

SARS-CoV-2-Induced Amyloidgenesis: Not One, but Three Hypotheses for Cerebral COVID-19 Outcomes

 ,
,  ,
, 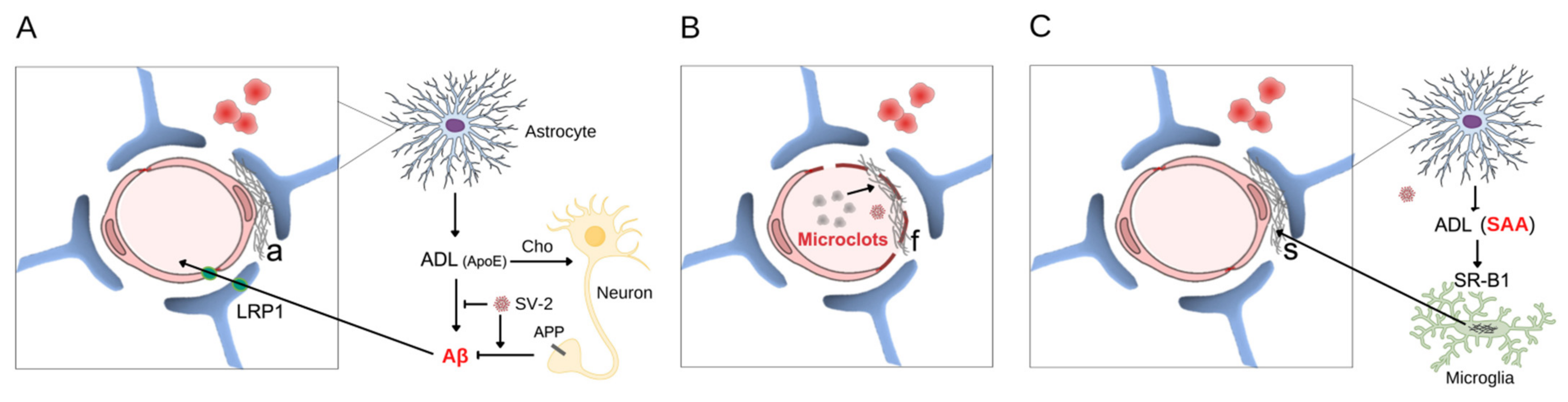{kind=link}
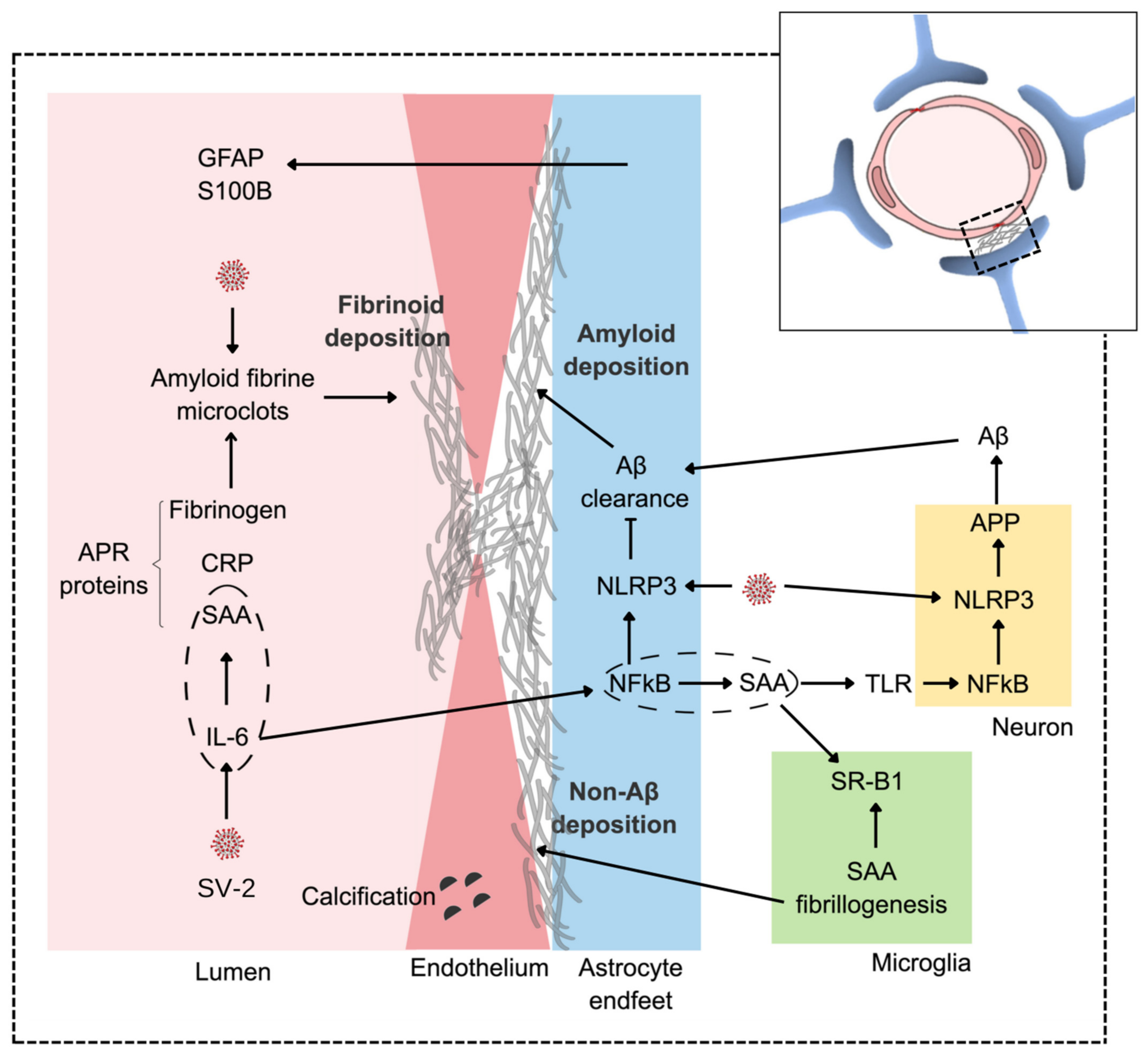{kind=link}
Abstract
1. Background
But What Is the Focus of This View?
2. Amyloid Beta Peptide Deposition
3. Amyloid Fibrin Clots
4. Serum Amyloid A Protein
5. Complementarity between Hypotheses and with Other Biochemical COVID-19 Changes
6. Concluding Remarks
- SARS-CoV-2 causes amyloidgenesis, and this may underlie the lasting effects of post-COVID-19;
- In the CNS, the effect is induced directly by viral proteins on fibrin and SAA fibrillogenesis, or indirectly on Aβ peptides, altering the production/clearance balance;
- The hypotheses discussed here of amyloidgenesis are complementary and converge, leading to ischemic and/or hemorrhagic events, which compromise the functionality of the CNS;
- These hypotheses reinforce the importance of astrocytes and the brain endothelium for brain integrity;
- Here, we emphasize perivascular amyloidosis induced by SARS-CoV-2, but it should be remembered that amyloidosis—as an unfolded protein disorder—is a ubiquitous phenomenon that underlies many diseases. Thus, it may be postulated that many other manifestations of COVID-19 may involve disorders of unfolded proteins;
- Infection by SARS-CoV-2, like that by some other pathogens, is capable of aggravating existing imbalances (co-morbidities) and/or even causing disorders in protein homeostasis. Thus, many non-communicable diseases (such as AD, diabetes mellitus, and stroke) can be exacerbated and even triggered by communicable disease agents;
- In the 19th century, diseases caused by infectious agents, due to vaccines and antibiotics, lost epidemiological relevance to non-communicable diseases. Apparently, the epidemiology of communicable diseases may recuperate its social and economic importance, shedding light on the dialectical essays on health by Richard Lewontin and Richard Levins in the book Biology Under Influence.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miners, S.; Kehoe, P.G.; Love, S. Cognitive Impact of COVID-19: Looking beyond the Short Term. Alzheimer’s Res. Ther. 2020, 12, 170. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.; Najjar, A.; Chong, D.J.; Pramanik, B.K.; Kirsch, C.; Kuzniecky, R.I.; Pacia, S.V.; Azhar, S. Central Nervous System Complications Associated with SARS-CoV-2 Infection: Integrative Concepts of Pathophysiology and Case Reports. J. Neuroinflamm. 2020, 17, 231. [Google Scholar] [CrossRef] [PubMed]
