
Metabolic Remodeling in Skeletal Muscle Atrophy as a Therapeutic Target

 ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
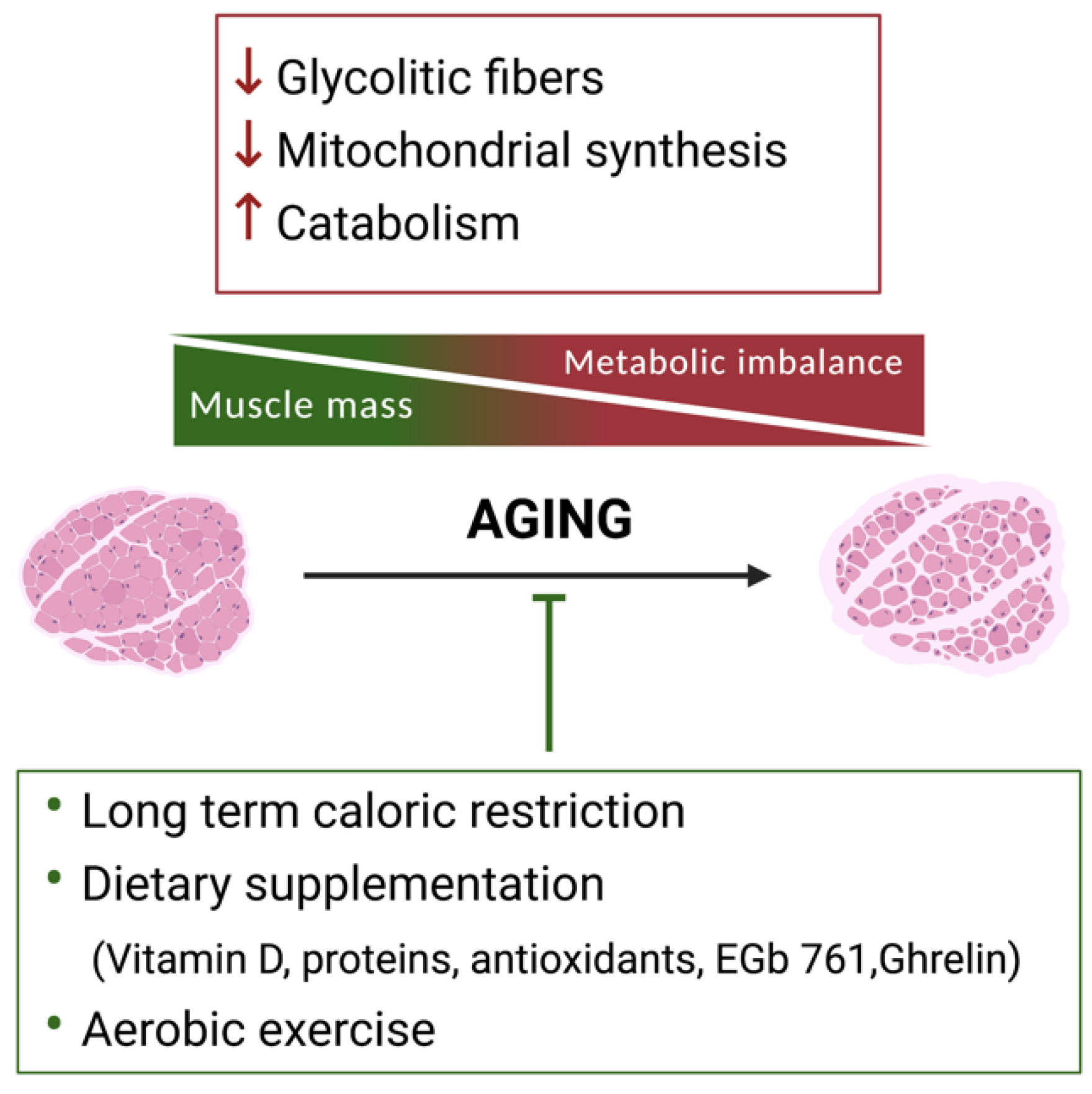
2. Skeletal Muscle Metabolic Reprogramming during Aging
2.1. The Use of Specific Nutrients to Counteract Sarcopenia
2.2. Exercise to Counteract Sarcopenia
3. Metabolic Alteration in Neurodegenerative Diseases
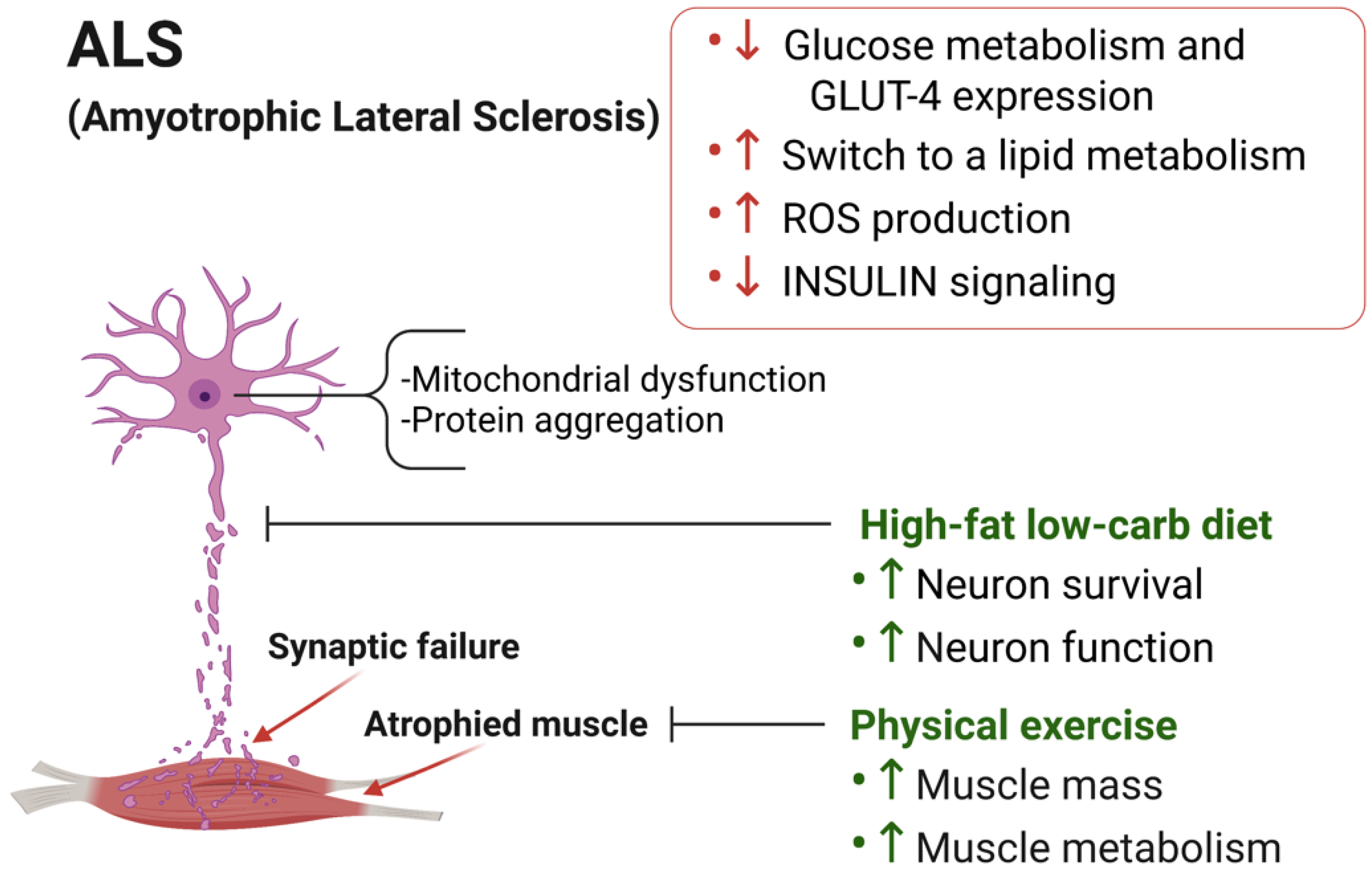
3.1. Skeletal Muscle Metabolic Alterations in ALS
3.2. Dietary Intervention to Counteract ALS Progression
3.3. Physical Exercise in ALS Progression
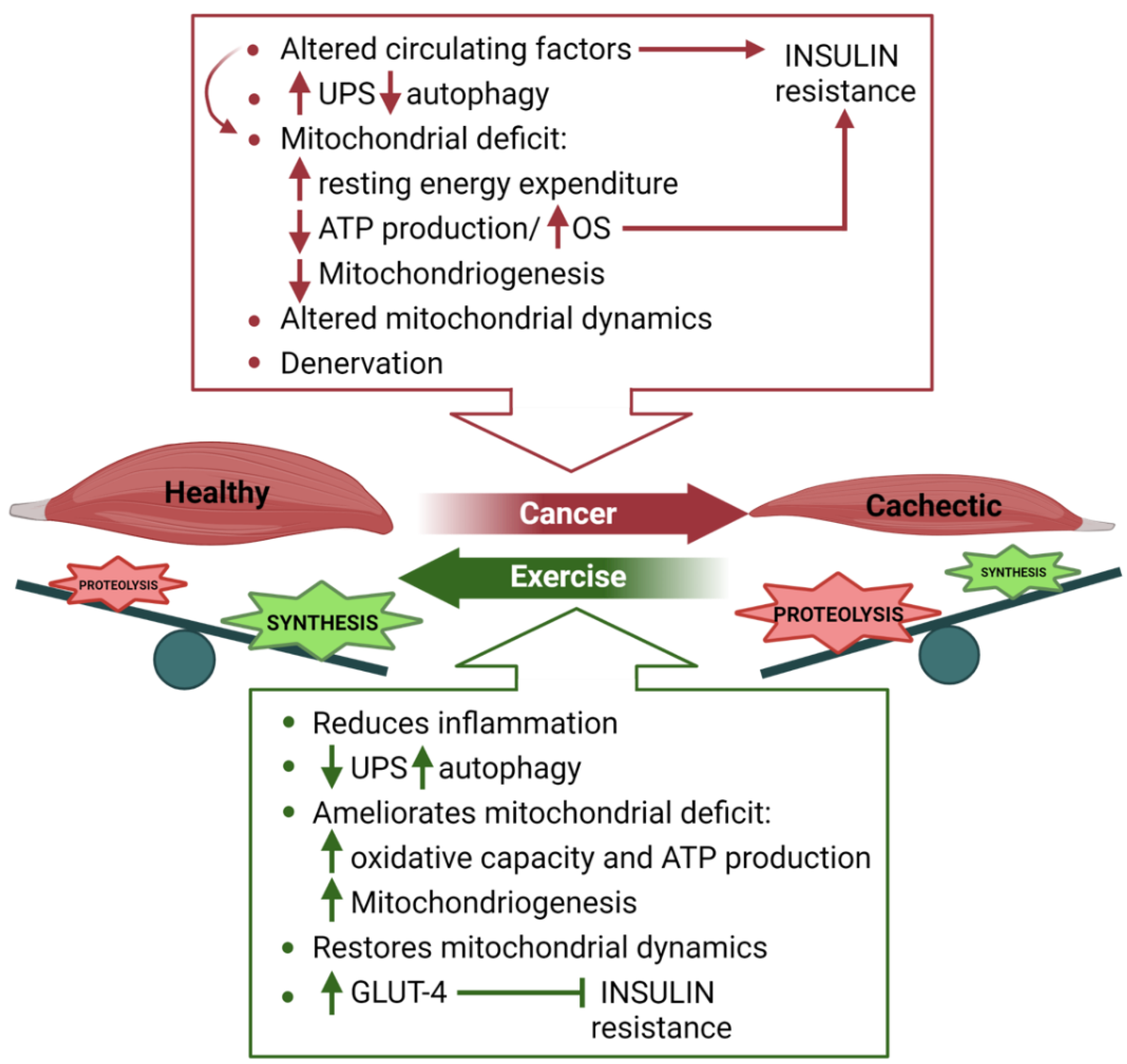
4. Skeletal Muscle Metabolism in Cancer Cachexia
4.1. Nutritional Support to Counteract Cancer Cachexia
4.2. Exercise to Fight Cancer-Induced Cachexia
5. Inter-or Transgenerational Effects of Nutrients and Exercise on Skeletal Muscle Mass
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schiaffino, S.; Reggiani, C. Fiber types in Mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Pette, D.; Staron, R.S. Myosin isoforms, muscle fiber types, and transitions. Microsc. Res. Tech. 2000, 50, 500–509. [Google Scholar] [CrossRef]
- Estévez, A.; Andree, K.; Johnston, I.A. Fast skeletal muscle transcriptome of the gilthead sea bream (Sparus aurata) determined by next generation sequencing. BMC Genom. 2012, 13, 181. [Google Scholar] [CrossRef]
- Zhu, J.; Lu, H.; Xia, B.; Li, Y.; Li, X.; Zhang, Q.; Yang, G. RNA-seq transcriptome analysis of extensor digitorum longus and soleus muscles in large white pigs. Mol. Genet. Genom. 2016, 291, 687–701. [Google Scholar] [CrossRef]
- Ma, J.; Wang, H.; Liu, R.; Jin, L.; Tang, Q.; Wang, X.; Jiang, A.; Hu, Y.; Li, Z.; Zhu, L.; et al. The miRNA Transcriptome Directly Reflects the Physiological and Biochemical Differences between Red, White, and Intermediate Muscle Fiber Types. Int. J. Mol. Sci. 2015, 16, 9635–9653. [Google Scholar] [CrossRef]
- Johnson, M.; Polgar, J.; Weightman, D.; Appleton, D. Data on the distribution of fibre types in thirty-six human muscles. An autopsy study. J. Neurol. Sci. 1973, 18, 111–129. [Google Scholar] [CrossRef]
- Ciciliot, S.; Rossi, A.C.; Dyar, K.A.; Blaauw, B.; Schiaffino, S. Muscle type and fiber type specificity in muscle wasting. Int. J. Biochem. Cell Biol. 2013, 45, 2191–2199. [Google Scholar] [CrossRef]
- Wang, Y.; Pessin, J.E. Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.P.; Wade, C.E. Metabolic consequences of muscle disuse atrophy. J. Nutr. 2005, 135, 1824S–1828S. [Google Scholar] [CrossRef]
- Peggion, C.; Massimino, M.L.; Biancotto, G.; Angeletti, R.; Reggiani, C.; Sorgato, M.C.; Bertoli, A.; Stella, R. Absolute quantification of myosin heavy chain isoforms by selected reaction monitoring can underscore skeletal muscle changes in a mouse model of amyotrophic lateral sclerosis. Anal. Bioanal. Chem. 2017, 409, 2143–2153. [Google Scholar] [CrossRef] [PubMed]
- Scaricamazza, S.; Salvatori, I.; Giacovazzo, G.; Loeffler, J.P.; Renè, F.; Rosina, M.; Quessada, C.; Proietti, D.; Heil, C.; Rossi, S.; et al. Skeletal-Muscle Metabolic Reprogramming in ALS-SOD1G93A Mice Predates Disease Onset and Is A Promising Therapeutic Target. iScience 2020, 23, 101087. [Google Scholar] [CrossRef]
