1. Introduction
Mass spectrometry is a versatile analytical method that is widely used in clinical applications, metabolomics, chemistry, biochemistry, and elsewhere [
1,
2,
3]. MS-based analyses are conducted using a variety of different methods and instruments with the commonality to separate and measure the mass-to-charge (m/z) ratio of molecules. MS is often used in combination with separation techniques such as gas chromatography (GC), liquid chromatography (LC), or ion mobility [
1,
4,
5]. The separation of compounds in a sample by polarity, size, shape, or other characteristics facilitates the identification and quantification of different substances in mixtures and complex matrices [
6]. The constant development improving individual aspects of MS, e.g., mass resolution, opens the door for novel applications.
One application particularly benefitting from higher resolution is metabolomics: the investigation of potentially all metabolites in a biological system to gain a better understanding of living organisms and to identify causes and potential cures for diseases. One of the aspects of metabolomics are metabolic flux experiments. In flux analysis, precursor molecules are labeled with isotopes that have a low natural abundance, provided to the biological system, and traced with respect to their conversion. Mostly, tracers containing
13C are used but
15N or
18O tracers are available as well. The techniques used to conduct such experiments include
2H/
13C NMR and MS [
7,
8].
GC-MS is frequently applied in flux analyses, generally using electron impact ionization (EI) because large databases with EI spectra allow for a broader targeted evaluation. A more recent alternative is high-resolution GC-APCI-MS [
9]. In comparison to EI, APCI is a soft ionization method leading to mainly [M+H]
+ and [M]
+ ion species, together with moderate fragmentations and rearrangements [
9].
The so monitored incorporation of tracer molecules into the metabolome of biological samples is a common tool used to investigate metabolic pathways and processes. The incorporation of the tracer, e.g.,
13C, is quantified by an analysis of the MID of a compound. Mass isotopologues (MI) are molecules with identical chemical structures but differences in mass due to the presence of different isotopes. The
rawMID of a compound is a vector of measured ion intensities in a sample. The ratio of these ion intensities allows for the inference of the fractional amount of individual isotopes. In the case of
13C, the natural abundance corresponds to a fractional amount of about 1.1%. Accurate determination of the isotope labeling of the metabolites is a prerequisite for metabolic flux calculations. To this end, MIDs need to be corrected for natural abundance and various tools have been developed previously to achieve this correction including MIDcor, IsoCorrectoR, and IsoCor [
10,
11,
12]. MIDcor corrects for the natural abundance of isotopes as well as for the potential overlap with other compounds, while IsoCorrectoR corrects for natural MID as well as for tracer purity (
Table S1).
In APCI experiments, we noticed (in addition to natural abundance and tracer impurity) additional effects requiring MID correction. Specifically, we observed reactions such as the loss of a proton, the loss of hydrogen, and the addition of water combined with the loss of CH
4, as described previously [
13]. The occurrence of such ionization products is molecule dependent and leads consequentially to superimposed MIDs, i.e.,
rawMIDs being an overlay of the same mass spectrum shifted by a small number of mass units. While the tools mentioned above allow researchers to correct MIDs for several effects, they do not provide a flexible framework to correct superimposed spectra resulting from different ionization adducts ([M]
+, [M+H]
+) and fragmentation reactions ([M+H
3O−CH
4]
+).
Here, we describe such a function, CorMID, and demonstrate its usefulness by evaluating the spectra of a compound library to confirm the amount of fragment occurrence in APCI-MS and by applying it to biological samples of a flux experiment to improve data preparation for flux analysis.
2. Results and Discussion
Monitoring the incorporation of
13C-Glucose or similar tracer molecules into the metabolome of biological samples is a common tool used to investigate metabolic pathways and processes [
8]. That includes unknown pathways, the speed and efficiency of metabolization, and the conversion of molecules normally not present in said biological systems. The
13C incorporation is quantified by an analysis of the MID of a compound. Mass isotopologues (MI) are molecules with identical chemical structures but differences in mass due to the presence of different isotopes, here either
12C or
13C. The mass difference between both carbon isotopes is 1.003355 Dalton. Neglecting the contribution of other chemical elements in a compound, the number of carbon atoms defines the number of possible MI. Glucose, which is frequently used as a tracer molecule, contains six carbon atoms, leading to seven possible MI ranging from 180.0634 Da (isotopologue M0, containing zero
13C and six
12C atoms) to 186.0835 Da (M6, containing
13C = 6 and
12C = 0). Often, fully labeled Glucose ([U-
13C]Glc) is provided to cells for uptake and its carbon atoms are distributed to glucose-derived metabolites via the metabolic pathways. The amount of labelling in a compound observed during the experiment is indicative of the amount produced from the tracer molecule, which ultimately facilitates the reconstruction of a metabolic network and the ability to infer knowledge about cellular processes.
