
Fecal Bile Acids and Neutral Sterols Are Associated with Latent Microbial Subgroups in the Human Gut

 , , ,
, , , 
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Collection of Biosamples and Participant Data
2.3. Metabolomics Analysis
2.4. 16S rRNA Gene Amplicon Sequencing and Amplicon Analysis
2.5. Calculation of Habitual Dietary Intake
2.6. Identification of Microbial Subgroups/Latent Dirichlet Allocation
2.7. Metabolite–Subgroup Associations
2.8. Descriptive Statistics and Figures
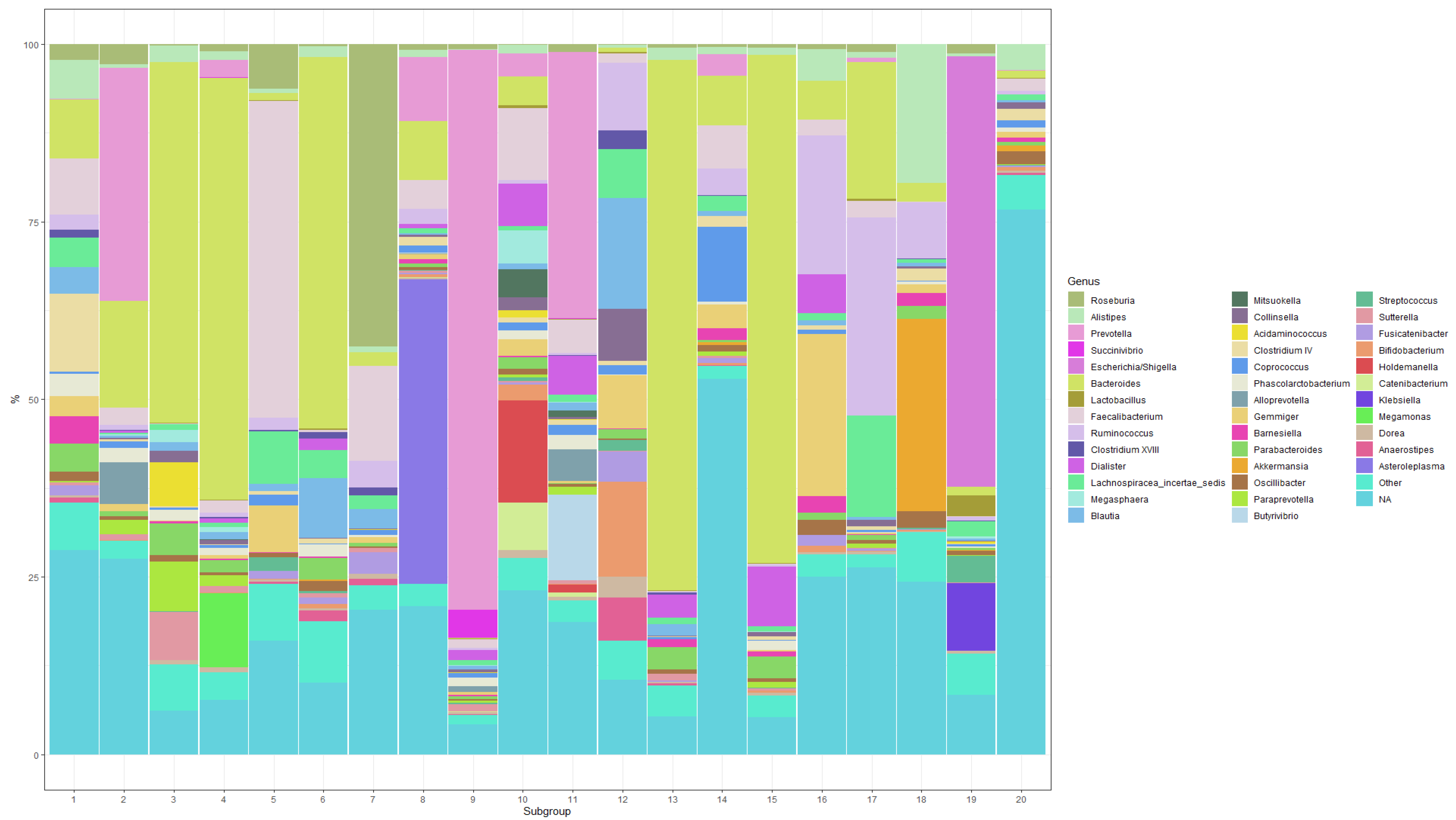
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grüner, N.; Mattner, J. Bile Acids and Microbiota: Multifaceted and Versatile Regulators of the Liver–Gut Axis. Int. J. Mol. Sci. 2021, 22, 1397. [Google Scholar] [CrossRef] [PubMed]
- McGlone, E.R.; Bloom, S.R. Bile acids and the metabolic syndrome. Ann. Clin. Biochem. 2019, 56, 326–337. [Google Scholar] [CrossRef]
- Joyce, S.A.; MacSharry, J.; Casey, P.G.; Kinsella, M.; Murphy, E.F.; Shanahan, F.; Hill, C.; Gahan, C.G.M. Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc. Natl. Acad. Sci. USA 2014, 111, 7421–7426. [Google Scholar] [CrossRef] [PubMed]
- Woting, A.; Blaut, M. The Intestinal Microbiota in Metabolic Disease. Nutrients 2016, 8, 202. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.A.; Theriot, C.M. Diversification of host bile acids by members of the gut microbiota. Gut Microbes 2020, 11, 158–171. [Google Scholar] [CrossRef]
- Ryan, P.M.; Stanton, C.; Caplice, N.M. Bile acids at the cross-roads of gut microbiome–host cardiometabolic interactions. Diabetol. Metab. Syndr. 2017, 9, 102. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile Acid Metabolism and Signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef]
- Li, T.; Chiang, J.Y.L. Bile Acid Signaling in Metabolic Disease and Drug Therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Jie, Z.; Xia, H.; Zhong, S.-L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The Gut Microbiome in Atherosclerotic Cardiovascular Disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef]
- Theriot, C.M.; Koenigsknecht, M.J.; Carlson, P.E., Jr.; Hatton, G.E.; Nelson, A.M.; Li, B.; Huffnagle, G.B.; Li, J.Z.; Young, V.B. Antibiotic-induced Shifts in the Mouse Gut Microbiome and Metabolome Increase Susceptibility to Clostridium Difficile Infection. Nat. Commun. 2014, 5, 3114. [Google Scholar] [CrossRef] [PubMed]
- Malinen, E.; Krogius-Kurikka, L.; Lyra, A.; Nikkilä, J.; Jääskeläinen, A.; Rinttilä, T.; Vilpponen-Salmela, T.; Wright, A.J.V.; Palva, A. Association of Symptoms with Gastrointestinal Microbiota in Irritable Bowel Syndrome. World J. Gastroenterol. 2010, 16, 4532–4540. [Google Scholar]
