
Guanosine Prevents Spatial Memory Impairment and Hippocampal Damage Following Amyloid-β1–42 Administration in Mice

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Drugs
2.3. Amyloid-Beta Infusion
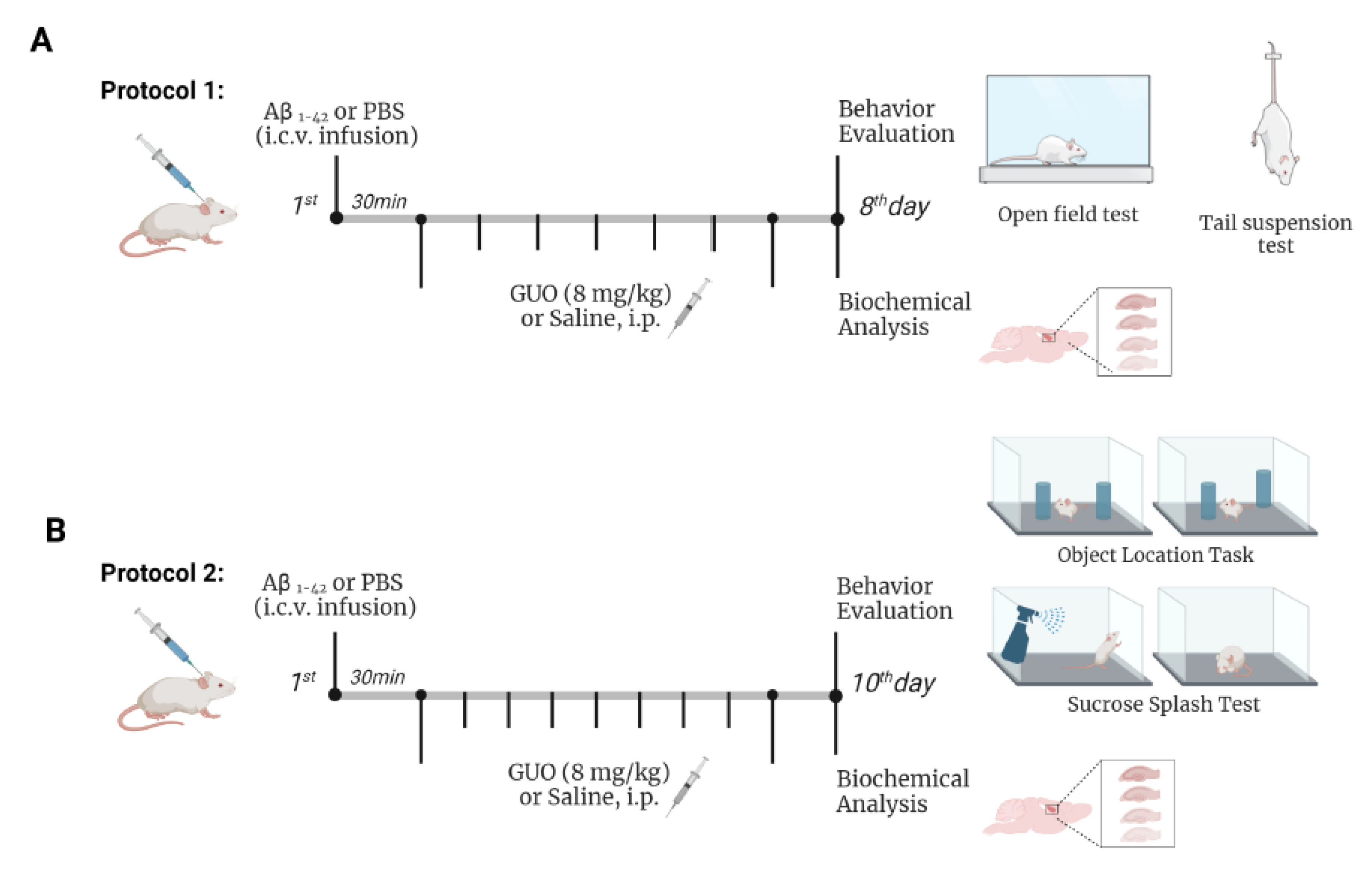
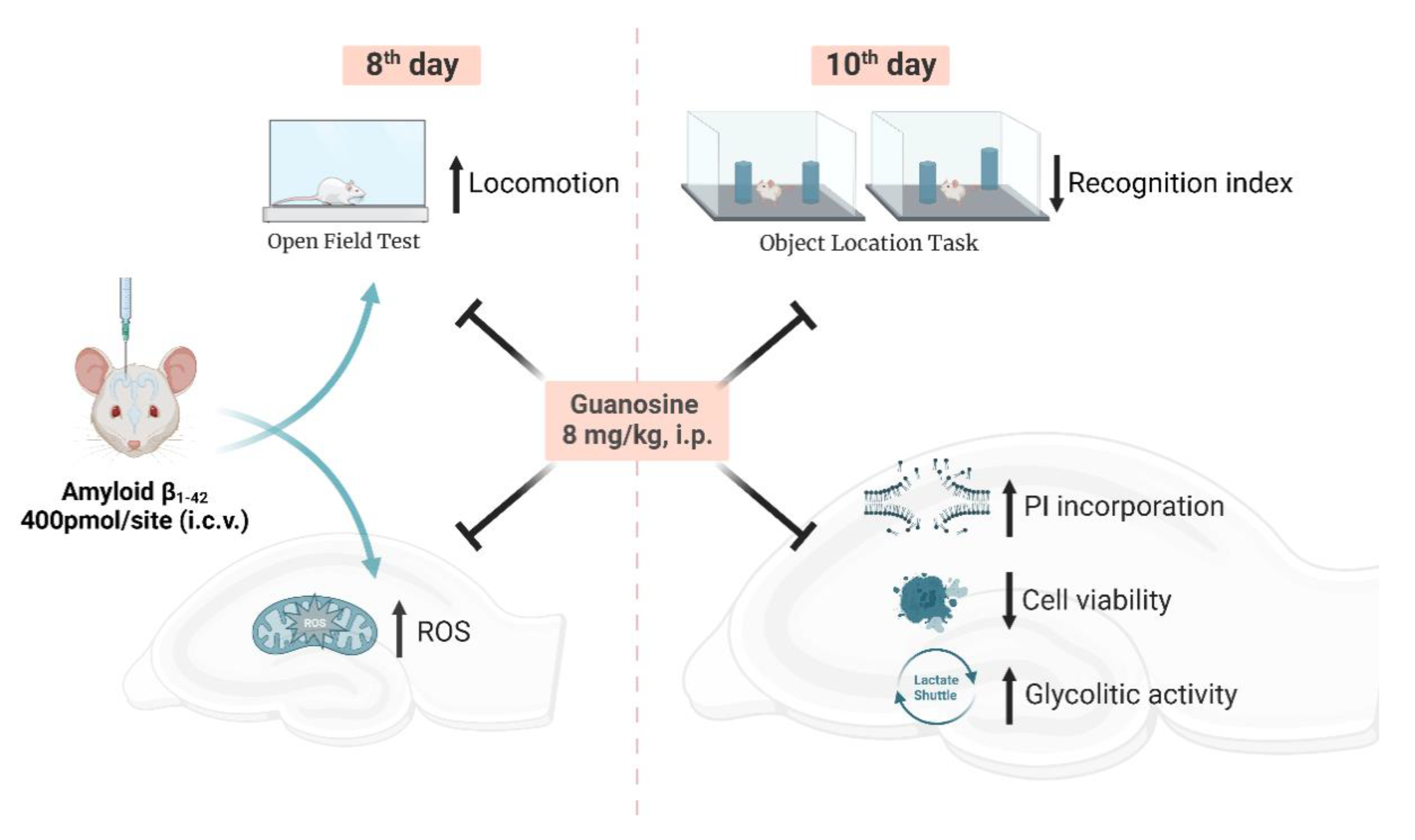
2.4. Experimental Design
2.5. Behavioral Analysis
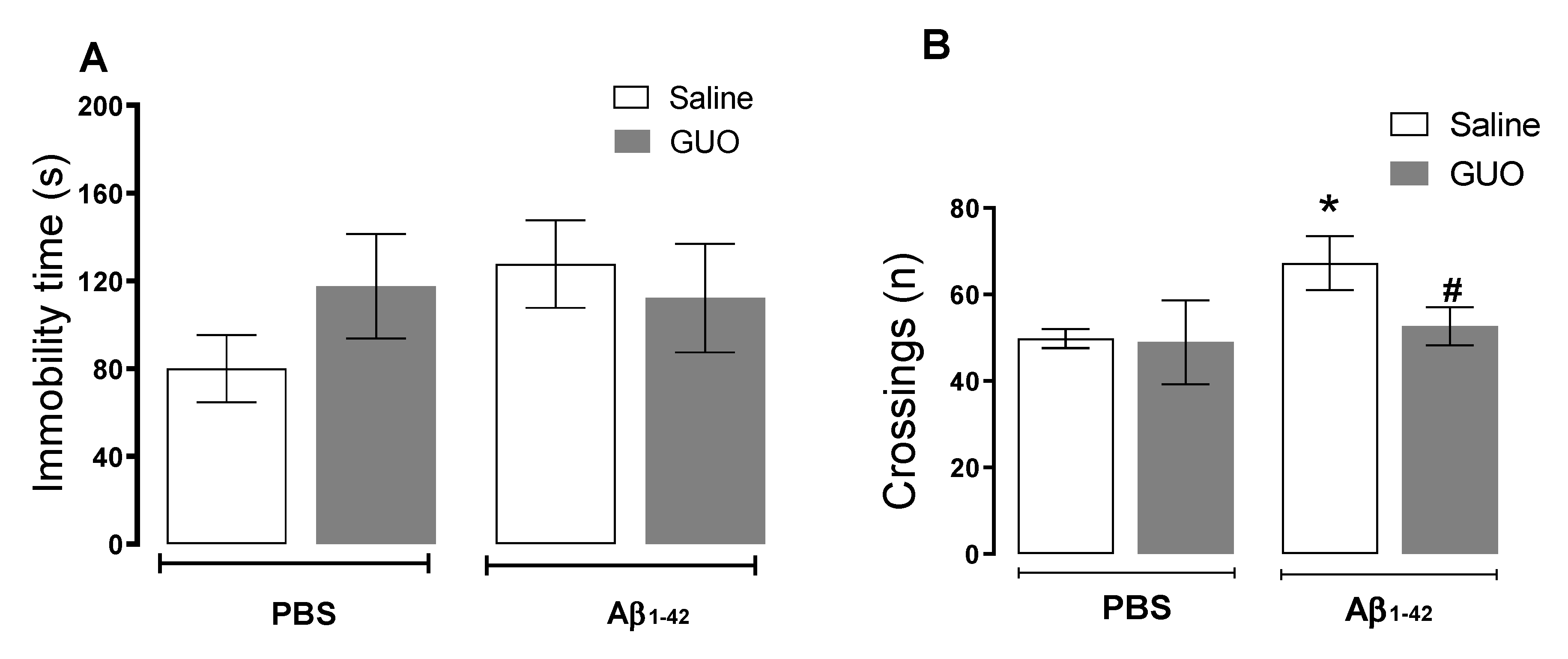
2.6. Tail Suspension Test (TST)
2.7. Open-Field Test (OFT)
2.8. Sucrose Splash Test (SST)
2.9. Object Location Task (OLT)
2.10. Biochemistry Analysis
2.10.1. Preparation of Brain Slices
2.10.2. Cellular Viability Evaluation
2.10.3. Propidium Iodide Incorporation
2.10.4. Reactive Oxygen Species (ROS) Generation
2.10.5. Mitochondrial Membrane Potential (ΔΨm) Measurement
2.10.6. L-[3H]Glutamate Release
2.10.7. Protein Measurement
2.11. Statistical Analysis
3. Results
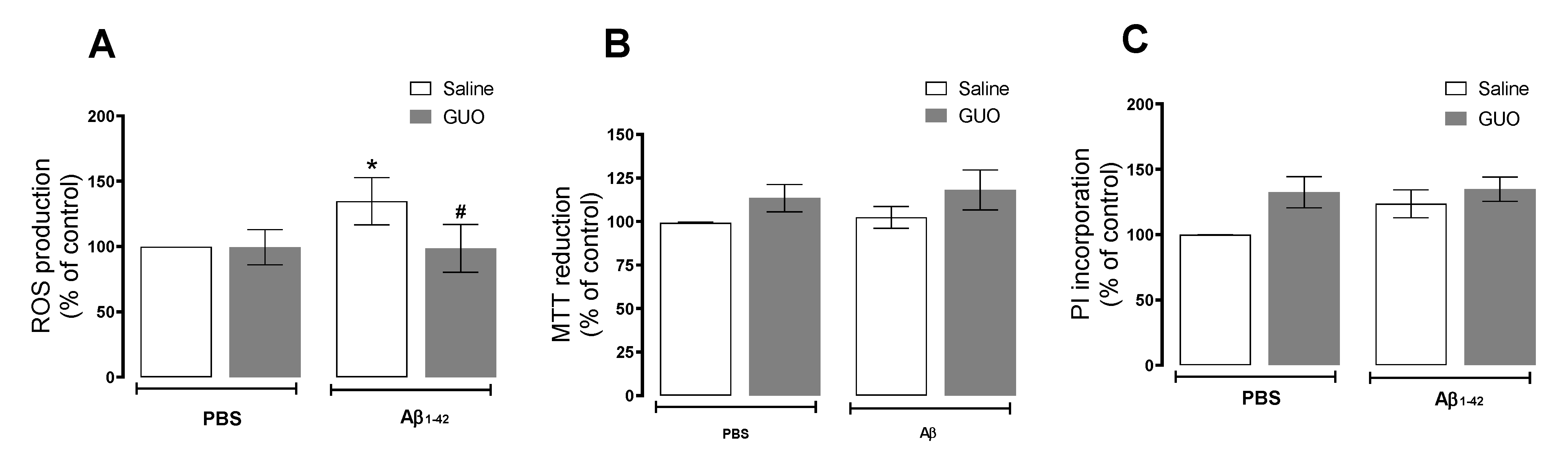
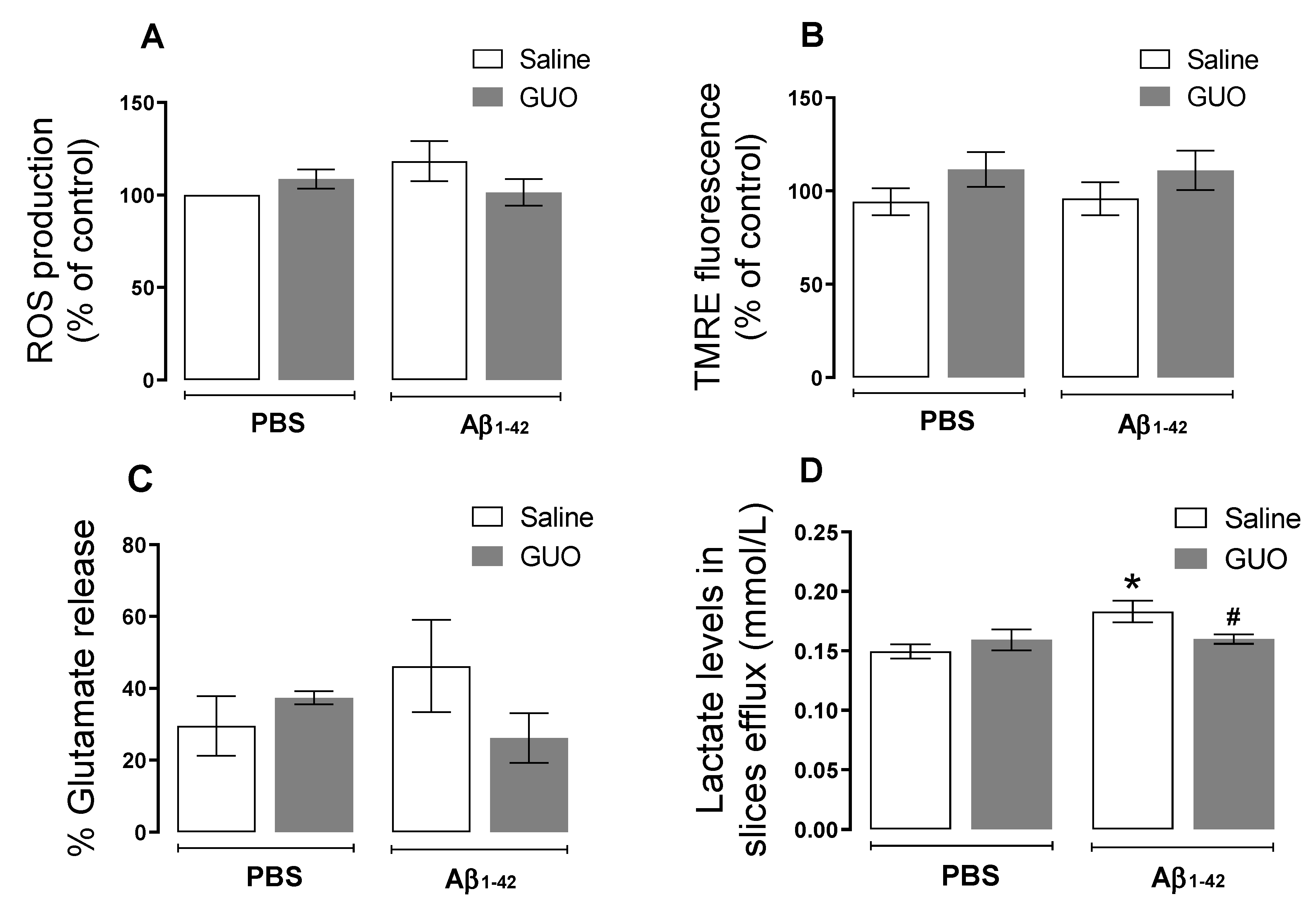
3.1. Guanosine Prevents Aβ1–42-Induced ROS Production
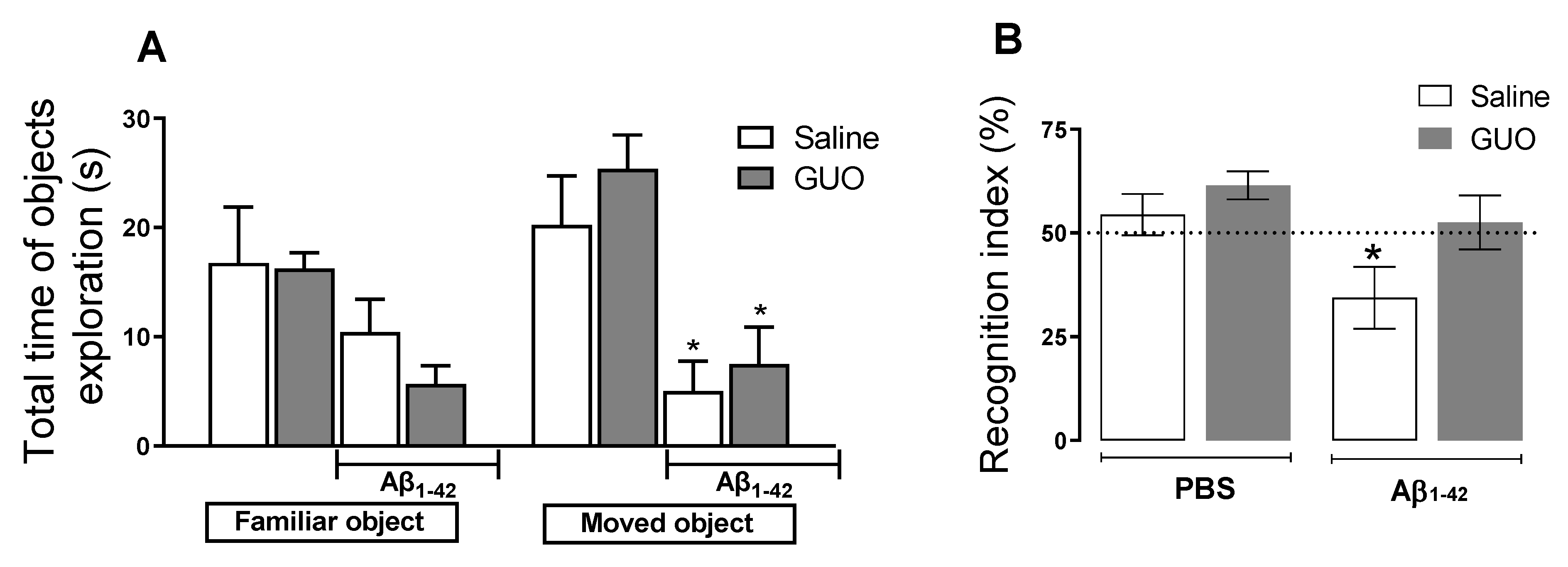
3.2. Guanosine Prevents Aβ1–42-Induced Short-Term Spatial Memory Impairment
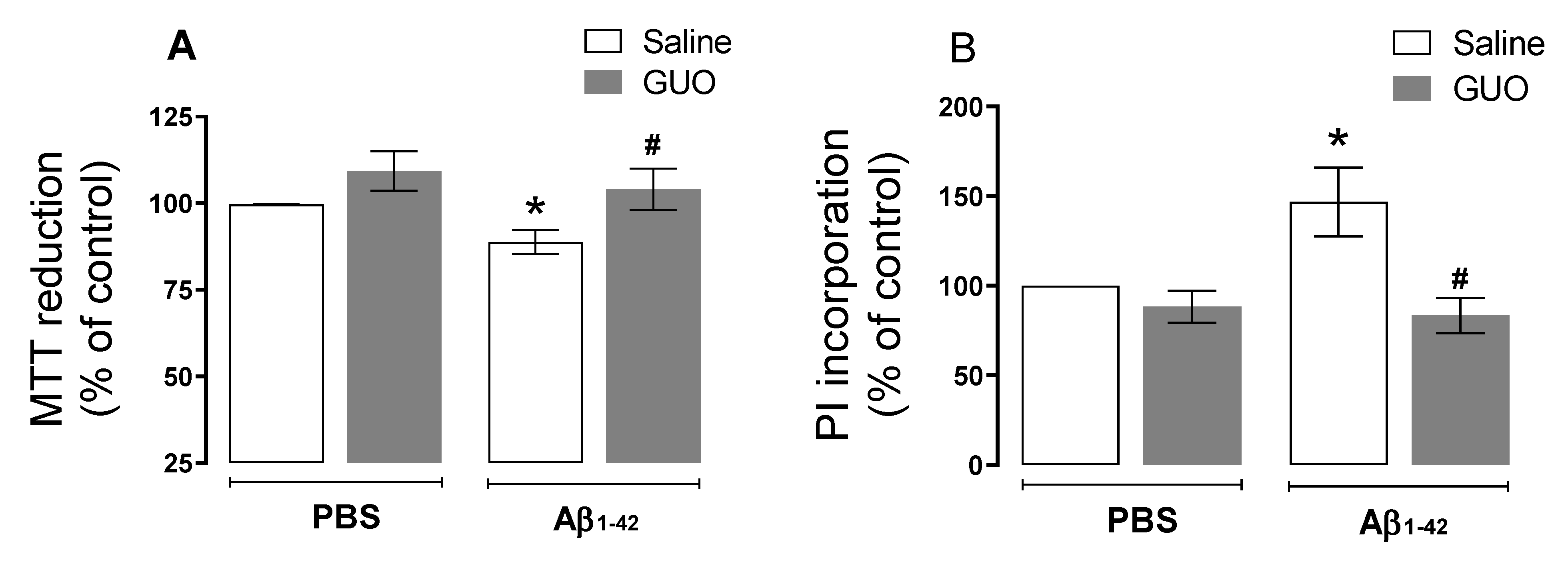
3.3. Guanosine Prevents Aβ1–42-Induced Hippocampal Slice Damage
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Karran, E.; de Strooper, B. The Amyloid Cascade Hypothesis: Are We Poised for Success or Failure? J. Neurochem. 2016, 139, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s Disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble Protein Oligomers in Neurodegeneration: Lessons from the Alzheimer’s Amyloid β-Peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Mancini, G.; Dias, C.; Lourenço, C.F.; Laranjinha, J.; de Bem, A.; Ledo, A. A High Fat/Cholesterol Diet Recapitulates Some Alzheimer’s Disease-Like Features in Mice: Focus on Hippocampal Mitochondrial Dysfunction. J. Alzheimer’s Dis. 2021, 82, 1619–1633. