
Cluster Analysis Statistical Spectroscopy for the Identification of Metabolites in 1H NMR Metabolomics

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
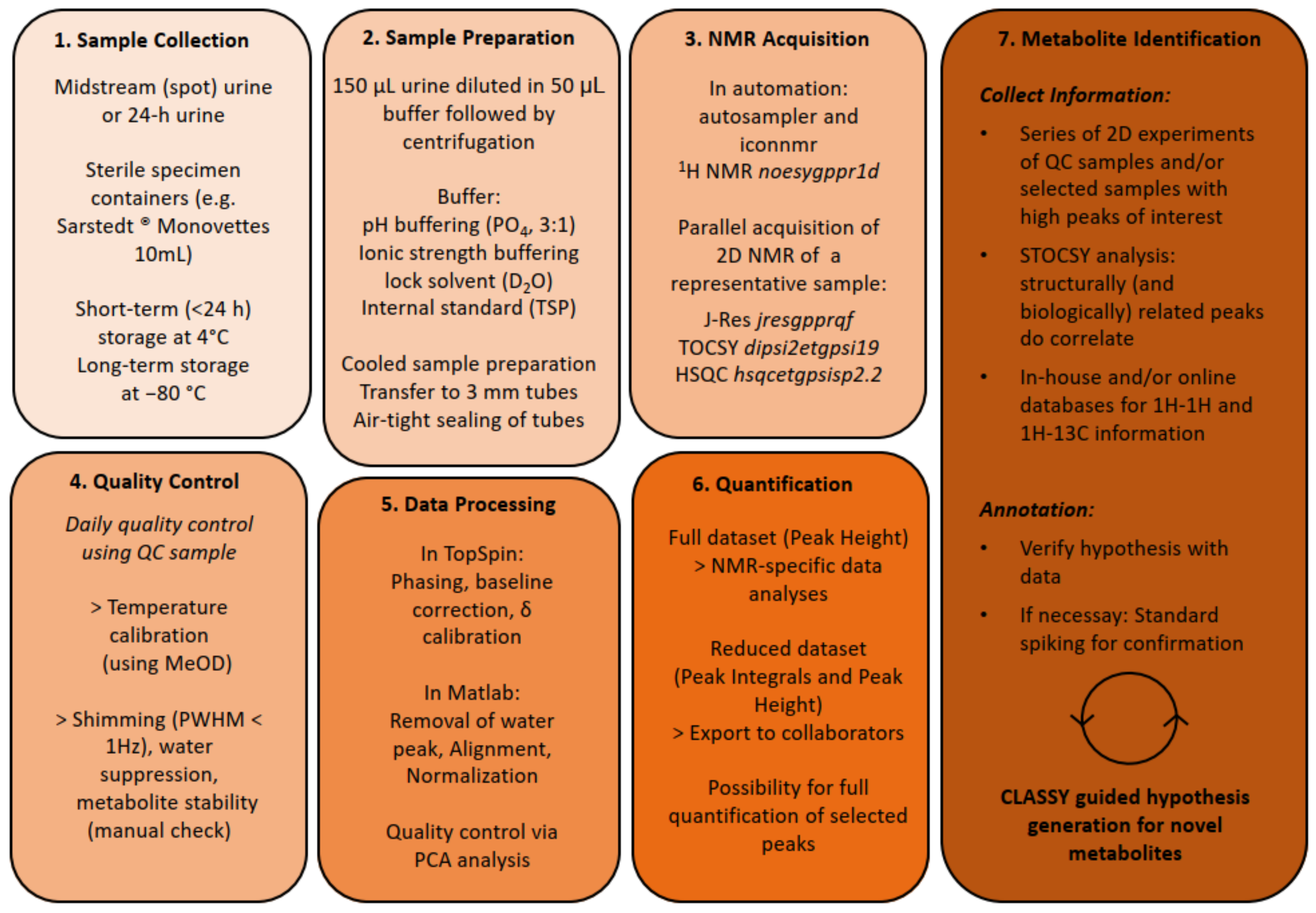
2. Materials and Methods
2.1. Dataset Description
2.2. Sample Preparation
2.3. NMR Analysis
2.4. NMR Data Processing
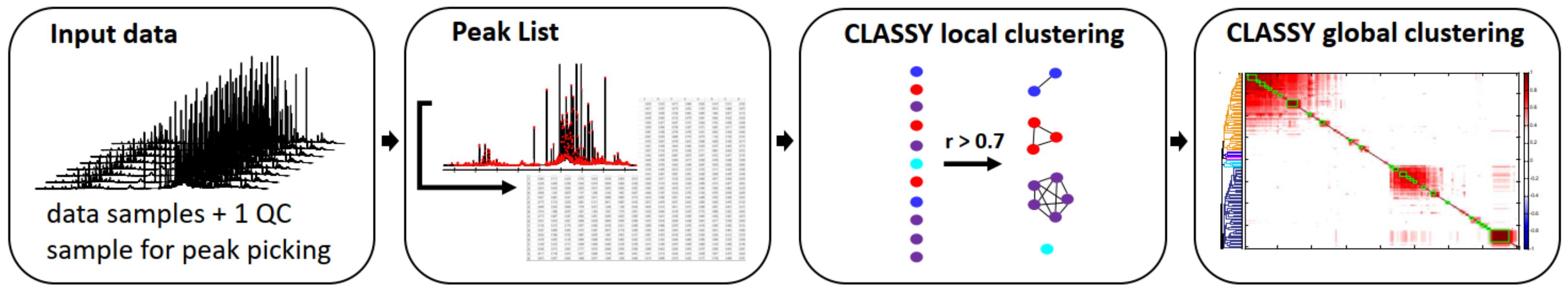
2.5. Clustering Approach
2.6. Metabolite Identification
2.7. Quality Assessment of the Dataset
3. Results and Discussion
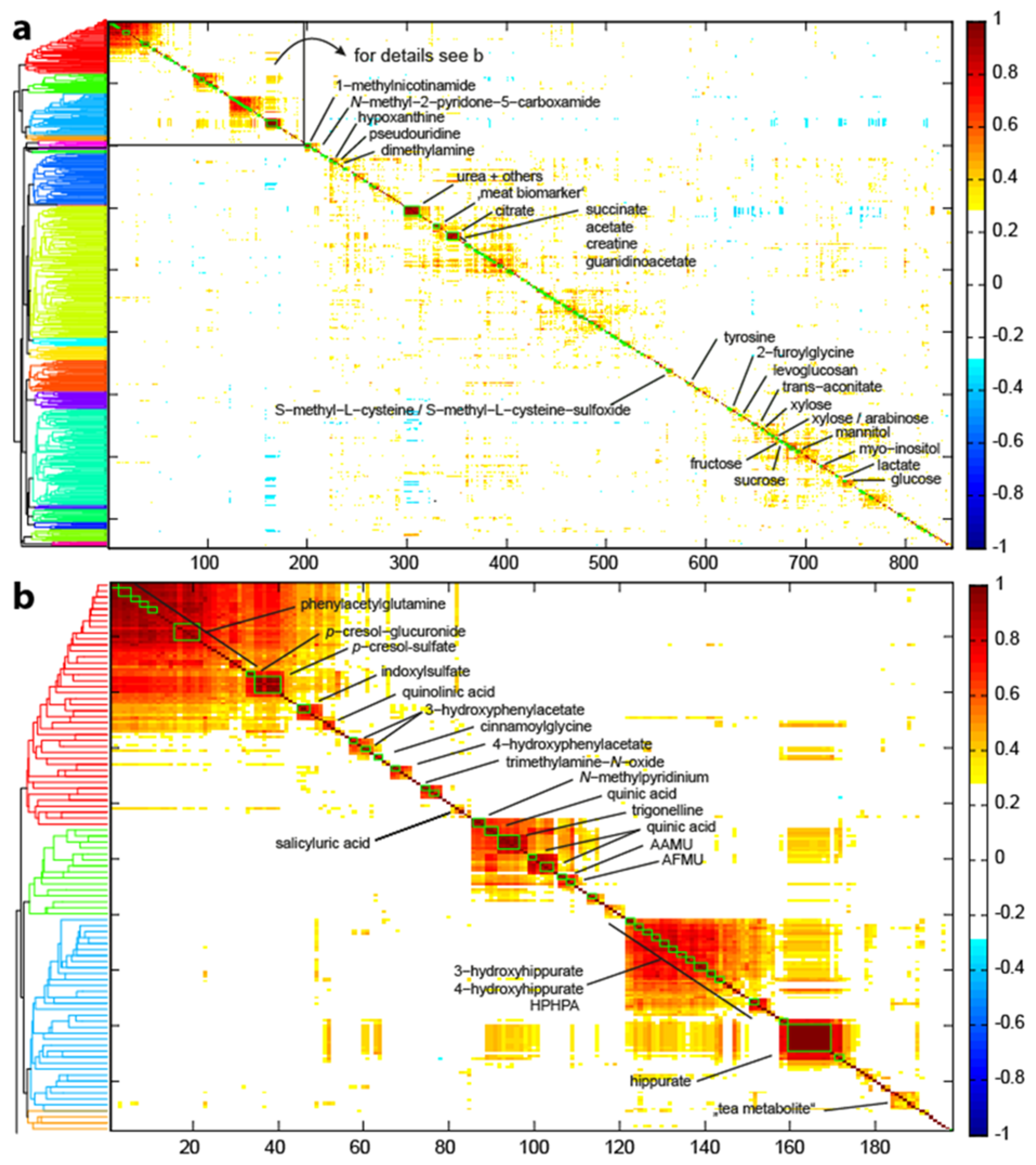
3.1. NMR Metabolomics Workflow and Metabolite Identification Approach
3.2. Metabolite Identification Approach
3.3. Validation and Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robinette, S.L.; Lindon, J.C.; Nicholson, J.K. Statistical Spectroscopic Tools for Biomarker Discovery and Systems Medicine. Anal. Chem. 2013, 85, 5297–5303. [Google Scholar] [CrossRef] [PubMed]
- Dona, A.C.; Kyriakides, M.; Scott, F.; Shephard, E.A.; Varshavi, D.; Veselkov, K.; Everett, J.R. A Guide to the Identification of Metabolites in NMR-Based Metabonomics/Metabolomics Experiments. Comput. Struct. Biotechnol. J. 2016, 14, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Bouatra, S.; Aziat, F.; Mandal, R.; Guo, A.C.; Wilson, M.R.; Knox, C.; Bjorndahl, T.C.; Krishnamurthy, R.; Saleem, F.; Liu, P.; et al. The Human Urine Metabolome. PLoS ONE 2013, 8, e73076. [Google Scholar] [CrossRef] [PubMed]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The Human Serum Metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef]
- Robinette, S.L.; Veselkov, K.A.; Bohus, E.; Coen, M.; Keun, H.C.; Ebbels, T.M.D.; Beckonert, O.; Holmes, E.C.; Lindon, J.C.; Nicholson, J.K. Cluster Analysis Statistical Spectroscopy Using Nuclear Magnetic Resonance Generated Metabolic Data Sets from Perturbed Biological Systems. Anal. Chem. 2009, 81, 6581–6589. [Google Scholar] [CrossRef]