- Cuevas-Tena, M.; Alegría, A.; Lagarda, M.J. Relationship between Dietary Sterols and Gut Microbiota: A Review. Eur. J. Lipid Sci. Technol. 2018, 120, 1800054. [Google Scholar] [CrossRef]
- Kenny, D.J.; Plichta, D.R.; Shungin, D.; Koppel, N.; Hall, B.; Fu, B.; Vasan, R.S.; Shaw, S.Y.; Vlamakis, H.; Balskus, E.P.; et al. Cholesterol Metabolism by Uncultured Human Gut Bacteria Influences Host Cholesterol Level. Cell Host Microbe 2020, 28, 245–257.e6. [Google Scholar] [CrossRef] [PubMed]
- Breuninger, T.A.; Wawro, N.; Breuninger, J.; Reitmeier, S.; Clavel, T.; Six-Merker, J.; Pestoni, G.; Rohrmann, S.; Rathmann, W.; Peters, A.; et al. Associations between habitual diet, metabolic disease, and the gut microbiota using latent Dirichlet allocation. Microbiome 2021, 9, 61. [Google Scholar] [CrossRef]
- Holle, R.; Happich, M.; Löwel, H.; Wichmann, H.E.; for the MONICA/KORA Study Group. KORA—A Research Platform for Population Based Health Research. Gesundheitswesen 2005, 67, 19–25. [Google Scholar] [CrossRef]
- Mitry, P.; Wawro, N.; Sharma, S.; Kriebel, J.; Artati, A.; Adamski, J.; Heier, M.; Meisinger, C.; Thorand, B.; Grallert, H.; et al. Associations between usual food intake and faecal sterols and bile acids: Results from the Cooperative Health Research in the Augsburg Region (KORA FF4) study. Br. J. Nutr. 2019, 122, 309–321. [Google Scholar] [CrossRef]
- Reitmeier, S.; Kießling, S.; Clavel, T.; List, M.; Almeida, E.L.; Ghosh, T.S.; Neuhaus, K.; Grallert, H.; Linseisen, J.; Skurk, T.; et al. Arrhythmic Gut Microbiome Signatures Predict Risk of Type-2 Diabetes. Cell Host Microbe 2020, 28, 258–272.e6. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Joseph, D.; Kapfhammer, M.; Giritli, S.; Horn, M.; Haller, D.; Clavel, T. IMNGS: A comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci. Rep. 2016, 6, 33721. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Reitmeier, S.; Hitch, T.C.; Fikas, N.; Hausmann, B.; Ramer-Tait, A.E.; Neuhaus, K.; Berry, D.; Haller, D.; Lagkouvardos, I.; Clavel, T. Handling of spurious sequences affects the outcome of high-throughput 16S rRNA gene amplicon profiling. ISME Commun. 2021, 1, 31. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Mitry, P.; Wawro, N.; Six-Merker, J.; Zoller, D.; Jourdan, C.; Meisinger, C.; Thierry, S.; Nöthlings, U.; Knüppel, S.; Boeing, H.; et al. Usual Dietary Intake Estimation Based on a Combination of Repeated 24-H Food Lists and a Food Frequency Questionnaire in the KORA FF4 Cross-Sectional Study. Front. Nutr. 2019, 6, 145. [Google Scholar] [CrossRef] [PubMed]