- Robinson-Agramonte, M.A.; Gonçalves, C.-A.; Noris-García, E.; Préndes Rivero, N.; Brigida, A.L.; Schultz, S.; Siniscalco, D.; García García, R.J. Impact of SARS-CoV-2 on Neuropsychiatric Disorders. World J. Psychiatry 2021, 11, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, Y.; Liu, M.; Niu, M.; Song, Z.; Yan, M.; Tian, J. Nervous System Diseases Are Associated with the Severity and Mortality of Patients with COVID-19: A Systematic Review and Meta-Analysis. Epidemiol. Infect. 2021, 149, e66. [Google Scholar] [CrossRef] [PubMed]
- Premraj, L.; Kannapadi, N.V.; Briggs, J.; Seal, S.M.; Battaglini, D.; Fanning, J.; Suen, J.; Robba, C.; Fraser, J.; Cho, S.-M. Mid and Long-Term Neurological and Neuropsychiatric Manifestations of Post-COVID-19 Syndrome: A Meta-Analysis. J. Neurol. Sci. 2022, 434, 120162. [Google Scholar] [CrossRef]
- Omidian, N.; Mohammadi, P.; Sadeghalvad, M.; Mohammadi-Motlagh, H.-R. Cerebral Microvascular Complications Associated with SARS-CoV-2 Infection: How Did It Occur and How Should It Be Treated? Biomed. Pharm. 2022, 154, 113534. [Google Scholar] [CrossRef]
- Mitra, J.; Kodavati, M.; Provasek, V.E.; Rao, K.S.; Mitra, S.; Hamilton, D.J.; Horner, P.J.; Vahidy, F.S.; Britz, G.W.; Kent, T.A.; et al. SARS-CoV-2 and the Central Nervous System: Emerging Insights into Hemorrhage-Associated Neurological Consequences and Therapeutic Considerations. Ageing Res. Rev. 2022, 80, 101687. [Google Scholar] [CrossRef]
- Sullivan, B.N.; Fischer, T. Age-Associated Neurological Complications of COVID-19: A Systematic Review and Meta-Analysis. Front. Aging Neurosci. 2021, 13, 653694. [Google Scholar] [CrossRef]
- Zhang, L.-K.; Sun, Y.; Zeng, H.; Wang, Q.; Jiang, X.; Shang, W.-J.; Wu, Y.; Li, S.; Zhang, Y.-L.; Hao, Z.-N.; et al. Calcium Channel Blocker Amlodipine Besylate Therapy Is Associated with Reduced Case Fatality Rate of COVID-19 Patients with Hypertension. Cell Discov. 2020, 6, 96. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Hillen, H. The Beta Amyloid Dysfunction (BAD) Hypothesis for Alzheimer’s Disease. Front. Neurosci. 2019, 13, 1154. [Google Scholar] [CrossRef]
- Picken, M.M. The Changing Concepts of Amyloid. Arch. Pathol. Lab. Med. 2001, 125, 38–43. [Google Scholar] [CrossRef]
- Picken, M.M. The Pathology of Amyloidosis in Classification: A Review. Acta Haematol. 2020, 143, 322–334. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, Y.; Ma, Q.-H.; Liu, Y. Alzheimer’s Disease: Amyloid-Based Pathogenesis and Potential Therapies. CST 2018, 2, 150–161. [Google Scholar] [CrossRef]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 150–161. [Google Scholar] [CrossRef]
- Charidimou, A.; Boulouis, G.; Gurol, M.E.; Ayata, C.; Bacskai, B.J.; Frosch, M.P.; Viswanathan, A.; Greenberg, S.M. Emerging Concepts in Sporadic Cerebral Amyloid Angiopathy. Brain 2017, 140, 1829–1850. [Google Scholar] [CrossRef]