- Cloarec, O.; Dumas, M.-E.; Craig, A.; Barton, R.H.; Trygg, J.; Hudson, J.; Blancher, C.; Gauguier, D.; Lindon, J.C.; Holmes, E.; et al. Statistical Total Correlation Spectroscopy: An Exploratory Approach for Latent Biomarker Identification from Metabolic 1 H NMR Data Sets. Anal. Chem. 2005, 77, 1282–1289. [Google Scholar] [CrossRef]
- Holle, R.; Happich, M.; Löwel, H.; Wichmann, H.E. MONICA/KORA Study Group KORA-A Research Platform for Population Based Health Research. Gesundh. (Bundesverb. Arzte Offentlichen Gesundh.) 2005, 67 (Suppl. 1), S19–S25. [Google Scholar] [CrossRef]
- Heinzmann, S.S.; Schmitt-Kopplin, P. Deep Metabotyping of the Murine Gastrointestinal Tract for the Visualization of Digestion and Microbial Metabolism. J. Proteome Res. 2015, 14, 2267–2277. [Google Scholar] [CrossRef]
- Veselkov, K.A.; Lindon, J.C.; Ebbels, T.M.D.; Crockford, D.; Volynkin, V.V.; Holmes, E.; Davies, D.B.; Nicholson, J.K. Recursive Segment-Wise Peak Alignment of Biological 1 H NMR Spectra for Improved Metabolic Biomarker Recovery. Anal. Chem. 2009, 81, 56–66. [Google Scholar] [CrossRef]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic Quotient Normalization as Robust Method to Account for Dilution of Complex Biological Mixtures. Application in 1 H NMR Metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef]
- Wong, J.W.H.; Cagney, G.; Cartwright, H.M. SpecAlign-Processing and Alignment of Mass Spectra Datasets. Bioinformatics 2005, 21, 2088–2090. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef]
- Gil, R.B.; Lehmann, R.; Schmitt-Kopplin, P.; Heinzmann, S.S. 1 H NMR-Based Metabolite Profiling Workflow to Reduce Inter-Sample Chemical Shift Variations in Urine Samples for Improved Biomarker Discovery. Anal. Bioanal. Chem. 2016, 408, 4683–4691. [Google Scholar] [CrossRef]