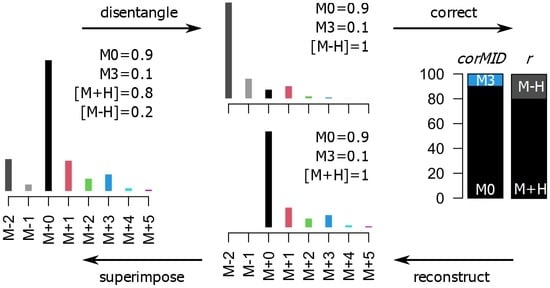
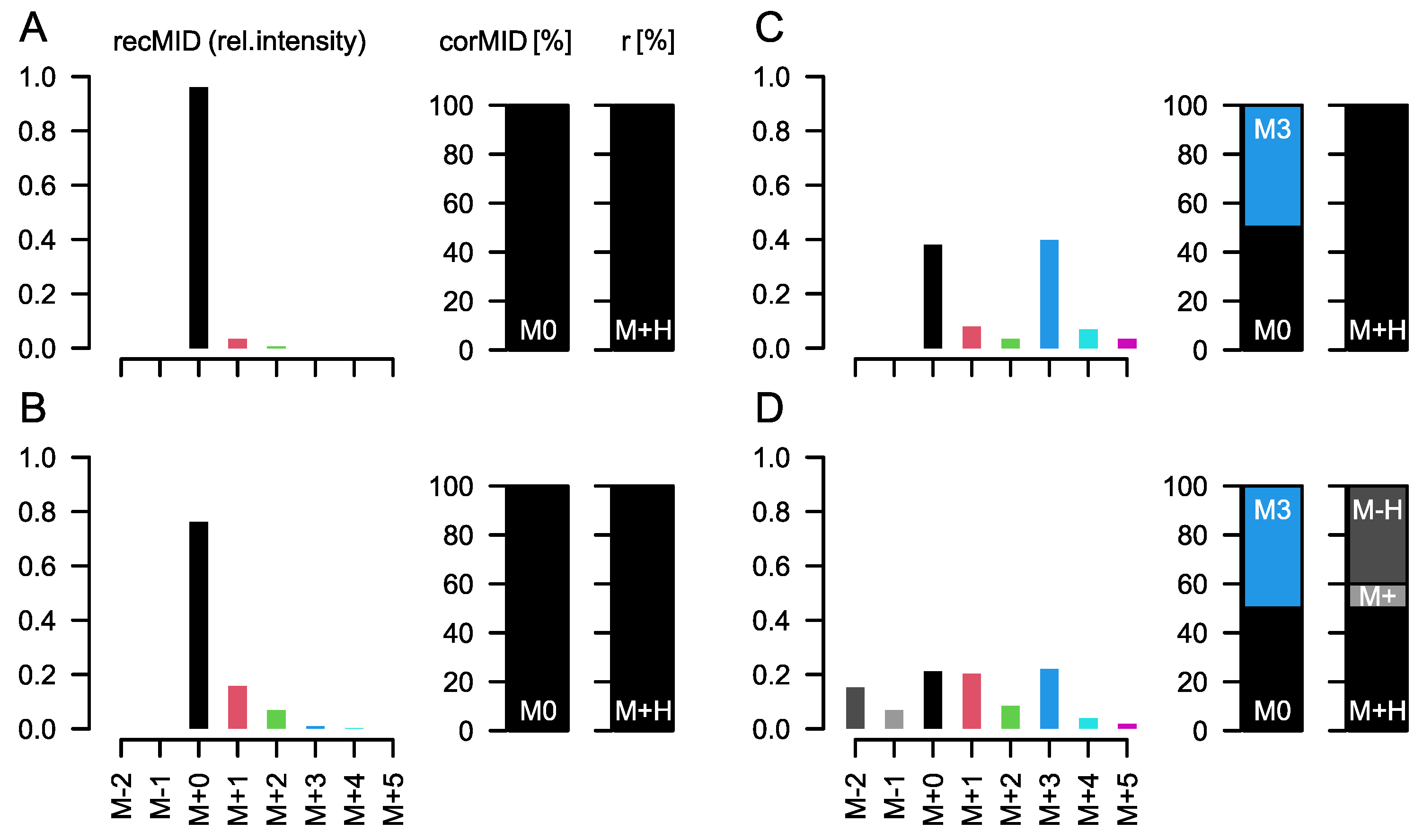
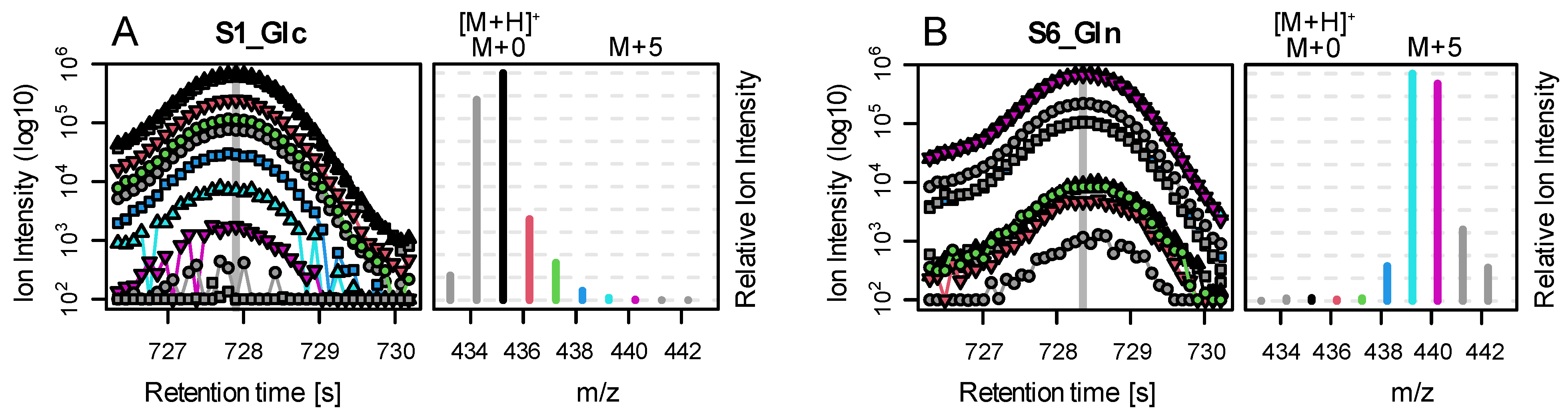
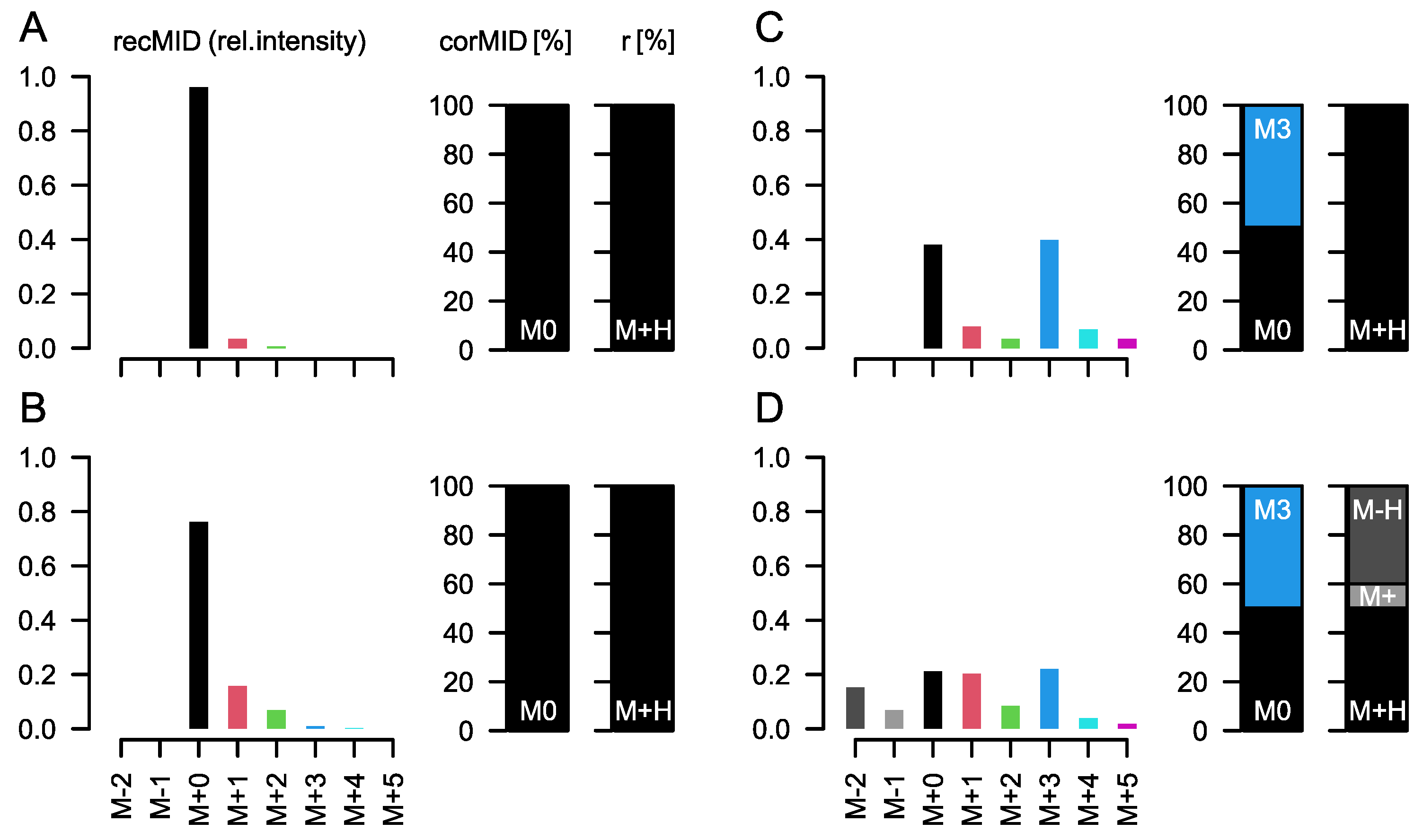
A key step during the necessary data processing is the correction for naturally abundant 13C, which is present in the environment at about 1.1%. As naturally abundant 13C is randomly distributed in compounds, we measure for an analyte containing three carbon atoms, e.g., lactic acid, about 3.2% and 0.04% of the intensity of the M+0 peak at M+1 and M+2, respectively (the measured intensity of M+3 is negligible). M+0 is the ion intensity of the monoisotopic peak of an analyte. M+i are the