- Slimani, N.; Deharveng, G.; Charrondière, R.U.; van Kappel, A.L.; Ocké, M.C.; Welch, A.; Lagiou, A.; van Liere, M.; Agudo, A.; Pala, V.; et al. Structure of the standardized computerized 24-h diet recall interview used as reference method in the 22 centers participating in the EPIC project. European Prospective Investigation into Cancer and Nutrition. Comput. Methods Programs Biomed. 1999, 58, 251–266. [Google Scholar] [CrossRef]
- Blei, D.M.; Ng, A.Y.; Jordan, M.I. Latent Dirichlet Allocation. J. Mach. Learn. Res. 2003, 3, 993–1022. [Google Scholar]
- Liu, L.; Tang, L.; Dong, W.; Yao, S.; Zhou, W. An overview of topic modeling and its current applications in bioinformatics. Springerplus 2016, 5, 1608. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Rajapakse, J.C.; The Alzheimer’s Disease Neuroimaging Initiative. Profiling heterogeneity of Alzheimer’s disease using white-matter impairment factors. Neuroimage Clin. 2018, 20, 1222–1232. [Google Scholar] [CrossRef]
- Yan, J.; Chuai, G.; Qi, T.; Shao, F.; Zhou, C.; Zhu, C.; Yang, J.; Yu, Y.; Shi, C.; Kang, N.; et al. MetaTopics: An integration tool to analyze microbial community profile by topic model. BMC Genom. 2017, 18, 962. [Google Scholar] [CrossRef]
- Maier, M.J. DirichletReg: Dirichlet Regression for Compositional Data in R; WU Vienna University of Economics and Business: Vienna, Austria, 2014. [Google Scholar]
- Haarman, B.C.M.B.; Lek, R.F.R.-V.d.; Nolen, W.A.; Mendes, R.; Drexhage, H.A.; Burger, H. Feature-expression Heat Maps—A New Visual Method to Explore Complex Associations between Two Variable Sets. J. Biomed. Inform. 2015, 53, 156–161. [Google Scholar] [CrossRef]
- Guzior, D.V.; Quinn, R.A. Review: Microbial transformations of human bile acids. Microbiome 2021, 9, 140. [Google Scholar] [CrossRef]
- Ovadia, C.; Perdones-Montero, A.; Spagou, K.; Smith, A.; Sarafian, M.H.; Gomez-Romero, M.; Bellafante, E.; Clarke, L.C.D.; Sadiq, F.; Nikolova, V.; et al. Enhanced Microbial Bile Acid Deconjugation and Impaired Ileal Uptake in Pregnancy Repress Intestinal Regulation of Bile Acid Synthesis. Hepatology 2019, 70, 276–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.-B.; Yi, S.-H.; Lee, B.H. Purification and Characterization of Three Different Types of Bile Salt Hydrolases from Bifidobacterium Strains. J. Dairy Sci. 2004, 87, 258–266. [Google Scholar] [CrossRef]