- Haslauer, K.E.; Schmitt-Kopplin, P.; Heinzmann, S.S. Data Processing Optimization in Untargeted Metabolomics of Urine Using Voigt Lineshape Model Non-Linear Regression Analysis. Metabolites 2021, 11, 285. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The Human Metabolome Database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Holmes, E.; Khan, F.; Kochhar, S.; Scanlan, P.; Shanahan, F.; Wilson, I.D.; Wang, Y. Rapid and Noninvasive Metabonomic Characterization of Inflammatory Bowel Disease. J. Proteome Res. 2007, 6, 546–551. [Google Scholar] [CrossRef]
- Jacobs, D.M.; Deltimple, N.; van Velzen, E.; van Dorsten, F.A.; Bingham, M.; Vaughan, E.E.; van Duynhoven, J. 1 H NMR Metabolite Profiling of Feces as a Tool to Assess the Impact of Nutrition on the Human Microbiome. NMR Biomed. 2008, 21, 615–626. [Google Scholar] [CrossRef]
- Diaz, S.O.; Barros, A.S.; Goodfellow, B.J.; Duarte, I.F.; Carreira, I.M.; Galhano, E.; Pita, C.; Almeida, M.d.C.; Gil, A.M. Following Healthy Pregnancy by Nuclear Magnetic Resonance (NMR) Metabolic Profiling of Human Urine. J. Proteome Res. 2013, 12, 969–979. [Google Scholar] [CrossRef]
- Tynkkynen, T.; Wang, Q.; Ekholm, J.; Anufrieva, O.; Ohukainen, P.; Vepsäläinen, J.; Männikkö, M.; Keinänen-Kiukaanniemi, S.; Holmes, M.V.; Goodwin, M.; et al. Proof of Concept for Quantitative Urine NMR Metabolomics Pipeline for Large-Scale Epidemiology and Genetics. Int. J. Epidemiol. 2019, 48, 978–993. [Google Scholar] [CrossRef]
- Furuta, T.; Horie, M.; Baba, S.; Nakagawa, H. Studies on Drug Metabolism by Use of Isotopes XXVI: Determination of Urinary Metabolites of Rutin in Humans. J. Pharm. Sci. 1981, 70, 780–782. [Google Scholar] [CrossRef]
- Ross, A.B.; Pere-Trépat, E.; Montoliu, I.; Martin, F.-P.J.; Collino, S.; Moco, S.; Godin, J.-P.; Cléroux, M.; Guy, P.A.; Breton, I.; et al. A Whole-Grain–Rich Diet Reduces Urinary Excretion of Markers of Protein Catabolism and Gut Microbiota Metabolism in Healthy Men after One Week. J. Nutr. 2013, 143, 766–773. [Google Scholar] [CrossRef]