- Galuske, R.A.W.; Drach, L.M.; Nichtweiss, M.; Marquardt, G.; Franz, K.; Bohl, J.; Schlote, W. Colocalization of Different Types of Amyloid in the Walls of Cerebral Blood Vessels of Patients Suffering from Cerebral Amyloid Angiopathy and Spontaneous Intracranial Hemorrhage: A Report of 5 Cases. Clin. Neuropathol. 2004, 23, 113–119. [Google Scholar]
- Nakamura, M.; Yamashita, T.; Ueda, M.; Obayashi, K.; Sato, T.; Ikeda, T.; Washimi, Y.; Hirai, T.; Kuwahara, Y.; Yamamoto, M.T.; et al. Neuroradiologic and Clinicopathologic Features of Oculoleptomeningeal Type Amyloidosis. Neurology 2005, 65, 1051–1056. [Google Scholar] [CrossRef]
- Tremblay, M.-E.; Madore, C.; Bordeleau, M.; Tian, L.; Verkhratsky, A. Neuropathobiology of COVID-19: The Role for Glia. Front. Cell Neurosci. 2020, 14, 592214. [Google Scholar] [CrossRef]
- Schultz, B.; Taday, J.; Menezes, L.; Cigerce, A.; Leite, M.C.; Gonçalves, C.-A. Calpain-Mediated Alterations in Astrocytes Before and During Amyloid Chaos in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 84, 1415–1430. [Google Scholar] [CrossRef]
- Beyer, L.; Stocker, H.; Rujescu, D.; Holleczek, B.; Stockmann, J.; Nabers, A.; Brenner, H.; Gerwert, K. Amyloid-beta Misfolding and GFAP Predict Risk of Clinical Alzheimer’s Disease Diagnosis within 17 Years. Alzheimer’s Dement. 2022. [Google Scholar] [CrossRef] [PubMed]
- Harpin, M.L.; Delaère, P.; Javoy-Agid, F.; Bock, E.; Jacque, C.; Delpech, B.; Villarroya, H.; Duyckaerts, C.; Hauw, J.J.; Baumann, N. Glial Fibrillary Acidic Protein and ΒA4 Protein Deposits in Temporal Lobe of Aging Brain and Senile Dementia of the Alzheimer Type: Relation with the Cognitive State and with Quantitative Studies of Senile Plaques and Neurofibrillary Tangles: GFAP in Aging Brain and SDAT. J. Neurosci. Res. 1990, 27, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Steardo, L.; Steardo, L.; Scuderi, C. Astrocytes and the Psychiatric Sequelae of COVID-19: What We Learned from the Pandemic. Neurochem. Res. 2022, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, C.-A.; Sesterheim, P.; Wartchow, K.M.; Bobermin, L.D.; Leipnitz, G.; Quincozes-Santos, A. Why Antidiabetic Drugs Are Potentially Neuroprotective during the Sars-CoV-2 Pandemic: The Focus on Astroglial UPR and Calcium-Binding Proteins. Front. Cell. Neurosci. 2022, 16, 905218. [Google Scholar] [CrossRef] [PubMed]
- Sashindranath, M.; Nandurkar, H.H. Endothelial Dysfunction in the Brain: Setting the Stage for Stroke and Other Cerebrovascular Complications of COVID-19. Stroke 2021, 52, 1895–1904. [Google Scholar] [CrossRef]
- Fraiman, P.; Godeiro Junior, C.; Moro, E.; Cavallieri, F.; Zedde, M. COVID-19 and Cerebrovascular Diseases: A Systematic Review and Perspectives for Stroke Management. Front. Neurol. 2020, 11, 574694. [Google Scholar] [CrossRef]