- Weingarden, A.R.; Chen, C.; Bobr, A.; Yao, D.; Lu, Y.; Nelson, V.M.; Sadowsky, M.J.; Khoruts, A. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G310–G319. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Paramsothy, S.; Nielsen, S.; Kamm, M.A.; Deshpande, N.P.; Faith, J.J.; Clemente, J.C.; Paramsothy, R.; Walsh, A.J.; Bogaerde, J.v.d.; Samuel, D.; et al. Specific Bacteria and Metabolites Associated with Response to Fecal Microbiota Transplantation in Patients with Ulcerative Colitis. Gastroenterology 2019, 156, 1440–1454.e2. [Google Scholar] [CrossRef]
- Veiga, P.; Juste, C.; Lepercq, P.; Saunier, K.; Béguet, F.; Gérard, P. Correlation between faecal microbial community structure and cholesterol-to-coprostanol conversion in the human gut. FEMS Microbiol. Lett. 2005, 242, 81–86. [Google Scholar] [CrossRef]
- Cuevas-Tena, M.; Pulgar, E.M.G.d.; Benítez-Páez, A.; Sanz, Y.; Alegría, A.; Lagarda, M.J. Plant sterols and human gut microbiota relationship: An in vitro colonic fermentation study. J. Funct. Foods 2018, 44, 322–329. [Google Scholar] [CrossRef]
- Cuevas-Tenaa, M.; Alegriaa, A.; Lagardaa, M.J.; Venema, K. Impact of plant sterols enrichment dose on gut microbiota from lean and obese subjects using TIM-2 in vitro fermentation model. J. Funct. Foods 2019, 54, 164–174. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Total | Men | Women | ||||
|---|---|---|---|---|---|---|
| n = 1280 | n = 636 | n = 634 | ||||
| Continuous variables | Mean | SD | Mean | SD | Mean | SD |
| Age (years) | 59.72 | 12.03 | 59.87 | 12.30 | 59.57 | 11.76 |
| Waist circumference (cm) | 96.91 | 13.80 | 102.21 | 11.45 | 91.51 | 13.90 |
| BMI (kg/m2) | 27.8 | 4.8 | 28.0 | 4.0 | 27.5 | 5.5 |
| Total energy (kcal/d) 1 | 1872.95 | 403.23 | 2120.43 | 351.85 | 1621.17 | 276.48 |
| Total fiber (g/d) 1 | 17.89 | 5.07 | 18.39 | 5.15 | 17.39 | 4.94 |
| Alcohol (g/d) 1 | 10.31 | 10.34 | 16.21 | 11.10 | 4.31 | 4.35 |
| Categorical variables | % | n | % | n | % | n |
| Education: | ||||||
| <13 years | 63.9 | 818 | 58.2 | 376 | 69.7 | 442 |
| ≥13 years | 36.1 | 462 | 41.8 | 270 | 30.3 | 192 |
| Physical activity: | ||||||
| Active | 58 | 742 | 55.7 | 360 | 60.3 | 382 |
| Inactive | 42 | 538 | 44.3 | 286 | 39.7 | 252 |
| Smoking: | ||||||
| Current | 15.4 | 197 | 16.6 | 107 | 14.2 | 90 |
| Ex | 35.9 | 459 | 42.1 | 272 | 29.5 | 187 |
| Never | 48.8 | 624 | 41.3 | 267 | 56.3 | 357 |
| Metabolite | Min | 25th %ile | Median | 75th %ile | Max | % Missing |
|---|---|---|---|---|---|---|
| stigmasterol | 0.002 | 0.033 | 0.051 | 0.078 | 0.663 | 4.53 |
| sitostanol | 0.001 | 0.029 | 0.052 | 0.079 | 0.441 | 7.42 |
| beta-sitosterol | 0.001 | 0.030 | 0.051 | 0.101 | 0.932 | 0.31 |
| campesterol | 0.001 | 0.028 | 0.051 | 0.104 | 1.139 | 1.17 |
| ergosterol | 0.001 | 0.025 | 0.054 | 0.119 | 10.336 | 4.21 |
| cholesterol | 0.002 | 0.024 | 0.055 | 0.142 | 1.507 | 0.23 |