ion intensities of the isotopologue peaks of the molecule with approximately i×1 Dalton mass difference to M+0.
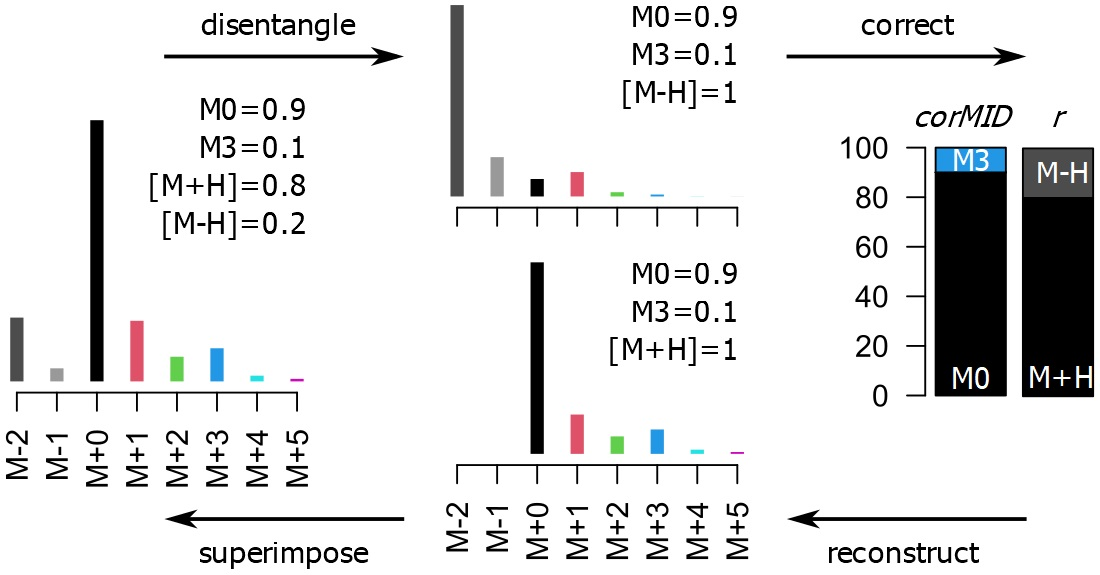
CorMID uses a fragment distribution vector (
r) and the corrected MID (
corMID) to determine a reconstructed vector of similar size (
recMID). By finding the
recMID that is closest to the observed vector of measured ion intensities (
rawMID), the correct
corMID is determined (
Figure S1). Correcting a
rawMID for natural abundance means subtracting the stochastically expected intensity of M+1 and M+2 before normalizing to the summed intensity, which would lead to a
corMID of {1, 0, 0} in this case.