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Sato, N.; Niisato, K.; Takeuchi, D.; Kurinami, H.; Shinohara, M.; Rakugi, H.; Kano, M.; Morishita, R. Validation of Aβ1-40 Administration into Mouse Cerebroventricles as an Animal Model for Alzheimer Disease. Brain Res. 2009, 1280, 137–147. [Google Scholar] [CrossRef]
- Lanznaster, D.; Mack, J.M.; Coelho, V.; Ganzella, M.; Almeida, R.F.; Dal-Cim, T.; Hansel, G.; Zimmer, E.R.; Souza, D.O.; Prediger, R.D.; et al. Guanosine Prevents Anhedonic-Like Behavior and Impairment in Hippocampal Glutamate Transport Following Amyloid-Β1–40 Administration in Mice. Mol. Neurobiol. 2017, 54, 5482–5496. [Google Scholar] [CrossRef]
- Ludka, F.K.; Cunha, M.P.; Dal-Cim, T.; Binder, L.B.; Constantino, L.C.; Massari, C.M.; Martins, W.C.; Rodrigues, A.L.S.; Tasca, C.I. Atorvastatin Protects from Aβ1–40-Induced Cell Damage and Depressive-Like Behavior via ProBDNF Cleavage. Mol. Neurobiol. 2017, 54, 6163–6173. [Google Scholar] [CrossRef]
- Rosa, J.M.; Pazini, F.L.; Cunha, M.P.; Colla, A.R.S.; Manosso, L.M.; Mancini, G.; Souza, A.C.G.; de Bem, A.F.; Prediger, R.D.; Rodrigues, A.L.S. Antidepressant Effects of Creatine on Amyloid β1–40-Treated Mice: The Role of GSK-3β/Nrf2 Pathway. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 86, 270–278. [Google Scholar] [CrossRef]
- Mancini, G.; Martins, W.C.; de Oliveira, J.; de Bem, A.F.; Tasca, C.I. Atorvastatin Improves Mitochondrial Function and Prevents Oxidative Stress in Hippocampus Following Amyloid-Β1–40 Intracerebroventricular Administration in Mice. Mol. Neurobiol. 2020, 57, 4187–4201. [Google Scholar] [CrossRef]