- Liang, H.W.; Mung, S.M.; Douglass, C.; Jude, E.B. COVID-19-Related Vasculopathy of the Brain. BMJ Case Rep. 2021, 14, e242028. [Google Scholar] [CrossRef]
- Ding, Q.; Shults, N.V.; Gychka, S.G.; Harris, B.T.; Suzuki, Y.J. Protein Expression of Angiotensin-Converting Enzyme 2 (ACE2) Is Upregulated in Brains with Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 1687. [Google Scholar] [CrossRef]
- Lim, K.-H.; Yang, S.; Kim, S.-H.; Joo, J.-Y. Elevation of ACE2 as a SARS-CoV-2 Entry Receptor Gene Expression in Alzheimer’s Disease. J. Infect. 2020, 81, e33–e34. [Google Scholar] [CrossRef]
- Kehoe, P.G.; Wong, S.; AL Mulhim, N.; Palmer, L.E.; Miners, J.S. Angiotensin-Converting Enzyme 2 Is Reduced in Alzheimer’s Disease in Association with Increasing Amyloid-β and Tau Pathology. Alz. Res. Ther. 2016, 8, 50. [Google Scholar] [CrossRef]
- Xiao, X.; Zhang, C.; Ma, X.; Miao, H.; Wang, J.; Liu, L.; Chen, S.; Zeng, R.; Chen, Y.; Bihl, J.C. Angiotensin-(1–7) Counteracts Angiotensin II-Induced Dysfunction in Cerebral Endothelial Cells via Modulating Nox2/ROS and PI3K/NO Pathways. Exp. Cell Res. 2015, 336, 58–65. [Google Scholar] [CrossRef]
- Duan, R.; Wang, S.-Y.; Wei, B.; Deng, Y.; Fu, X.-X.; Gong, P.-Y.; Yan, E.; Sun, X.-J.; Cao, H.-M.; Shi, J.-Q.; et al. Angiotensin-(1–7) Analogue AVE0991 Modulates Astrocyte-Mediated Neuroinflammation via LncRNA SNHG14/MiR-223-3p/NLRP3 Pathway and Offers Neuroprotection in a Transgenic Mouse Model of Alzheimer’s Disease. J. Inflamm. Res. 2021, 14, 7007–7019. [Google Scholar] [CrossRef]
- Camacho, E.; LoPresti, M.A.; Bruce, S.; Lin, D.; Abraham, M.; Appelboom, G.; Taylor, B.; McDowell, M.; DuBois, B.; Sathe, M.; et al. The Role of Age in Intracerebral Hemorrhages. J. Clin. Neurosci. 2015, 22, 1867–1870. [Google Scholar] [CrossRef]
- Heneka, M.T.; Golenbock, D.; Latz, E.; Morgan, D.; Brown, R. Immediate and Long-Term Consequences of COVID-19 Infections for the Development of Neurological Disease. Alzheimer’s Res. Ther. 2020, 12, 69. [Google Scholar] [CrossRef]
- Guan, Y.; Han, F. Key Mechanisms and Potential Targets of the NLRP3 Inflammasome in Neurodegenerative Diseases. Front. Integr. Neurosci. 2020, 14, 37. [Google Scholar] [CrossRef]
- Sita, G.; Graziosi, A.; Hrelia, P.; Morroni, F. NLRP3 and Infections: β-Amyloid in Inflammasome beyond Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 6984. [Google Scholar] [CrossRef]
- He, X.; Li, L.; Xian, W.; Li, M.; Zhang, L.; Xu, J.; Pei, Z.; Zheng, H.; Hu, X. Chronic Colitis Exacerbates NLRP3-Dependent Neuroinflammation and Cognitive Impairment in Middle-Aged Brain. J. Neuroinflamm. 2021, 18, 153. [Google Scholar] [CrossRef]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.B.; Cooper, M.A.; O’Neill, L.A.J.; Lynch, M.A. Inhibiting the NLRP3 Inflammasome with MCC950 Promotes Non-Phlogistic Clearance of Amyloid-β and Cognitive Function in APP/PS1 Mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef]