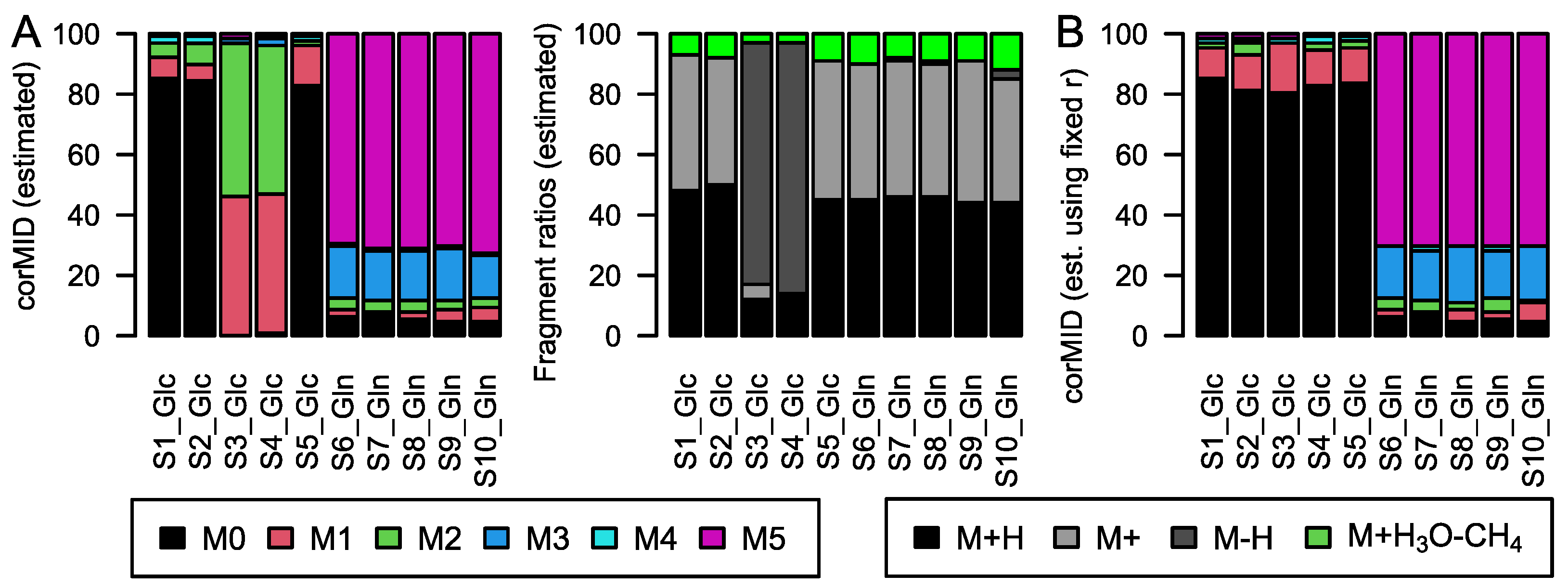
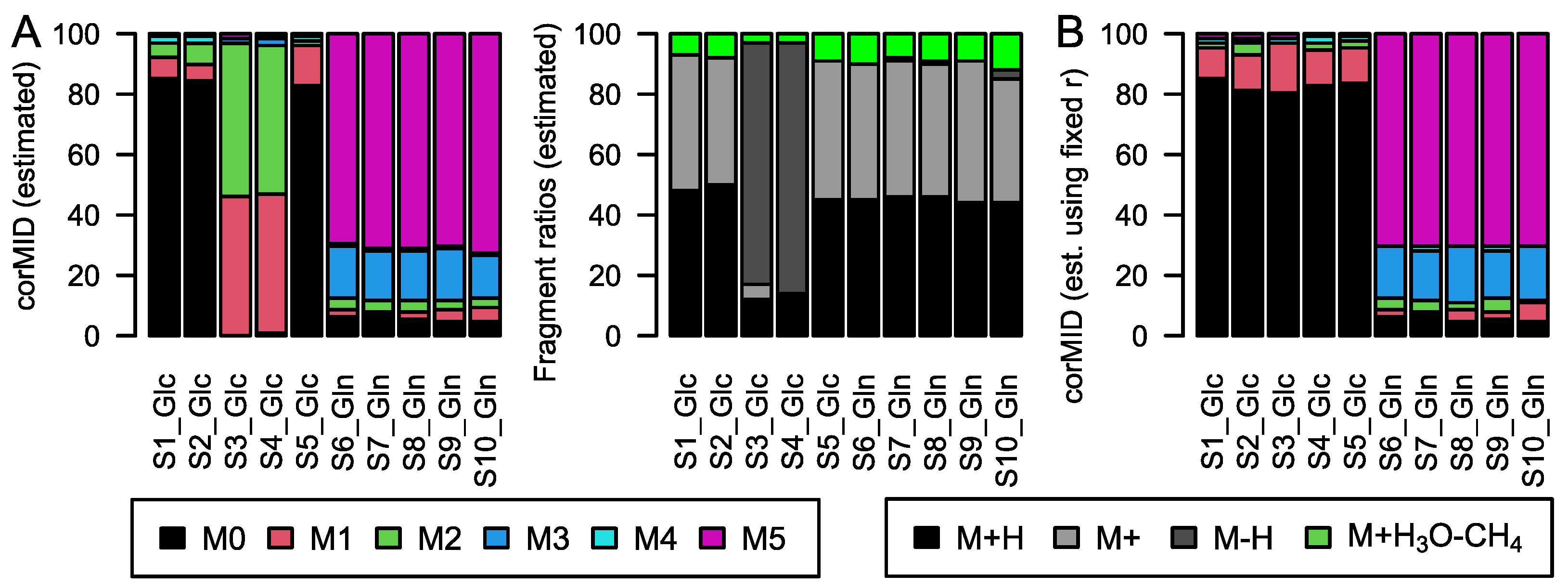
While this correction seems trivial, several effects complicate the calculations in real experiments: derivatization, fragment and adduct formation, and combinations of the latter two. Derivatization is the process where, e.g., tri-methyl-silyl groups (TMS) are attached to molecules of biological origin during sample preparation to facilitate volatilization. Each TMS group contains 3 C with a stochastic labelling of 1.1%. Furthermore, it contains 1 Si, which is either present as
28Si (92.2%),
29Si (4.7%), or
30Si (3.1%). While isotopes of other atomic species can mostly be neglected due to their low abundance (e.g.,
15N~0.004%), TMS molecules contribute strongly to a compound’s MID and must be considered during calculations (
Figure 1B). Incorporation of
13C from a tracer molecule will affect only the carbon of biological origin and not the carbon contained in TMS groups (
Figure 1C). Fragmentation describes the process where APCI results in the formation of different in-source fragments. Generally, the protonated version [M+H]
+ is observed. However, [M+H]
+ ions are often prone to proton losses, leading to [M]
+ and [M-H]
+ ions. The fractional distribution of these ion entities is dependent on the molecule and instrument conditions, but it is stable within measurement series (own observation). A fourth fragment that is often present in spectra of carbonic acids is formed by attaching water and by dissociating a methane molecule [M+H
3O−CH
4]
+. The resulting ion is in close proximity to the M+2 isotopologue (H
2O ~18 Da, CH
4 ~16 Da, difference to M+2 ~−30 mDa).