- Piermartiri, T.C.B.; Figueiredo, C.P.; Rial, D.; Duarte, F.S.; Bezerra, S.C.; Mancini, G.; de Bem, A.F.; Prediger, R.D.S.; Tasca, C.I. Atorvastatin Prevents Hippocampal Cell Death, Neuroinflammation and Oxidative Stress Following Amyloid-Β1-40 Administration in Mice: Evidence for Dissociation between Cognitive Deficits and Neuronal Damage. Exp. Neurol. 2010, 226, 274–284. [Google Scholar] [CrossRef]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A Systemic View of Alzheimer Disease—Insights from Amyloid-β Metabolism beyond the Brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Lanznaster, D.; Dal-Cim, T.; Piermartiri, T.C.B.; Tasca, C.I. Guanosine: A Neuromodulator with Therapeutic Potential in Brain Disorders. Aging Dis. 2016, 7, 657–679. [Google Scholar] [CrossRef] [PubMed]
- Bettio, L.E.B.; Gil-Mohapel, J.; Rodrigues, A.L.S. Guanosine and Its Role in Neuropathologies. Purinergic Signal. 2016, 12, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Tasca, C.I.; Lanznaster, D.; Oliveira, K.A.; Fernández-Dueñas, V.; Ciruela, F. Neuromodulatory Effects of Guanine-Based Purines in Health and Disease. Front. Cell. Neurosci. 2018, 12, 376. [Google Scholar] [CrossRef]
- Dos Santos-Rodrigues, A.; Grañé-Boladeras, N.; Bicket, A.; Coe, I.R. Nucleoside Transporters in the Purinome. Neurochem. Int. 2014, 73, 229–237. [Google Scholar] [CrossRef]
- Zimmermann, H.; Braun, N. Extracellular Metabolism of Nucleotides in the Nervous System. J. Auton. Pharmacol. 1996, 16, 397–400. [Google Scholar] [CrossRef]
- Ciccarelli, R.; Ballerini, P.; Sabatino, G.; Rathbone, M.P.; D’Onofrio, M.; Caciagli, F.; di Iorio, P. Involvement of Astrocytes in Purine-Mediated Reparative Processes in the Brain. Int. J. Dev. Neurosci. 2001, 19, 395–414. [Google Scholar] [CrossRef]
- Schmidt, A.P.; Böhmer, A.E.; Schallenberger, C.; Antunes, C.; Tavares, R.G.; Wofchuk, S.T.; Elisabetsky, E.; Souza, D.O. Mechanisms Involved in the Antinociception Induced by Systemic Administration of Guanosine in Mice. Br. J. Pharmacol. 2010, 159, 1247–1263. [Google Scholar] [CrossRef]