- Ucci, S.; Renzini, A.; Russi, V.; Mangialardo, C.; Cammarata, I.; Cavioli, G.; Santaguida, M.G.; Virili, C.; Centanni, M.; Adamo, S.; et al. Thyroid hormone protects from fasting-induced skeletal muscle atrophy by promoting metabolic adaptation. Int. J. Mol. Sci. 2019, 20, 5754. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Saltin, B. Exercise as medicine—Evidence for prescribing exercise as therapy in 26 different chronic diseases. Scand. J. Med. Sci. Sports 2015, 25, 1–72. [Google Scholar] [CrossRef]
- Haba, Y.; Fujimura, T.; Oyama, K.; Kinoshita, J.; Miyashita, T.; Fushida, S.; Harada, S.; Ohta, T. Effect of Oral Branched-Chain Amino Acids and Glutamine Supplementation on Skeletal Muscle Atrophy After Total Gastrectomy in Rat Model. J. Surg. Res. 2019, 243, 281–288. [Google Scholar] [CrossRef]
- Moro, T.; Ebert, S.M.; Adams, C.M.; Rasmussen, B.B. Amino Acid Sensing in Skeletal Muscle. Trends Endocrinol. Metab. 2016, 27, 796–806. [Google Scholar] [CrossRef]
- Sánchez Riera, C.; Lozanoska-ochser, B.; Testa, S.; Fornetti, E.; Bouché, M.; Madaro, L. Muscle diversity, heterogeneity, and gradients: Learning from sarcoglycanopathies. Int. J. Mol. Sci. 2021, 22, 2502. [Google Scholar] [CrossRef]
- Gomes, M.J.; Martinez, P.F.; Pagan, L.U.; Damatto, R.L.; Cezar, M.D.M.; Lima, A.R.R.; Okoshi, K.; Okoshi, M.P. Skeletal muscle aging: Influence of oxidative stress and physical exercise. Oncotarget 2017, 8, 20428–20440. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Leeuwenburgh, C. Skeletal muscle apoptosis, sarcopenia and frailty at old age. Exp. Gerontol. 2006, 41, 1234–1238. [Google Scholar] [CrossRef] [PubMed]
- Hiona, A.; Leeuwenburgh, C. The role of mitochondrial DNA mutations in aging and sarcopenia: Implications for the mitochondrial vicious cycle theory of aging. Exp. Gerontol. 2008, 43, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Blatteis, C.M. Age-dependent changes in temperature regulation—A mini review. Gerontology 2012, 58, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Roubenoff, R. Catabolism of aging: Is it an inflammatory process? Curr. Opin. Clin. Nutr. Metab. Care 2003, 6, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.J.; Yu, L.J. Oxidative stress, molecular inflammation and sarcopenia. Int. J. Mol. Sci. 2010, 11, 1509–1526. [Google Scholar] [CrossRef]
- Brinster, R.L.; Troike, D.E. Requirements for blastocyst development in vitro. J. Anim. Sci. 1979, 49, 26–34. [Google Scholar] [CrossRef]
- Jansen, S.; Pantaleon, M.; Kaye, P.L. Characterization and regulation of monocarboxylate cotransporters Slc16a7 and Slc16a3 in preimplantation mouse embryos. Biol. Reprod. 2008, 79, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.L.; Leese, H.J. Role of glucose in mouse preimplantation embryo development. Mol. Reprod. Dev. 1995, 40, 436–443. [Google Scholar] [CrossRef]
- Peterson, C.M.; Johannsen, D.L.; Ravussin, E. Skeletal muscle mitochondria and aging: A review. J. Aging Res. 2012, 2012, 194821. [Google Scholar] [CrossRef]
- Conley, K.E.; Amara, C.E.; Jubrias, S.A.; Marcinek, D.J. Mitochondrial function, fibre types and ageing: New insights from human muscle in vivo. Exp. Physiol. 2007, 92, 333–339. [Google Scholar] [CrossRef]
- Johnson, M.L.; Robinson, M.M.; Nair, S.K. Skeletal muscle aging and the mitochondrion. Trends Endocrinol. Metab. 2013, 24, 247–256. [Google Scholar] [CrossRef]