Several software packages exist to correct for naturally abundant
13C, mostly assuming the presence of either a single or two ion entities ([M+H]
+ and [M]
+) [
10,
11]. However, as described above, many molecules give rise to several fragment ions which overlap with respect to their MIDs. The measured intensity at [M+H]
+ is the sum of [M+H]
+, [M]
++1 and [M-H]
++2. This is due to the mass resolution (R = m/Δm) of current quadrupole time-of-flight instruments being around 35,000. CorMID is designed to mitigate that disadvantage. Devices with a higher resolution, e.g., Fourier-transform ion cyclotron resonance instruments, (R ~1 × 10
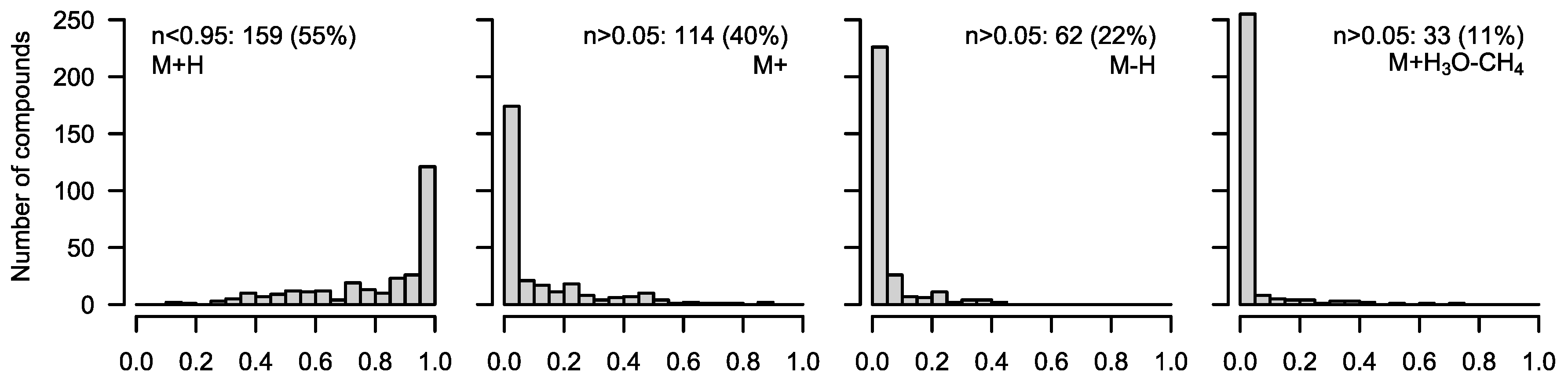
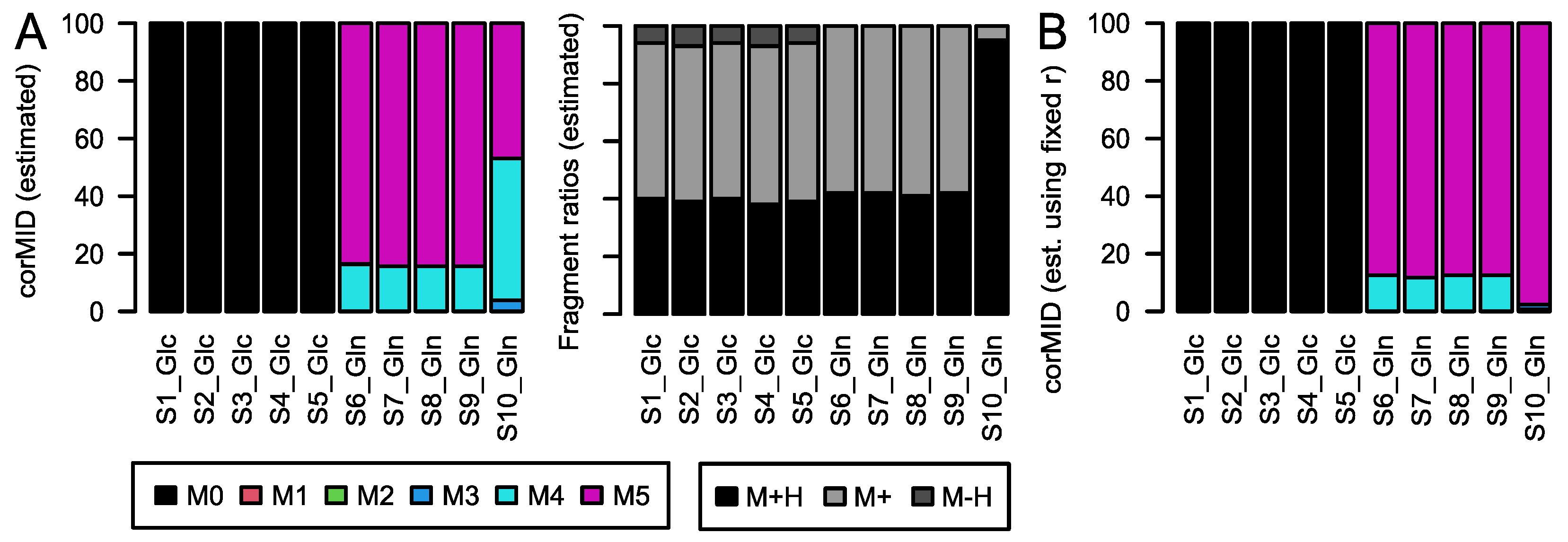
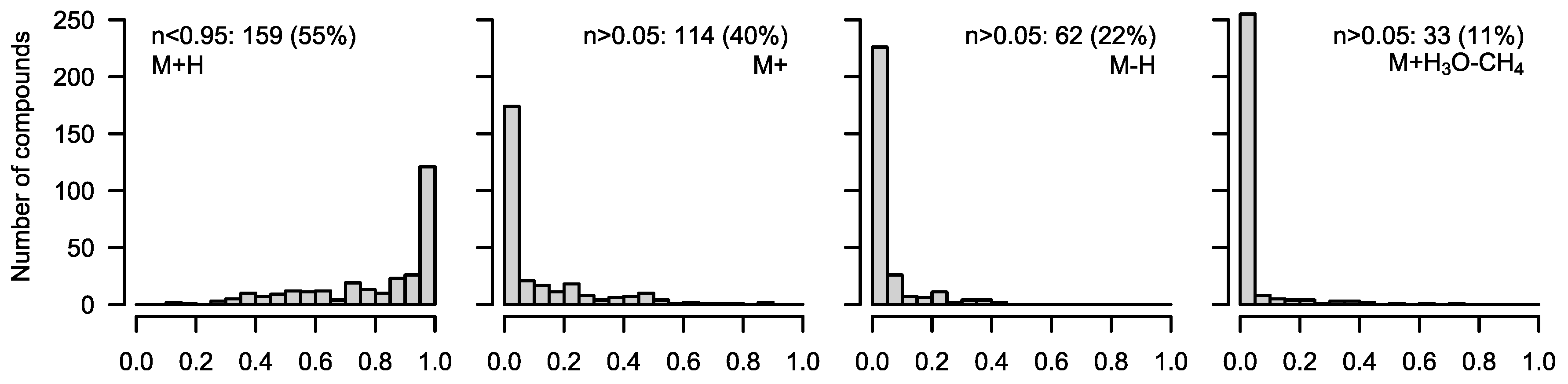
6) will resolve the isotopic fine structure of the compound and allow for the separation of these peaks; here, CorMID is not used. Neglecting the contribution of multiple fragments will lead to severe errors in the correction of natural abundance and calculation of tracer incorporation. As an example, Glucose (Glc) forms approximately 10% [M-H]
+ within the APCI spectrum. While this is not critical in standard metabolomics experiments, where the intensity of [M+H]
+ would be compared over samples, this will lead to a 10% [M+H]
++4 peak in an experiment where fully labeled Glc is used as a tracer. Not accounting for the [M-H]
+ fragment in consequence will lead to a
corMID of {0, 0, 0, 0.1, 0, 0.9) or M4 = 10% and M6 = 90%. We have programmed a function CorMID that estimates the ratio (fractional distribution) of potential fragments and the corresponding corrected MID, solely based on the measured
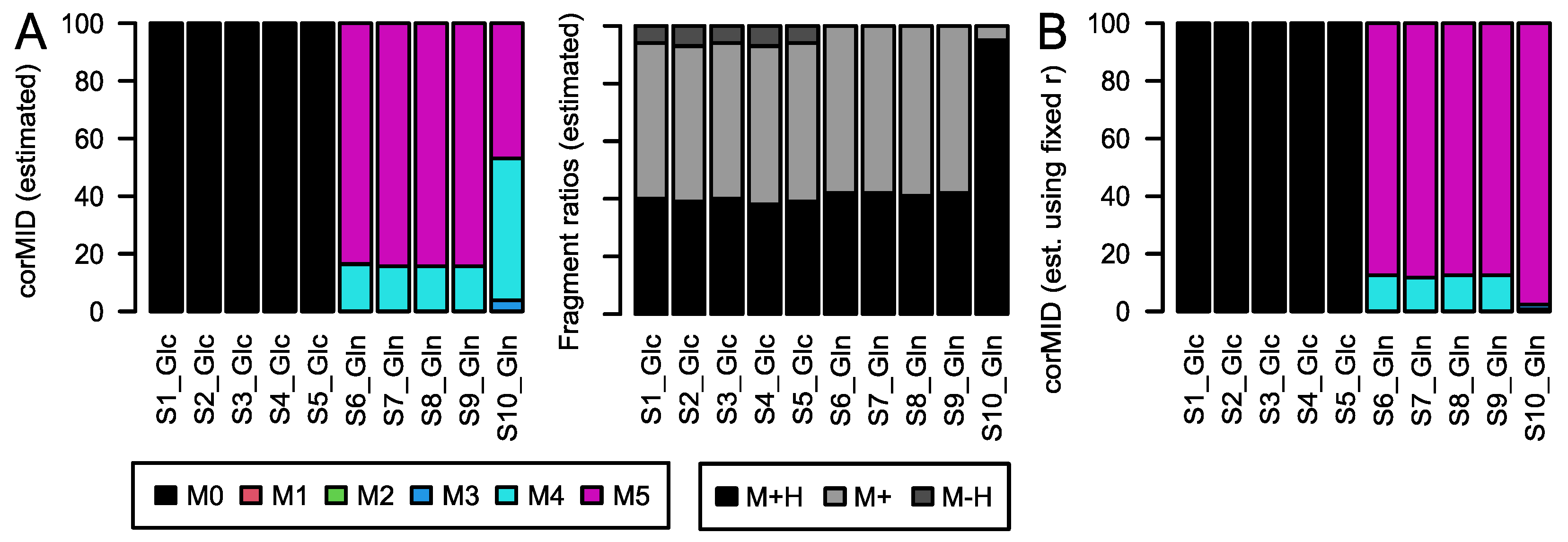