- Dal-Cim, T.; Poluceno, G.G.; Lanznaster, D.; de Oliveira, K.A.; Nedel, C.B.; Tasca, C.I. Guanosine Prevents Oxidative Damage and Glutamate Uptake Impairment Induced by Oxygen/Glucose Deprivation in Cortical Astrocyte Cultures: Involvement of A 1 and A 2A Adenosine Receptors and PI3K, MEK and PKC Pathways. Purinergic Signal. 2019, 15, 465–476. [Google Scholar] [CrossRef]
- Thomaz, D.T.; Andreguetti, R.R.; Binder, L.B.; da Luz Scheffer, D.; Corrêa, A.W.; Silva, F.R.M.B.; Tasca, C.I. Guanosine Neuroprotective Action in Hippocampal Slices Subjected to Oxygen and Glucose Deprivation Restores ATP Levels, Lactate Release and Glutamate Uptake Impairment: Involvement of Nitric Oxide. Neurochem. Res. 2020, 45, 2217–2229. [Google Scholar] [CrossRef]
- Dobrachinski, F.; Gerbatin, R.R.; Sartori, G.; Golombieski, R.M.; Antoniazzi, A.; Nogueira, C.W.; Royes, L.F.; Fighera, M.R.; Porciúncula, L.O.; Cunha, R.A.; et al. Guanosine Attenuates Behavioral Deficits After Traumatic Brain Injury by Modulation of Adenosinergic Receptors. Mol. Neurobiol. 2019, 56, 3145–3158. [Google Scholar] [CrossRef] [PubMed]
- Courtes, A.A.; Gonçalves, D.F.; Hartmann, D.D.; da Rosa, P.C.; Cassol, G.; Royes, L.F.F.; de Carvalho, N.R.; Soares, F.A.A. Guanosine Protects against Behavioural and Mitochondrial Bioenergetic Alterations after Mild Traumatic Brain Injury. Brain Res. Bull. 2020, 163, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.S.; Nonose, Y.; Rohden, F.; Lukasewicz Ferreira, P.C.; Fontella, F.U.; Rocha, A.; Brochier, A.W.; Apel, R.V.; de Lima, T.M.; Seminotti, B.; et al. Guanosine Neuroprotection of Presynaptic Mitochondrial Calcium Homeostasis in a Mouse Study with Amyloid-β Oligomers. Mol. Neurobiol. 2020, 57, 4790–4809. [Google Scholar] [CrossRef] [PubMed]
- Rathbone, M.; Pilutti, L.; Caciagli, F.; Jiang, S. Neurotrophic Effects of Extracellular Guanosine. Nucleosides. Nucleotides Nucleic Acids 2008, 27, 666–672. [Google Scholar] [CrossRef]