- Chalmers, R.A.; Valman, H.B.; Liberman, M.M. Measurement of 4-Hydroxyphenylacetic Aciduria as a Screening Test for Small-Bowel Disease. Clin. Chem. 1979, 25, 1791–1794. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Liu, D.; Wang, Y.; Zeng, T.; Peng, Y. Urinary 3-(3-Hydroxyphenyl)-3-Hydroxypropionic Acid, 3-Hydroxyphenylacetic Acid, and 3-Hydroxyhippuric Acid Are Elevated in Children with Autism Spectrum Disorders. BioMed. Res. Int. 2016, 2016, 1–8. [Google Scholar] [CrossRef]
- Heyes, M.P.; Saito, K.; Crowley, J.S.; Davis, L.E.; Demitrack, M.A.; Der, M.; Dilling, L.A.; Elia, J.; Kruesi, M.J.P.; Lackner, A.; et al. Quinolinic Acid and Kynurenine Pathway Metabolism in Inflammatory and Non-Inflammatory Neurological Disease. Brain 1992, 115, 1249–1273. [Google Scholar] [CrossRef]
- Cashman, J.; Camp, K.; Fakharzadeh, S.; Fennessey, P.; Hines, R.; Mamer, O.; Mitchell, S.; Preti, G.; Schlenk, D.; Smith, R. Biochemical and Clinical Aspects of the Human Flavin-Containing Monooxygenase Form 3 (FMO3) Related to Trimethylaminuria. Curr. Drug Metab. 2003, 4, 151–170. [Google Scholar] [CrossRef]
- Hoskins, J.A.; Holliday, S.B.; Greenway, A.M. The Metabolism of Cinnamic Acid by Healthy and Phenylketonuric Adults: A Kinetic Study. Biol. Mass Spectrom. 1984, 11, 296–300. [Google Scholar] [CrossRef]
- Bar, N.; Korem, T.; Weissbrod, O.; Zeevi, D.; Rothschild, D.; Leviatan, S.; Kosower, N.; Lotan-Pompan, M.; Weinberger, A.; le Roy, C.I.; et al. A Reference Map of Potential Determinants for the Human Serum Metabolome. Nature 2020, 588, 135–140. [Google Scholar] [CrossRef]
- Lawrence, J.R. Urinary Excretion of Salicyluric and Salicylic Acids by Non-Vegetarians, Vegetarians, and Patients Taking Low Dose Aspirin. J. Clin. Pathol. 2003, 56, 651–653. [Google Scholar] [CrossRef]
- Nyéki, Á.; Buclin, T.; Biollaz, J.; Decosterd, L.A. NAT2 and CYP1A2 Phenotyping with Caffeine: Head-to-Head Comparison of AFMU vs. AAMU in the Urine Metabolite Ratios. Br. J. Clin. Pharmacol. 2003, 55, 62–67. [Google Scholar] [CrossRef]