| coprostanol | 0.0005 | 0.030 | 0.054 | 0.086 | 0.530 | 4.53 |
| cholate | 0.0002 | 0.015 | 0.051 | 0.210 | 64.810 | 3.20 |
| glycochenodeoxycholate | 0.003 | 0.024 | 0.052 | 0.129 | 12.007 | 6.02 |
| glycocholate | 0.001 | 0.021 | 0.052 | 0.152 | 12.721 | 1.09 |
| 12-dehydrocholate | 0.002 | 0.017 | 0.053 | 0.225 | 61.283 | 17.42 |
| 3b-hydroxy-5-cholenoic acid | 0.001 | 0.031 | 0.053 | 0.090 | 1.066 | 12.58 |
| 6-oxolithocholate | 0.002 | 0.030 | 0.052 | 0.089 | 0.738 | 14.45 |
| 7,12-diketolithocholate | 0.001 | 0.023 | 0.051 | 0.129 | 23.395 | 21.25 |
| 7-ketodeoxycholate | 0.001 | 0.018 | 0.053 | 0.207 | 53.246 | 10.31 |
| dehydrolithocholate | 0.0004 | 0.024 | 0.051 | 0.092 | 0.465 | 1.41 |
| deoxycholate | 0.0003 | 0.020 | 0.054 | 0.110 | 1.809 | 1.95 |
| glycodeoxycholate | 0.001 | 0.022 | 0.052 | 0.129 | 11.315 | 8.83 |
| glycoursodeoxycholate | 0.003 | 0.025 | 0.055 | 0.135 | 6.540 | 18.36 |
| hyocholate | 0.002 | 0.028 | 0.052 | 0.106 | 2.646 | 9.22 |
| isoursodeoxycholate | 0.001 | 0.023 | 0.052 | 0.135 | 8.105 | 0.47 |
| lithocholate | 0.001 | 0.031 | 0.051 | 0.082 | 0.723 | 1.09 |
| ursocholate | 0.002 | 0.025 | 0.050 | 0.195 | 54.447 | 0.63 |
| ursodeoxycholate | 0.001 | 0.021 | 0.048 | 0.125 | 6.654 | 1.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breuninger, T.A.; Wawro, N.; Freuer, D.; Reitmeier, S.; Artati, A.; Grallert, H.; Adamski, J.; Meisinger, C.; Peters, A.; Haller, D.; et al. Fecal Bile Acids and Neutral Sterols Are Associated with Latent Microbial Subgroups in the Human Gut. Metabolites 2022, 12, 846. https://doi.org/10.3390/metabo12090846
Breuninger TA, Wawro N, Freuer D, Reitmeier S, Artati A, Grallert H, Adamski J, Meisinger C, Peters A, Haller D, et al. Fecal Bile Acids and Neutral Sterols Are Associated with Latent Microbial Subgroups in the Human Gut. Metabolites. 2022; 12(9):846. https://doi.org/10.3390/metabo12090846
Chicago/Turabian StyleBreuninger, Taylor A., Nina Wawro, Dennis Freuer, Sandra Reitmeier, Anna Artati, Harald Grallert, Jerzy Adamski, Christa Meisinger, Annette Peters, Dirk Haller, and et al. 2022. "Fecal Bile Acids and Neutral Sterols Are Associated with Latent Microbial Subgroups in the Human Gut" Metabolites 12, no. 9: 846. https://doi.org/10.3390/metabo12090846
APA StyleBreuninger, T. A., Wawro, N., Freuer, D., Reitmeier, S., Artati, A., Grallert, H., Adamski, J., Meisinger, C., Peters, A., Haller, D., & Linseisen, J. (2022). Fecal Bile Acids and Neutral Sterols Are Associated with Latent Microbial Subgroups in the Human Gut. Metabolites, 12(9), 846. https://doi.org/10.3390/metabo12090846