- Chojnowski, K.; Opielka, M.; Nazar, W.; Kowianski, P.; Smolenski, R.T. Neuroprotective Effects of Guanosine in Ischemic Stroke-Small Steps towards Effective Therapy. Int. J. Mol. Sci. 2021, 22, 6898. [Google Scholar] [CrossRef]
- Piermartiri, T.C.B.; dos Santos, B.; Barros-Aragão, F.G.Q.; Prediger, R.D.; Tasca, C.I. Guanosine Promotes Proliferation in Neural Stem Cells from Hippocampus and Neurogenesis in Adult Mice. Mol. Neurobiol. 2020, 57, 3814–3826. [Google Scholar] [CrossRef]
- Steru, L.; Chermat, R.; Thierry, B.; Simon, P. The Tail Suspension Test: A New Method for Screening Antidepressants in Mice. Psychopharmacology 1985, 85, 367–370. [Google Scholar] [CrossRef]
- Mantovani, M.; Pértile, R.; Calixto, J.B.; Santos, A.R.S.; Rodrigues, A.L.S. Melatonin Exerts an Antidepressant-like Effect in the Tail Suspension Test in Mice: Evidence for Involvement of N-Methyl-D-Aspartate Receptors and the L-Arginine-Nitric Oxide Pathway. Neurosci. Lett. 2003, 343, 1–4. [Google Scholar] [CrossRef]
- Rodrigues, A.L.S.; Rocha, J.B.T.; Mello, C.F.; Souza, D.O. Effect of Perinatal Lead Exposure on Rat Behaviour in Open-Field and Two-Wky Avoidance Tasks. Pharmacol. Toxicol. 1996, 79, 150–156. [Google Scholar] [CrossRef]
- Molz, S.; Tharine, D.C.; Decker, H.; Tasca, C.I. GMP Prevents Excitotoxicity Mediated by NMDA Receptor Activation but Not by Reversal Activity of Glutamate Transporters in Rat Hippocampal Slices. Brain Res. 2008, 1231, 113–120. [Google Scholar] [CrossRef]
- Piermartiri, T.C.B.; Vandresen-Filho, S.; de Araújo Herculano, B.; Martins, W.C.; Dal’Agnolo, D.; Stroeh, E.; Carqueja, C.L.; Boeck, C.R.; Tasca, C.I. Atorvastatin Prevents Hippocampal Cell Death Due to Quinolinic Acid-Induced Seizures in Mice by Increasing Akt Phosphorylation and Glutamate Uptake. Neurotox. Res. 2009, 16, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Dal-Cim, T.; Ludka, F.K.; Martins, W.C.; Reginato, C.; Parada, E.; Egea, J.; Lõpez, M.G.; Tasca, C.I. Guanosine Controls Inflammatory Pathways to Afford Neuroprotection of Hippocampal Slices under Oxygen and Glucose Deprivation Conditions. J. Neurochem. 