- Wang, H.; Lu, J.; Zhao, X.; Qin, R.; Song, K.; Xu, Y.; Zhang, J.; Chen, Y. Alzheimer’s Disease in Elderly COVID-19 Patients: Potential Mechanisms and Preventive Measures. Neurol. Sci. 2021, 42, 4913–4920. [Google Scholar] [CrossRef]
- Ziff, O.J.; Ashton, N.J.; Mehta, P.R.; Brown, R.; Athauda, D.; Heaney, J.; Heslegrave, A.J.; Benedet, A.L.; Blennow, K.; Checkley, A.M.; et al. Amyloid Processing in COVID-19-Associated Neurological Syndromes. J. Neurochem. 2022, 161, 146–157. [Google Scholar] [CrossRef]
- Wang, Q.; Davis, P.B.; Gurney, M.E.; Xu, R. COVID-19 and Dementia: Analyses of Risk, Disparity, and Outcomes from Electronic Health Records in the US. Alzheimer’s Dement. 2021, 17, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Laubscher, G.J.; Pretorius, E. A Central Role for Amyloid Fibrin Microclots in Long COVID/PASC: Origins and Therapeutic Implications. Biochem. J. 2022, 479, 537–559. [Google Scholar] [CrossRef] [PubMed]
- Undas, A. How to Assess Fibrinogen Levels and Fibrin Clot Properties in Clinical Practice? Semin. Thromb. Hemost. 2016, 42, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Pieters, M.; Wolberg, A.S. Fibrinogen and Fibrin: An Illustrated Review. Res. Pract. Thromb. Haemost. 2019, 3, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Mbotwe, S.; Bester, J.; Robinson, C.J.; Kell, D.B. Acute Induction of Anomalous and Amyloidogenic Blood Clotting by Molecular Amplification of Highly Substoichiometric Levels of Bacterial Lipopolysaccharide. J. R. Soc. Interface 2016, 13, 20160539. [Google Scholar] [CrossRef]
- Pretorius, E.; Briedenhann, S.; Marx, J.; Franz, R.C. Structural Changes in the Fibrin Network of a Pretoria Family with Dysfibrinogenemia: A Scanning Electron Microscopical Study. Ultrastruct. Pathol. 2006, 30, 167–176. [Google Scholar] [CrossRef]
- Pretorius, E.; Page, M.J.; Hendricks, L.; Nkosi, N.B.; Benson, S.R.; Kell, D.B. Both Lipopolysaccharide and Lipoteichoic Acids Potently Induce Anomalous Fibrin Amyloid Formation: Assessment with Novel AmytrackerTM Stains. J. R. Soc. Interface 2018, 15, 20170941. [Google Scholar] [CrossRef]
- Swanepoel, A.C.; Lindeque, B.G.; Swart, P.J.; Abdool, Z.; Pretorius, E. Estrogen Causes Ultrastructural Changes of Fibrin Networks during the Menstrual Cycle: A Qualitative Investigation: Estrogen Influence Fibrin Network Morphology. Microsc. Res. Tech. 2014, 77, 594–601. [Google Scholar] [CrossRef]