- Crupi, A.N.; Nunnelee, J.S.; Taylor, D.J.; Thomas, A.; Vit, P.; Riera, C.E.; Gottlieb, R.A.; Goodridge, H.S. Oxidative muscles have better mitochondrial homeostasis than glycolytic muscles throughout life and maintain mitochondrial function during aging. Aging 2018, 10, 3327–3352. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Sjödin, B.; Karlsson, J. Histochemical and biochemical changes in human skeletal muscle with age in sedentary males, age 22–65 years. Acta Physiol. Scand. 1978, 103, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L. Morphological and functional characteristics of the ageing skeletal muscle in man. A cross-sectional study. Acta Physiol. Scand. Suppl. 1978, 457, 1–36. [Google Scholar] [PubMed]
- Coggan, A.R.; Spina, R.J.; King, D.S.; Rogers, M.A.; Rogers, M.A.; Brown, M.; Nemeth, P.M.; Holloszy, J.O. Histochemical and enzymatic comparison of the gastrocnemius muscle of young and elderly men and women. J. Gerontol. 1992, 47, B71–B76. [Google Scholar] [CrossRef] [PubMed]
- Klitgaard, H.; Mantoni, M.; Schiaffino, S.; Ausoni, S.; Gorza, L.; Laurent-Winter, C.; Schnohr, P.; Saltin, B. Function, morphology and protein expression of ageing skeletal muscle: A cross-sectional study of elderly men with different training backgrounds. Acta Physiol. Scand. 1990, 140, 41–54. [Google Scholar] [CrossRef]
- Lexell, J.; Taylor, C.C.; Sjöström, M. What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J. Neurol. Sci. 1988, 84, 275–294. [Google Scholar] [CrossRef]
- Frontera, W.R.; Hughes, V.A.; Fielding, R.A.; Fiatarone, M.A.; Evans, W.J.; Roubenoff, R. Aging of skeletal muscle: A 12-yr longitudinal study. J. Appl. Physiol. 2000, 88, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.L. Muscle fibre type adaptation in the elderly human muscle. Scand. J. Med. Sci. Sports 2003, 13, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J.; McComas, A.J.; Petito, F. Physiological changes in ageing muscles. J. Neurol. Neurosurg. Psychiatry 1973, 36, 174–182. [Google Scholar] [CrossRef]
- Flück, M.; Hoppeler, H. Molecular basis of skeletal muscle plasticity—from gene to form and function. Rev. Physiol. Biochem. Pharmacol. 2003, 146, 159–216. [Google Scholar] [CrossRef]
- Seene, T.; Kaasik, P.; Riso, E.M. Review on aging, unloading and reloading: Changes in skeletal muscle quantity and quality. Arch. Gerontol. Geriatr. 2012, 54, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Henze, H.; Jung, M.J.; Ahrens, H.E.; Steiner, S.; von Maltzahn, J. Skeletal muscle aging—Stem cells in the spotlight. Mech. Ageing Dev. 2020, 189, 111283. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Finley, L.W.S.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Moussaieff, A.; Kogan, N.M.; Aberdam, D. Concise Review: Energy Metabolites: Key Mediators of the Epigenetic State of Pluripotency. Stem Cells 2015, 33, 2374–2380. [Google Scholar] [CrossRef] [PubMed]
- Ryall, J.G.; Cliff, T.; Dalton, S.; Sartorelli, V. Metabolic Reprogramming of Stem Cell Epigenetics. Cell Stem Cell 2015, 17, 651–662. [Google Scholar] [CrossRef]
- Harvey, A.; Caretti, G.; Moresi, V.; Renzini, A.; Adamo, S. Interplay between Metabolites and the Epigenome in Regulating Embryonic and Adult Stem Cell Potency and Maintenance. Stem Cell Rep. 2019, 13, 573–589. [Google Scholar] [CrossRef]
- Yamakawa, H.; Kusumoto, D.; Hashimoto, H.; Yuasa, S. Stem cell aging in skeletal muscle regeneration and disease. Int. J. Mol. Sci. 2020, 21, 1830. [Google Scholar] [CrossRef]