2013, 126, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Molz, S.; Dal-Cim, T.; Budni, J.; Martín-de-Saavedra, M.D.; Egea, J.; Romero, A.; del Barrio, L.; Rodrigues, A.L.S.; López, M.G.; Tasca, C.I. Neuroprotective Effect of Guanosine against Glutamate-Induced Cell Death in Rat Hippocampal Slices Is Mediated by the Phosphatidylinositol-3 Kinase/Akt/Glycogen Synthase Kinase 3β Pathway Activation and Inducible Nitric Oxide Synthase Inhibition. J. Neurosci. Res. 2011, 89, 1400–1408. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Findley, C.A.; Bartke, A.; Hascup, K.N.; Hascup, E.R. Amyloid Beta-Related Alterations to Glutamate Signaling Dynamics During Alzheimer’s Disease Progression. ASN Neuro 2019, 11, 1759091419855541. [Google Scholar] [CrossRef]
- Tait, S.W.G.; Green, D.R. Mitochondria and Cell Signalling. J. Cell Sci. 2012, 125, 807–815. [Google Scholar] [CrossRef]
- Stowe, D.F.; Camara, A.K.S. Mitochondrial Reactive Oxygen Species Production in Excitable Cells: Modulators of Mitochondrial and Cell Function. Antioxid. Redox Signal. 2009, 11, 1373–1414. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative Stress, Dysfunctional Glucose Metabolism and Alzheimer Disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Bicca, M.A.; Figueiredo, C.P.; Piermartiri, T.C.; Meotti, F.C.; Bouzon, Z.L.; Tasca, C.I.; Medeiros, R.; Calixto, J.B. The Selective and Competitive N-Methyl-D-Aspartate Receptor Antagonist, (-)-6-Phosphonomethyl-Deca-Hydroisoquinoline-3-Carboxylic Acid, Prevents Synaptic Toxicity Induced by Amyloid-β in Mice. Neuroscience 2011, 192, 631–641. [Google Scholar] [CrossRef]