- Page, M.J.; Thomson, G.J.A.; Nunes, J.M.; Engelbrecht, A.-M.; Nell, T.A.; de Villiers, W.J.S.; de Beer, M.C.; Engelbrecht, L.; Kell, D.B.; Pretorius, E. Serum Amyloid A Binds to Fibrin(Ogen), Promoting Fibrin Amyloid Formation. Sci. Rep. 2019, 9, 3102. [Google Scholar] [CrossRef]
- Grobbelaar, L.M.; Venter, C.; Vlok, M.; Ngoepe, M.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. SARS-CoV-2 Spike Protein S1 Induces Fibrin(Ogen) Resistant to Fibrinolysis: Implications for Microclot Formation in COVID-19. Biosci. Rep. 2021, 41, BSR20210611. [Google Scholar] [CrossRef]
- Riley, R.S.; Gilbert, A.R.; Dalton, J.B.; Pai, S.; McPherson, R.A. Widely Used Types and Clinical Applications of D-Dimer Assay. Lab. Med. 2016, 47, 90–102. [Google Scholar] [CrossRef]
- Janes, F.; Gigli, G.L.; Kuris, F.; Morassi, M.; Costa, P.; Nesi, L.; Giacomello, R.; Mazzacane, F.; Leuci, E.; Cavallini, A.; et al. Failure of Therapeutic Anticoagulation in COVID-19 Patients With Acute Ischemic Stroke. A Retrospective Multicenter Study. Front. Neurol. 2022, 13, 834469. [Google Scholar] [CrossRef]
- Gonçalves, C.; Sesterheim, P. Serum Amyloid A Protein Has Been Undervalued as a Biomarker of COVID-19. Diabetes Metab. Res. Rev. 2021, 37, e3376. [Google Scholar] [CrossRef]
- Han, C.Y.; Tang, C.; Guevara, M.E.; Wei, H.; Wietecha, T.; Shao, B.; Subramanian, S.; Omer, M.; Wang, S.; O’Brien, K.D.; et al. Serum Amyloid A Impairs the Antiinflammatory Properties of HDL. J. Clin. Investig. 2015, 126, 266–281. [Google Scholar] [CrossRef]
- Sack, G.H. Serum Amyloid A—A Review. Mol. Med. 2018, 24, 46. [Google Scholar] [CrossRef]
- Barbierato, M.; Borri, M.; Facci, L.; Zusso, M.; Skaper, S.D.; Giusti, P. Expression and Differential Responsiveness of Central Nervous System Glial Cell Populations to the Acute Phase Protein Serum Amyloid A. Sci. Rep. 2017, 7, 12158. [Google Scholar] [CrossRef]
- Soriano, S.; Moffet, B.; Wicker, E.; Villapol, S. Serum Amyloid A Is Expressed in the Brain After Traumatic Brain Injury in a Sex-Dependent Manner. Cell Mol. Neurobiol. 2020, 40, 1199–1211. [Google Scholar] [CrossRef]
- Lin, A.; Liu, J.; Gong, P.; Chen, Y.; Zhang, H.; Zhang, Y.; Yu, Y. Serum Amyloid A Inhibits Astrocyte Migration via Activating P38 MAPK. J. Neuroinflamm. 2020, 17, 254. [Google Scholar] [CrossRef]
- Miida, T.; Yamada, T.; Seino, U.; Ito, M.; Fueki, Y.; Takahashi, A.; Kosuge, K.; Soda, S.; Hanyu, O.; Obayashi, K.; et al. Serum Amyloid A (SAA)-Induced Remodeling of CSF-HDL. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2006, 1761, 424–433. [Google Scholar] [CrossRef]
- Kindy, M.S.; Yu, J.; Guo, J.-T.; Zhu, H. Apolipoprotein Serum Amyloid A in Alzheimer’s Disease. J. Alzheimer’s Dis. 1999, 1, 155–167. [Google Scholar] [CrossRef]