- Bernet, J.D.; Doles, J.D.; Hall, J.K.; Kelly Tanaka, K.; Carter, T.A.; Olwin, B.B. P38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat. Med. 2014, 20, 265–271. [Google Scholar] [CrossRef]
- Solanas, G.; Peixoto, F.O.; Perdiguero, E.; Jardí, M.; Ruiz-Bonilla, V.; Datta, D.; Symeonidi, A.; Castellanos, A.; Welz, P.-S.; Caballero, J.M.; et al. Aged Stem Cells Reprogram Their Daily Rhythmic Functions to Adapt to Stress. Cell 2017, 170, 678–692.e20. [Google Scholar] [CrossRef]
- Nieuwenhuizen, W.F.; Weenen, H.; Rigby, P.; Hetherington, M.M. Older adults and patients in need of nutritional support: Review of current treatment options and factors influencing nutritional intake. Clin. Nutr. 2010, 29, 160–169. [Google Scholar] [CrossRef]
- Robinson, S.; Cooper, C.; Aihie Sayer, A. Nutrition and sarcopenia: A review of the evidence and implications for preventive strategies. J. Aging Res. 2012, 2012, 510801. [Google Scholar] [CrossRef]
- Sarkar, T.J.; Quarta, M.; Mukherjee, S.; Colville, A.; Paine, P.; Doan, L.; Tran, C.M.; Chu, C.R.; Horvath, S.; Qi, L.S.; et al. Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat. Commun. 2020, 11, 1545. [Google Scholar] [CrossRef]
- Kaiser, M.J.; Bandinelli, S.; Lunenfeld, B. Frailty and the role of nutrition in older people: A review of the current literature. Acta Biomed. 2010, 81 (Suppl. 1), 37–45. [Google Scholar]
- Kupisz-Urbańska, M.; Płudowski, P.; Marcinowska-Suchowierska, E. Vitamin D deficiency in older patients—Problems of sarcopenia, drug interactions, management in deficiency. Nutrients 2021, 13, 1247. [Google Scholar] [CrossRef]
- Abiri, B.; Vafa, M. Vitamin D and Muscle Sarcopenia in Aging. Methods Mol. Biol. 2020, 2138, 29–47. [Google Scholar] [PubMed]
- Rondanelli, M.; Klersy, C.; Terracol, G.; Talluri, J.; Maugeri, R.; Guido, D.; Faliva, M.A.; Solerte, B.S.; Fioravanti, M.; Lukaski, H.; et al. Whey protein, amino acids, and Vitamin D supplementation with physical activity increases fat-free mass and strength, functionality, and quality of life and decreases inflammation in sarcopenic elderly. Am. J. Clin. Nutr. 2016, 103, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Verlaan, S.; Maier, A.B.; Bauer, J.M.; Bautmans, I.; Brandt, K.; Donini, L.M.; Maggio, M.; McMurdo, M.E.T.; Mets, T.; Seal, C.; et al. Sufficient levels of 25-hydroxyvitamin D and protein intake required to increase muscle mass in sarcopenic older adults—The PROVIDE study. Clin. Nutr. 2018, 37, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Wilson, J.M.; Lee, S.R. Dietary implications on mechanisms of sarcopenia: Roles of protein, amino acids and antioxidants. J. Nutr. Biochem. 2010, 21, 1–13. [Google Scholar] [CrossRef]
- Drummond, M.J.; Dickinson, J.M.; Fry, C.S.; Walker, D.K.; Gundermann, D.M.; Reidy, P.T.; Timmerman, K.L.; Markofski, M.M.; Paddon-Jones, D.; Rasmussen, B.B.; et al. Bed rest impairs skeletal muscle amino acid transporter expression, mTORC1 signaling, and protein synthesis in response to essential amino acids in older adults. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1113–E1122. [Google Scholar] [CrossRef] [PubMed]
- Bidon, C.; Lachuer, J.; Molgó, J.; Wierinckx, A.; de la Porte, S.; Pignol, B.; Christen, Y.; Meloni, R.; Koenig, H.; Biguet, N.F.; et al. The extract of Ginkgo biloba EGb 761 reactivates a juvenile profile in the skeletal muscle of sarcopenic rats by transcriptional reprogramming. PLoS ONE 2009, 4, e7998. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Liang, T.; Wang, G.; Li, Z. Ghrelin, A gastrointestinal hormone, regulates energy balance and lipid metabolism. Biosci. Rep. 2018, 38, BSR20181061. [Google Scholar] [CrossRef]
