–70% [57,58] | Remaining [57,58] | Remaining [57,58] | balanced | ||
| HfrEF | Early HF: no change [48,59,60] Late HF: ↓ [48,59,60] | Early HF: ↑ [48,59,60] Late HF: ↓ [60,61,62,63,64,65,66] | Progressing HF: ↑ [35,65,67,68,69,70,71,72,73,74,75] | Loss of electrons [76] Accumulation of ROS [76] | early HF: ↓ [48,77,78,79] | CO +40% [80] EF +8% [80] |
| BV-HF | ↓ [81] | ↑ [82,83,84,85] | Progressing HF: ↑ [69,86,87,88] | Loss of electrons [89,90] Accumulation of ROS [91,92] | ↓ [89,93] | CO +27% [87] PVR—18% [87] |
| SV-HF | ↓ [94] | ↓ [95] | ? | Loss of electrons [96] Accumulation of ROS [76,97,98] | ? | ? |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renaud, D.; Scholl-Bürgi, S.; Karall, D.; Michel, M. Comparative Metabolomics in Single Ventricle Patients after Fontan Palliation: A Strong Case for a Targeted Metabolic Therapy. Metabolites 2023, 13, 932. https://doi.org/10.3390/metabo13080932
Renaud D, Scholl-Bürgi S, Karall D, Michel M. Comparative Metabolomics in Single Ventricle Patients after Fontan Palliation: A Strong Case for a Targeted Metabolic Therapy. Metabolites. 2023; 13(8):932. https://doi.org/10.3390/metabo13080932
Chicago/Turabian StyleRenaud, David, Sabine Scholl-Bürgi, Daniela Karall, and Miriam Michel. 2023. "Comparative Metabolomics in Single Ventricle Patients after Fontan Palliation: A Strong Case for a Targeted Metabolic Therapy" Metabolites 13, no. 8: 932. https://doi.org/10.3390/metabo13080932
APA StyleRenaud, D., Scholl-Bürgi, S., Karall, D., & Michel, M. (2023). Comparative Metabolomics in Single Ventricle Patients after Fontan Palliation: A Strong Case for a Targeted Metabolic Therapy. Metabolites, 13(8), 932. https://doi.org/10.3390/metabo13080932