- Souza, L.C.; Jesse, C.R.; Antunes, M.S.; Ruff, J.R.; de Oliveira Espinosa, D.; Gomes, N.S.; Donato, F.; Giacomeli, R.; Boeira, S.P. Indoleamine-2,3-Dioxygenase Mediates Neurobehavioral Alterations Induced by an Intracerebroventricular Injection of Amyloid-Β1-42 Peptide in Mice. Brain. Behav. Immun. 2016, 56, 363–377. [Google Scholar] [CrossRef]
- Sonnewald, U.; Westergaard, N.; Schousboe, A. Glutamate Transport and Metabolism in Astrocytes. Glia 1997, 21, 56–63. [Google Scholar] [CrossRef]
- McKenna, M.C.; Sonnewald, U.; Huang, X.; Stevenson, J.; Zielke, H.R. Exogenous Glutamate Concentration Regulates the Metabolic Fate of Glutamate in Astrocytes. J. Neurochem. 1996, 66, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Whitelaw, B.S.; Robinson, M.B. Inhibitors of Glutamate Dehydrogenase Block Sodium-Dependent Glutamate Uptake in Rat Brain Membranes. Front. Endocrinol. 2013, 4, 123. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Magistretti, P.J. Glutamate Uptake into Astrocytes Stimulates Aerobic Glycolysis: A Mechanism Coupling Neuronal Activity to Glucose Utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial Patterns of Neuroimaging Biomarker Change in Individuals from Families with Autosomal Dominant Alzheimer’s Disease: A Longitudinal Study. Lancet. Neurol. 2018, 17, 241–250. [Google Scholar] [CrossRef]
- Lanznaster, D.; Massari, C.M.; Marková, V.; Šimková, T.; Duroux, R.; Jacobson, K.A.; Fernández-Dueñas, V.; Tasca, C.I.; Ciruela, F. Adenosine A1–A2A Receptor-Receptor Interaction: Contribution to Guanosine-Mediated Effects. Cells 2019, 8, 1630. [Google Scholar] [CrossRef]
- Dal-Cim, T.; Martins, W.C.; Santos, A.R.S.; Tasca, C.I. Guanosine Is Neuroprotective against Oxygen/Glucose Deprivation in Hippocampal Slices via Large Conductance Ca2+-Activated K+ Channels, Phosphatidilinositol-3 Kinase/Protein Kinase B Pathway Activation and Glutamate Uptake. Neuroscience 2011, 183, 212–220. [Google Scholar] [CrossRef]