rawMID and the sum formula of the compound. In the following paragraphs, we show the problem, and explain and test the algorithm on a large set of standard compounds as well as on a biological data set from a cell culture experiment using fully labeled Glc and glutamine (Gln) as tracer molecules.
3. Materials and Methods
The algorithm behind CorMID is a greedy minimization of a residual error (
err) between an observed vector of measured ion intensities (
rawMID) and a reconstructed vector of similar size (
recMID) calculated out of a fragment distribution vector (
r) and the corrected MID (
corMID) (
Figure S1). The size of
r is generally dependent on the number of fragments considered, with currently
k = 4 possible fragments defined and
. The size of vector
corMID is dependent on
n, the number of biological carbon atoms within the molecule. It is of size
n + 1, with
and
. When
M0 = 1, the compound is fully unlabeled and fully labelled when
Mn = 1. For every
r and
corMID, we can reconstruct a vector
recMID of similar size as
rawMID to compute
. The reconstruction is performed by converting
corMID into a matrix and by multiplying this matrix with
r,
recMID =
frec(
corMID,
n,
r)·
r. The matrix dimensions are dependent on the measured intensities in
rawMID, the number of biological carbon atoms
n, and the specified fragments in
r. To this end, the function
frec() will prepend and append
corMID with 0 values depending on the present fragments. The following multiplication with
r will yield a vector of size
rawMID. For example, if
r contains only two fragments [M]
+ and [M+H]
+,
n = 2 and
rawMID contains four consecutive ion intensities starting at [M]
+, the result of
frec() would be the matrix {
corMID,0}{0,
corMID}. Because the size of
corMID is
n + 1, multiplying this matrix with
r will lead to
recMID being of a size similar to
rawMID. In fact,
recMID and
rawMID are a superimposed
corMID after being shifted according to
r.