- Seyhanli, E.S.; Lok, U.; Gulacti, U.; Buyukaslan, H.; Atescelik, M.; Yildiz, M.; Onur, M.R.; Goktekin, M.C.; Aydın, S. Assessment of serum and urine ghrelin levels in patients with acute stroke. Int. J. Clin. Exp. Med. 2015, 8, 722–729. [Google Scholar]
- Porporato, P.E.; Filigheddu, N.; Reano, S.; Ferrara, M.; Angelino, E.; Gnocchi, V.F.; Prodam, F.; Ronchi, G.; Fagoonee, S.; Fornaro, M.; et al. Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice. J. Clin. Investig. 2013, 123. [Google Scholar] [CrossRef]
- Amitani, M.; Amitani, H.; Cheng, K.C.; Kairupan, T.S.; Sameshima, N.; Shimoshikiryo, I.; Mizuma, K.; Rokot, N.T.; Nerome, Y.; Owaki, T.; et al. The role of ghrelin and ghrelin signaling in aging. Int. J. Mol. Sci. 2017, 18, 1511. [Google Scholar] [CrossRef]
- Brook, M.S.; Wilkinson, D.J.; Phillips, B.E.; Perez-Schindler, J.; Philp, A.; Smith, K.; Atherton, P.J. Skeletal muscle homeostasis and plasticity in youth and ageing: Impact of nutrition and exercise. Acta Physiol. 2016, 216, 15–41. [Google Scholar] [CrossRef] [PubMed]
- Bowen, T.S.; Schuler, G.; Adams, V. Skeletal muscle wasting in cachexia and sarcopenia: Molecular pathophysiology and impact of exercise training. J. Cachexia Sarcopenia Muscle 2015, 6, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Hornberger, T.A.; Chu, W.K.; Mak, Y.W.; Hsiung, J.W.; Huang, S.A.; Chien, S. The role of phospholipase D and phosphatidic acid in the mechanical activation of mTOR signaling in skeletal muscle. Proc. Natl. Acad. Sci. USA 2006, 103, 4741–4746. [Google Scholar] [CrossRef]
- O’neil, T.K.; Duffy, L.R.; Frey, J.W.; Hornberger, T.A. The role of phosphoinositide 3-kinase and phosphatidic acid in the regulation of mammalian target of rapamycin following eccentric contractions. J. Physiol. 2009, 587, 3691–3701. [Google Scholar] [CrossRef]
- Klossner, S.; Durieux, A.C.; Freyssenet, D.; Flueck, M. Mechano-transduction to muscle protein synthesis is modulated by FAK. Eur. J. Appl. Physiol. 2009, 106, 389–398. [Google Scholar] [CrossRef]
- Jones, A.M.; Carter, H. The effect of endurance training on parameters of aerobic fitness. Sports Med. 2000, 29, 373–386. [Google Scholar] [CrossRef]
- Di Donato, D.M.; West, D.W.D.; Churchward-Venne, T.A.; Breen, L.; Baker, S.K.; Phillips, S.M. Influence of aerobic exercise intensity on myofibrillar and mitochondrial protein synthesis in young men during early and late postexercise recovery. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1025–E1032. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhang, H.; Zeng, Z.; Wu, L.; Zhang, Y.; Guo, Y.; Lv, J.; Wang, C.; Fan, J.; Chen, N. Lifelong Aerobic Exercise Alleviates Sarcopenia by Activating Autophagy and Inhibiting Protein Degradation via the AMPK/PGC-1α Signaling Pathway. Metabolites 2021, 11, 323. [Google Scholar] [CrossRef]
- Rooyackers, O.E.; Adey, D.B.; Ades, P.A.; Nair, K.S. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc. Natl. Acad. Sci. USA 1996, 93, 15364–15369. [Google Scholar] [CrossRef] [PubMed]
- Muddapu, V.R.; Dharshini, S.A.P.; Chakravarthy, V.S.; Gromiha, M.M. Neurodegenerative Diseases—Is Metabolic Deficiency the Root Cause? Front. Neurosci. 2020, 14, 213. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Pradat, P.F.; Ludolph, A.C.; Loeffler, J.P. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 2011, 10, 75–82. [Google Scholar] [CrossRef]