- Jang, S.; Jang, W.Y.; Choi, M.; Lee, J.; Kwon, W.; Yi, J.; Park, S.J.; Yoon, D.; Lee, S.; Kim, M.O.; et al. Serum Amyloid A1 Is Involved in Amyloid Plaque Aggregation and Memory Decline in Amyloid Beta Abundant Condition. Transgenic Res. 2019, 28, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, J.; Dohgu, S.; Takata, F.; Iwao, T.; Kimura, I.; Tomohiro, M.; Aono, K.; Kataoka, Y.; Yamauchi, A. Serum Amyloid A-Induced Blood-Brain Barrier Dysfunction Associated with Decreased Claudin-5 Expression in Rat Brain Endothelial Cells and Its Inhibition by High-Density Lipoprotein in Vitro. Neurosci. Lett. 2020, 738, 135352. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, X.; Li, Z.; Li, D.; Yang, J. Prognostic Value of Serum Amyloid A in COVID-19: A Meta-Analysis. Medicine 2022, 101, e28880. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Huang, Y.; Guo, Y.; Yin, M.; Chen, X.; Xiao, L.; Deng, G. Association of Inflammatory Markers with the Severity of COVID-19: A Meta-Analysis. Int. J. Infect. Dis. 2020, 96, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Huang, W.-J.; Lan, M.-Q.; Gan, F.-R.; Liu, Y.-Y.; Sun, L.; Chen, J.-L.; Sun, Y.-F.; Tao, C.-M. Association between Serum Amyloid A Levels and Predicting Disase Severity in COVID-19 Patients: A Systematic Review and Meta-Analysis. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 4627–4638. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.M.; Pei, S.F.; Chong, Y.Z.; Guo, Y.; Gao, X.L.; Tang, Q.Y.; Li, Y.; Feng, F.M. CRP, SAA, LDH, and DD Predict Poor Prognosis of Coronavirus Disease (COVID-19): A Meta-Analysis from 7739 Patients. Scand. J. Clin. Lab. Investig. 2021, 81, 679–686. [Google Scholar] [CrossRef]
- Zhao, Y.; He, X.; Shi, X.; Huang, C.; Liu, J.; Zhou, S.; Heng, C.-K. Association between Serum Amyloid A and Obesity: A Meta-Analysis and Systematic Review. Inflamm. Res. 2010, 59, 323–334. [Google Scholar] [CrossRef]
- Jana, A.K.; Greenwood, A.B.; Hansmann, U.H.E. Presence of a SARS-CoV-2 Protein Enhances Amyloid Formation of Serum Amyloid A. J. Phys. Chem. B 2021, 125, 9155–9167. [Google Scholar] [CrossRef]
- Galkin, A.P. Hypothesis: AA Amyloidosis Is a Factor Causing Systemic Complications after Coronavirus Disease. Prion 2021, 15, 53–55. [Google Scholar] [CrossRef]
- Mahley, R.W. Central Nervous System Lipoproteins: ApoE and Regulation of Cholesterol Metabolism. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1305–1315. [Google Scholar] [CrossRef]
- Nechipurenko, Y.D.; Semyonov, D.A.; Lavrinenko, I.A.; Lagutkin, D.A.; Generalov, E.A.; Zaitceva, A.Y.; Matveeva, O.V.; Yegorov, Y.E. The Role of Acidosis in the Pathogenesis of Severe Forms of COVID-19. Biology 2021, 10, 852. [Google Scholar] [CrossRef]