The minimization of err has a unique solution when either r or corMID are fixed. To provide an example, let us assume that r is known and stable for a specific compound with two carbon atoms of biological origin. The optimal corMID fitting to this r and a specific rawMID can then be obtained by testing all possible combinations of corMID, i.e., all vectors M. In practice, the solution is approached by testing possible M using a widely spread set of potential solutions initially (M = {1,0,0}, {0,1,0}, {0,0,1}, {0.5,0.5,0}, {0.5,0,0.5} and {0,0.5,0.5}) to find the best fit (minimal err). In this example, a step size of 0.5 was used to build the set of solutions. In each following iteration, a new set of potential solutions is tested based on the best fit of the previous round and decreasing the step size.
If both
r and
corMID need to be estimated in parallel, the above approach is performed in a nested fashion, i.e., testing each
corMID hypothesis
M with a solution set of different
r. This can lead to wrong
corMID estimates as, e.g., the constructed
recMID vector in the above setting (
n = 2, two fragments for r) with
corMID = {
0,
1,
0} and
r = {
1,
0} is exactly the same as for
corMID = {
1,
0,
0} and
r = {
0,
1}. When
r can neither be estimated from non-labeled control samples nor be estimated based on statistical evaluation of replicate measurements, the function CorMID offers the option to set a penalization parameter, which will apply a weighting factor on
err based on the amount of [M+H] estimated for