- Heinzmann, S.S.; Merrifield, C.A.; Rezzi, S.; Kochhar, S.; Lindon, J.C.; Holmes, E.; Nicholson, J.K. Stability and Robustness of Human Metabolic Phenotypes in Response to Sequential Food Challenges. J. Proteome Res. 2012, 11, 643–655. [Google Scholar] [CrossRef]
- Gil, R.B.; Ortiz, A.; Sanchez-Niño, M.D.; Markoska, K.; Schepers, E.; Vanholder, R.; Glorieux, G.; Schmitt-Kopplin, P.; Heinzmann, S.S. Increased Urinary Osmolyte Excretion Indicates Chronic Kidney Disease Severity and Progression Rate. Nephrol. Dial. Transplant. 2018, 33, 2156–2164. [Google Scholar] [CrossRef] [PubMed]
- Lagkouvardos, I.; Kläring, K.; Heinzmann, S.S.; Platz, S.; Scholz, B.; Engel, K.H.; Schmitt-Kopplin, P.; Haller, D.; Rohn, S.; Skurk, T.; et al. Gut Metabolites and Bacterial Community Networks during a Pilot Intervention Study with Flaxseeds in Healthy Adult Men. Mol. Nutr. Food Res. 2015, 59, 1614–1628. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heinzmann, S.S.; Waldenberger, M.; Peters, A.; Schmitt-Kopplin, P. Cluster Analysis Statistical Spectroscopy for the Identification of Metabolites in 1H NMR Metabolomics. Metabolites 2022, 12, 992. https://doi.org/10.3390/metabo12100992
Heinzmann SS, Waldenberger M, Peters A, Schmitt-Kopplin P. Cluster Analysis Statistical Spectroscopy for the Identification of Metabolites in 1H NMR Metabolomics. Metabolites. 2022; 12(10):992. https://doi.org/10.3390/metabo12100992
Chicago/Turabian StyleHeinzmann, Silke S., Melanie Waldenberger, Annette Peters, and Philippe Schmitt-Kopplin. 2022. "Cluster Analysis Statistical Spectroscopy for the Identification of Metabolites in 1H NMR Metabolomics" Metabolites 12, no. 10: 992. https://doi.org/10.3390/metabo12100992
APA StyleHeinzmann, S. S., Waldenberger, M., Peters, A., & Schmitt-Kopplin, P. (2022). Cluster Analysis Statistical Spectroscopy for the Identification of Metabolites in 1H NMR Metabolomics. Metabolites, 12(10), 992. https://doi.org/10.3390/metabo12100992