- Osman, W.; Al Fahdi, F.; Al Salmi, I.; Al Khalili, H.; Gokhale, A.; Khamis, F. Serum Calcium and Vitamin D Levels: Correlation with Severity of COVID-19 in Hospitalized Patients in Royal Hospital, Oman. Int. J. Infect. Dis. 2021, 107, 153–163. [Google Scholar] [CrossRef]
- Eugenín, J.; Vecchiola, A.; Murgas, P.; Arroyo, P.; Cornejo, F.; von Bernhardi, R. Expression Pattern of Scavenger Receptors and Amyloid-β Phagocytosis of Astrocytes and Microglia in Culture Are Modified by Acidosis: Implications for Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 53, 857–873. [Google Scholar] [CrossRef]
- Claus, S.; Meinhardt, K.; Aumüller, T.; Puscalau-Girtu, I.; Linder, J.; Haupt, C.; Walther, P.; Syrovets, T.; Simmet, T.; Fändrich, M. Cellular Mechanism of Fibril Formation from Serum Amyloid A1 Protein. EMBO Rep. 2017, 18, 1352–1366. [Google Scholar] [CrossRef]
- Martha, J.W.; Wibowo, A.; Pranata, R. Hypocalcemia Is Associated with Severe COVID-19: A Systematic Review and Meta-Analysis. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 337–342. [Google Scholar] [CrossRef]
- Dillinger, J.G.; Benmessaoud, F.A.; Pezel, T.; Voicu, S.; Sideris, G.; Chergui, N.; Hamzi, L.; Chauvin, A.; Leroy, P.; Gautier, J.F.; et al. Coronary Artery Calcification and Complications in Patients With COVID-19. JACC Cardiovasc. Imaging 2020, 13, 2468–2470. [Google Scholar] [CrossRef]
- Nair, A.V.; Kumar, D.; Yadav, S.K.; Nepal, P.; Jacob, B.; Al-Heidous, M. Utility of Visual Coronary Artery Calcification on Non-Cardiac Gated Thoracic CT in Predicting Clinical Severity and Outcome in COVID-19. Clin. Imaging 2021, 74, 123–130. [Google Scholar] [CrossRef]
- Yuan, C.; Ni, L.; Zhang, C.; Hu, X.; Wu, X. Vascular Calcification: New Insights into Endothelial Cells. Microvasc. Res. 2021, 134, 104105. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonçalves, C.-A.; Bobermin, L.D.; Sesterheim, P.; Netto, C.A. SARS-CoV-2-Induced Amyloidgenesis: Not One, but Three Hypotheses for Cerebral COVID-19 Outcomes. Metabolites 2022, 12, 1099. https://doi.org/10.3390/metabo12111099
Gonçalves C-A, Bobermin LD, Sesterheim P, Netto CA. SARS-CoV-2-Induced Amyloidgenesis: Not One, but Three Hypotheses for Cerebral COVID-19 Outcomes. Metabolites. 2022; 12(11):1099. https://doi.org/10.3390/metabo12111099
Chicago/Turabian StyleGonçalves, Carlos-Alberto, Larissa Daniele Bobermin, Patricia Sesterheim, and Carlos Alexandre Netto. 2022. "SARS-CoV-2-Induced Amyloidgenesis: Not One, but Three Hypotheses for Cerebral COVID-19 Outcomes" Metabolites 12, no. 11: 1099. https://doi.org/10.3390/metabo12111099
APA StyleGonçalves, C.-A., Bobermin, L. D., Sesterheim, P., & Netto, C. A. (2022). SARS-CoV-2-Induced Amyloidgenesis: Not One, but Three Hypotheses for Cerebral COVID-19 Outcomes. Metabolites, 12(11), 1099. https://doi.org/10.3390/metabo12111099