- Joardar, A.; Manzo, E.; Zarnescu, D.C. Metabolic Dysregulation in Amyotrophic Lateral Sclerosis: Challenges and Opportunities. Curr. Genet. Med. Rep. 2017, 5, 108–114. [Google Scholar] [CrossRef]
- Steyn, F.J.; Li, R.; Kirk, S.E.; Tefera, T.W.; Xie, T.Y.; Tracey, T.J.; Kelk, D.; Wimberger, E.; Garton, F.C.; Roberts, L.; et al. Altered skeletal muscle glucose-fatty acid flux in amyotrophic lateral sclerosis. Brain Commun. 2020, 2, fcaa154. [Google Scholar] [CrossRef] [PubMed]
- Palamiuc, L.; Schlagowski, A.; Ngo, S.T.; Vernay, A.; Dirrig-Grosch, S.; Henriques, A.; Boutillier, A.; Zoll, J.; Echaniz-Laguna, A.; Loeffler, J.; et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 2015, 7, 526–546. [Google Scholar] [CrossRef]
- Desseille, C.; Deforges, S.; Biondi, O.; Houdebine, L.; D’Amico, D.; Lamazière, A.; Caradeuc, C.; Bertho, G.; Bruneteau, G.; Weill, L.; et al. Specific physical exercise improves energetic metabolism in the skeletal muscle of amyotrophic-lateral- sclerosis mice. Front. Mol. Neurosci. 2017, 10, 332. [Google Scholar] [CrossRef]
- Smittkamp, S.E.; Morris, J.K.; Bomhoff, G.L.; Chertoff, M.E.; Geiger, P.C.; Stanford, J.A. SOD1-G93A mice exhibit muscle-fiber-type-specific decreases in glucose uptake in the absence of whole-body changes in metabolism. Neurodegener. Dis. 2013, 13, 29–37. [Google Scholar] [CrossRef]
- White, M.F. IRS proteins and the common path to diabetes. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E413–E422. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Boncompagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal Muscle Is a Primary Target of SOD1G93A-Mediated Toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Lepore, E.; Martini, M.; Barberi, L.; Nunn, A.; Scicchitano, B.M.; Musarò, A. Metabolic changes associated with muscle expression of SOD1G93A. Front. Physiol. 2018, 9, 831. [Google Scholar] [CrossRef] [PubMed]
- Begani Provinciali, G.; Pieroni, N.; Bukreeva, I.; Fratini, M.; Massimi, L.; Maugeri, L.; Palermo, F.; Bardelli, F.; Mittone, A.; Bravin, A.; et al. X-ray phase contrast tomography for the investigation of amyotrophic lateral sclerosis. J. Synchrotron Radiat. 2020, 27, 1042–1048. [Google Scholar] [CrossRef]
- Tefera, T.W.; Steyn, F.J.; Ngo, S.T.; Borges, K. CNS glucose metabolism in Amyotrophic Lateral Sclerosis: A therapeutic target? Cell Biosci. 2021, 11, 14. [Google Scholar] [CrossRef]
- Park, Y.; Park, J.; Kim, Y.; Baek, H.; Kim, S.H. Association between nutritional status and disease severity using the amyotrophic lateral sclerosis (ALS) functional rating scale in ALS patients. Nutrition 2015, 31, 1362–1367. [Google Scholar] [CrossRef]
- Paganoni, S.; Deng, J.; Jaffa, M.; Cudkowicz, M.E.; Wills, A.M. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve 2011, 44, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Reich-Slotky, R.; Andrews, J.; Cheng, B.; Buchsbaum, R.; Levy, D.; Kaufmann, P.; Thompson, J.L.P. Body mass index (BMI) as predictor of ALSFRS-R score decline in ALS patients. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 212–216. [Google Scholar] [CrossRef]