r. Penalizing solutions with small amounts of [M+H] is justified based on
Figure 3. For further details, please see the package documentation.
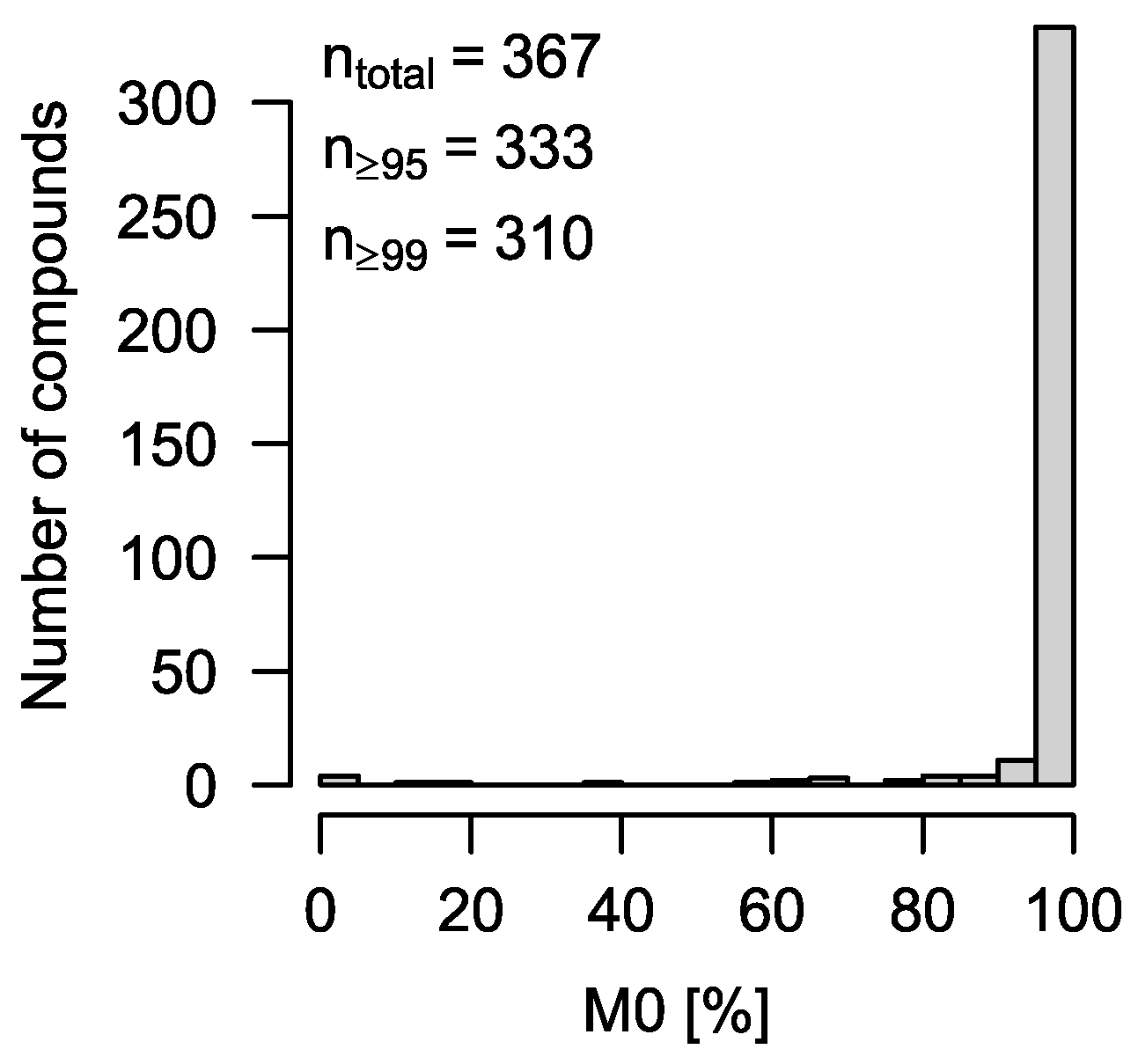
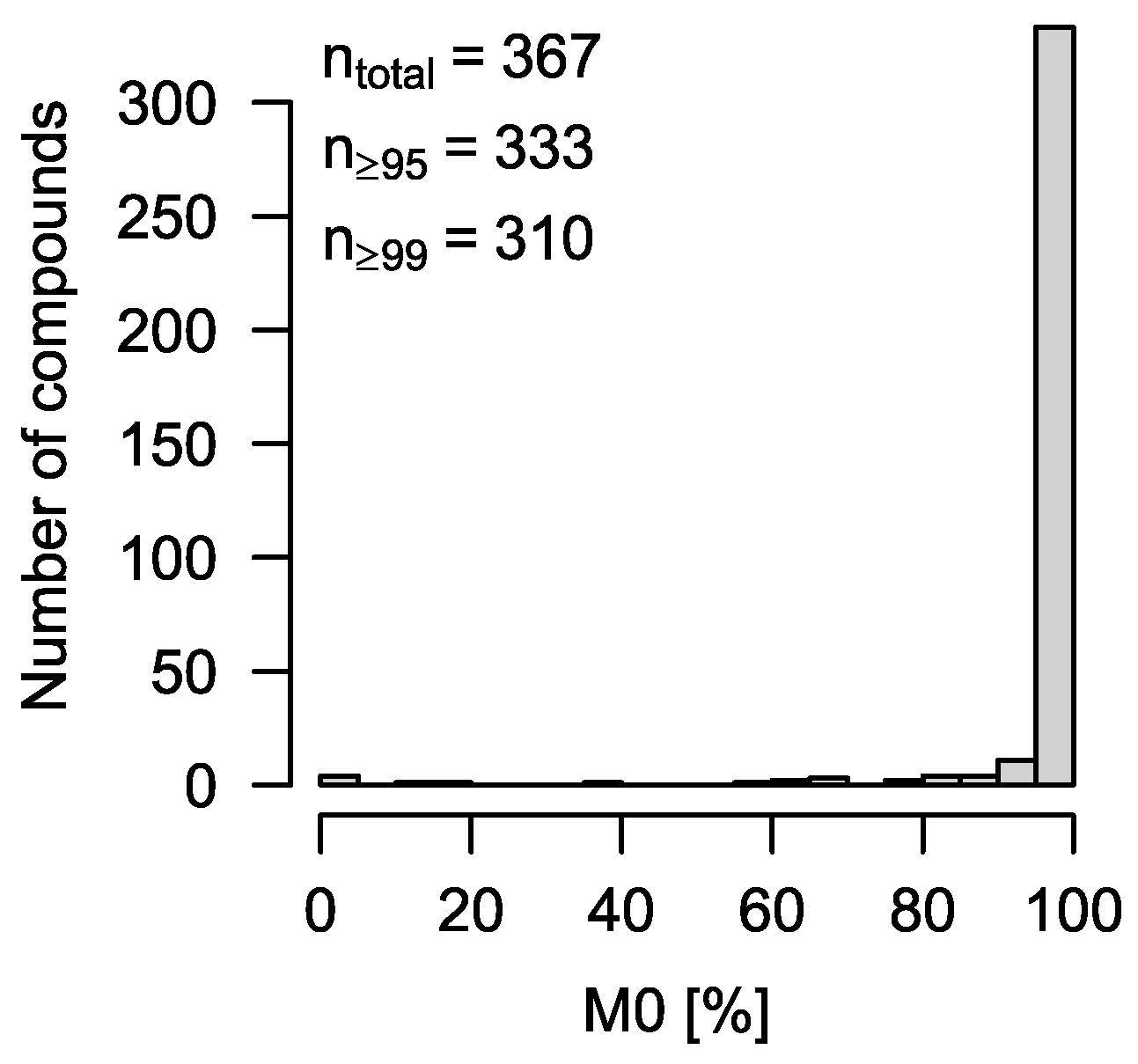
To test CorMID systematically, the MSMLS metabolite library containing 604 unique compounds was purchased from Sigma-Aldrich (Hamburg, Germany). Metabolite standards (5 µg) were reconstituted by adding 100 µL 5% methanol or 100 µL 3:3:1 (
v/
v/
v) chloroform/methanol/water, respectively, to each well of 96-well plates according to the manufacturer’s instructions; 15–24 metabolites were combined each into
n = 28 different master mixes, aliquoted, and dried down in an Alpha 2–4 vacuum rotator (Christ, Osterode, Germany). All samples were derivatized to substitute reactive protons with tri-methyl-silyl groups and measured on an Impact II Q-TOF MS (Bruker, Bremen, Germany) coupled with an Agilent 7890 B gas chromatograph (Agilent, Waldbronn, Germany) via a GC-APCI II source (Bruker, Bremen, Germany). Data processing was carried out as previously described [
14].
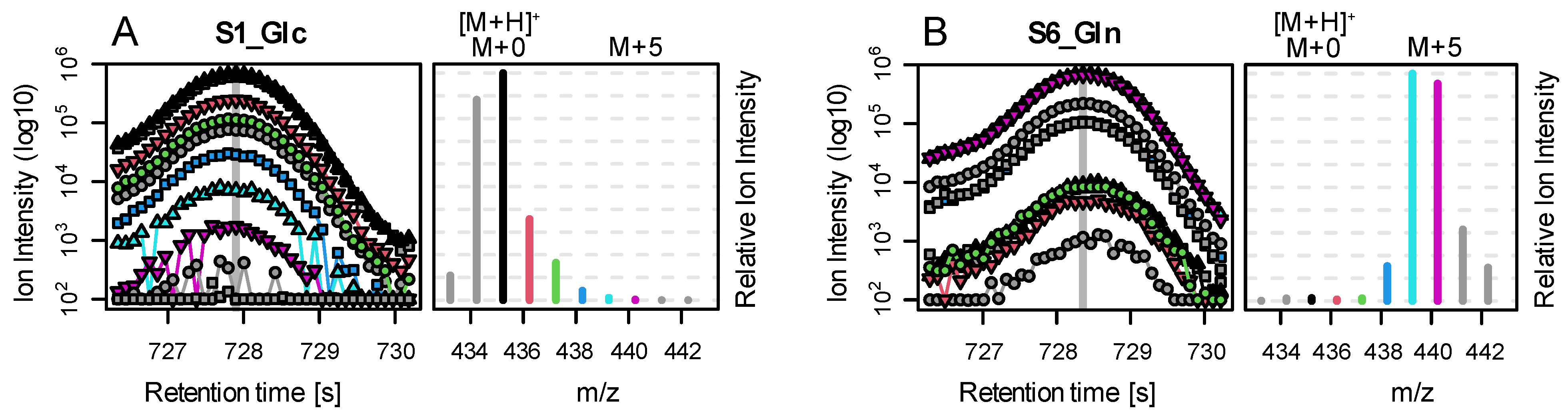
To test CorMID on complex biological samples, we used previously published data from a flux experiment in colon cancer cell cultures applying glucose and glutamine as tracer molecules to incorporate labelled carbon. Here, SW620 and SW480 cells (ATCC, Manassas, VA, USA) were grown, propagated, treated, harvested, and prepared as previously described [
15].





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}