- Vogel-Ciernia, A.; Wood, M.A. Examining Object Location and Object Recognition Memory in Mice. Curr. Protoc. Neurosci. 2014, 69, 8.31.1–8.31.17. [Google Scholar] [CrossRef]
- Denninger, J.K.; Smith, B.M.; Kirby, E.D. Novel Object Recognition and Object Location Behavioral Testing in Mice on a Budget. J. Vis. Exp. 2018, 2018, e58593. [Google Scholar] [CrossRef]
- Tarozzi, A.; Bartolini, M.; Piazzi, L.; Valgimigli, L.; Amorati, R.; Bolondi, C.; Djemil, A.; Mancini, F.; Andrisano, V.; Rampa, A. From the Dual Function Lead AP2238 to AP2469, a Multi-Target-Directed Ligand for the Treatment of Alzheimer’s Disease. Pharmacol. Res. Perspect. 2014, 2, 23. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, V.; Binder, L.B.; Marques, N.F.; Constantino, L.C.; Mancini, G.; Tasca, C.I. Guanosine Prevents Spatial Memory Impairment and Hippocampal Damage Following Amyloid-β1–42 Administration in Mice. Metabolites 2022, 12, 1207. https://doi.org/10.3390/metabo12121207
Coelho V, Binder LB, Marques NF, Constantino LC, Mancini G, Tasca CI. Guanosine Prevents Spatial Memory Impairment and Hippocampal Damage Following Amyloid-β1–42 Administration in Mice. Metabolites. 2022; 12(12):1207. https://doi.org/10.3390/metabo12121207
Chicago/Turabian StyleCoelho, Victor, Luisa Bandeira Binder, Naiani Ferreira Marques, Leandra Celso Constantino, Gianni Mancini, and Carla Inês Tasca. 2022. "Guanosine Prevents Spatial Memory Impairment and Hippocampal Damage Following Amyloid-β1–42 Administration in Mice" Metabolites 12, no. 12: 1207. https://doi.org/10.3390/metabo12121207
APA StyleCoelho, V., Binder, L. B., Marques, N. F., Constantino, L. C., Mancini, G., & Tasca, C. I. (2022). Guanosine Prevents Spatial Memory Impairment and Hippocampal Damage Following Amyloid-β1–42 Administration in Mice. Metabolites, 12(12), 1207. https://doi.org/10.3390/metabo12121207