- Longinetti, E.; Mariosa, D.; Larsson, H.; Ye, W.; Ingre, C.; Almqvist, C.; Lichtenstein, P.; Piehl, F.; Fang, F. Neurodegenerative and psychiatric diseases among families with amyotrophic lateral sclerosis. Neurology 2017, 89, 578–585. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Sparks, L.M. Metabolic Flexibility in Health and Disease. Cell Metab. 2017, 25, 1027–1036. [Google Scholar] [CrossRef]
- Fergani, A.; Oudart, H.; De Aguilar, J.L.G.; Fricker, B.; René, F.; Hocquette, J.F.; Meininger, V.; Dupuis, L.; Loeffler, J.P. Increased peripheral lipid clearance in an animal model of amyotrophic lateral sclerosis. J. Lipid Res. 2007, 48, 1571–1580. [Google Scholar] [CrossRef]
- Turner, N.; Cooney, G.J.; Kraegen, E.W.; Bruce, C.R. Fatty acid metabolism, energy expenditure and insulin resistance in muscle. J. Endocrinol. 2014, 220, T61–T79. [Google Scholar] [CrossRef] [PubMed]
- Steyn, F.J.; Ioannides, Z.A.; Van Eijk, R.P.A.; Heggie, S.; Thorpe, K.A.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; Van Den Berg, L.H.; et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef]
- Wills, A.M.; Hubbard, J.; Macklin, E.A.; Glass, J.; Tandan, R.; Simpson, E.P.; Brooks, B.; Gelinas, D.; Mitsumoto, H.; Mozaffar, T.; et al. Hypercaloric enteral nutrition in patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet 2014, 383, 2065–2072. [Google Scholar] [CrossRef]
- Veldink, J.H.; Kalmijn, S.; Groeneveld, G.J.; Wunderink, W.; Koster, A.; De Vries, J.H.M.; Van Der Luyt, J.; Wokke, J.H.J.; Van Den Berg, L.H. Intake of polyunsaturated fatty acids and vitamin E reduces the risk of developing amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2007, 78, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.C.; O’Reilly, É.J.; Falcone, G.J.; McCullough, M.L.; Park, Y.; Kolonel, L.N.; Ascherio, A. Dietary ω-3 polyunsaturated fatty acid intake and risk for amyotrophic lateral sclerosis. JAMA Neurol. 2014, 71, 1102–1110. [Google Scholar] [CrossRef]
- Ludolph, A.C.; Dorst, J.; Dreyhaupt, J.; Weishaupt, J.H.; Kassubek, J.; Weiland, U.; Meyer, T.; Petri, S.; Hermann, A.; Emmer, A.; et al. Effect of High-Caloric Nutrition on Survival in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2020, 87, 206–216. [Google Scholar] [CrossRef]
- Zhao, Z.; Sui, Y.; Gao, W.; Cai, B.; Fan, D. Effects of diet on adenosine monophosphate-activated protein kinase activity and disease progression in an amyotrophic lateral sclerosis model. J. Int. Med. Res. 2015, 43, 67–79. [Google Scholar] [CrossRef]
- Coughlan, K.S.; Halang, L.; Woods, I.; Prehn, J.H.M. A high-fat jelly diet restores bioenergetic balance and extends lifespan in the presence of motor dysfunction and lumbar spinal cord motor neuron loss in TDP-43A315T mutant C57BL6/J mice. DMM Dis. Model. Mech. 2016, 9, 1029–1037. [Google Scholar] [CrossRef]
- Pedersen, W.A.; Mattson, M.P. No benefit of dietary restriction on disease onset or progression in amyotrophic lateral sclerosis Cu/Zn-superoxide dismutase mutant mice. Brain Res. 1999, 833, 117–120. [Google Scholar] [CrossRef]
- Gracies, J.M. Pathophysiology of spastic paresis. II: Emergence of muscle overactivity. Muscle Nerve 2005, 31, 552–571. [Google Scholar] [CrossRef]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.L.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; Baik, M.J.; Gurney, M.; Brown, R.H.; Beal, M.F. Metabolic dysfunction in familial, but not sporadic, amyotrophic lateral sclerosis. J. Neurochem. 1998, 71, 281–287. [Google Scholar] [CrossRef]
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar] [CrossRef]
- Zhao, W.; Varghese, M.; Vempati, P.; Dzhun, A.; Cheng, A.; Wang, J.; Lange, D.; Bilski, A.; Faravelli, I.; Pasinetti, G.M. Caprylic Triglyceride as a Novel Therapeutic Approach to Effectively Improve the Performance and Attenuate the Symptoms Due to the Motor Neuron Loss in ALS Disease. PLoS ONE 2012, 7, e49191. [Google Scholar] [CrossRef] [PubMed]
- Romijn, J.A.; Coyle, E.F.; Sidossis, L.S.; Gastaldelli, A.; Horowitz, J.F.; Endert, E.; Wolfe, R.R. Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am. J. Physiol. 1993, 265, E380–E391. [Google Scholar] [CrossRef]
- Van Loon, L.J.; Greenhaff, P.L.; Constantin-Teodosiu, D.; Saris, W.H.; Wagenmakers, A.J. The effects of increasing exercise intensity on muscle fuel utilisation in humans. J. Physiol. 2001, 536, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Memme, J.M.; Erlich, A.T.; Phukan, G.; Hood, D.A. Exercise and mitochondrial health. J. Physiol. 2021, 599, 803–817. [Google Scholar] [CrossRef]
- Kurtzke, J.F. Risk factors in amyotrophic lateral sclerosis. Adv. Neurol. 1991, 56, 245–270. [Google Scholar] [PubMed]
- Strickland, D.; Smith, S.A.; Dolliff, G.; Goldman, L.; Roelofs, R.I. Amyotrophic lateral sclerosis and occupational history: A pilot case- control study. Arch. Neurol. 1996, 53, 730–733. [Google Scholar] [CrossRef]
- Scarmeas, N.; Shih, T.; Stern, Y.; Ottman, R.; Rowland, L.P. Premorbid weight, body mass, and varsity athletics in ALS. Neurology 2002, 59, 773–775. [Google Scholar] [CrossRef]
- Julian, T.H.; Glascow, N.; Barry, A.D.F.; Moll, T.; Harvey, C.; Klimentidis, Y.C.; Newell, M.; Zhang, S.; Snyder, M.P.; Cooper-Knock, J.; et al. Physical exercise is a risk factor for amyotrophic lateral sclerosis: Convergent evidence from Mendelian randomisation, transcriptomics and risk genotypes. EBioMedicine 2021, 68, 103397. [Google Scholar] [CrossRef]
- Veldink, J.H.; Kalmijn, S.; Van Der Hout, A.H.; Lemmink, H.H.; Groeneveld, G.J.; Lummen, C.; Scheffer, H.; Wokke, J.H.J.; Van Den Berg, L.H. SMN genotypes producing less SMN protein increase susceptibility to and severity of sporadic ALS. Neurology 2005, 65, 820–825. [Google Scholar] [CrossRef]
- Arnon, R.; Aharoni, R. Neurogenesis and neuroprotection in the CNS—Fundamental elements in the effect of glatiramer acetate on treatment of autoimmune neurological disorders. Mol. Neurobiol. 2007, 36, 245–253. [Google Scholar] [CrossRef]
- Huisman, M.H.; Seelen, M.; de Jong, S.W.; Dorresteijn, K.R.I.S.; van Doormaal, P.T.C.; van der Kooi, A.J.; de Visser, M.; Jurgen Schelhaas, H.; van den Berg, L.H.; Herman Veldink, J.; et al. Lifetime physical activity and the risk of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.C.; Alves, M.; Nogueira, A.; Evangelista, T.; Carvalho, J.; Coelho, A.; De Carvalho, M.; Sales-Luís, M.L. Can amyotrophic lateral sclerosis patients with respiratory insufficiency exercise? J. Neurol. Sci. 1999, 169, 69–75. [Google Scholar] [CrossRef]
- Drory, V.E.; Goltsman, E.; Goldman Reznik, J.; Mosek, A.; Korczyn, A.D. The value of muscle exercise in patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2001, 191, 133–137. [Google Scholar] [CrossRef]
- Bello-Haas, V.D.; Florence, J.M.; Kloos, A.D.; Scheirbecker, J.; Lopate, G.; Hayes, S.M.; Pioro, E.P.; Mitsumoto, H. A randomized controlled trial of resistance exercise in individuals with ALS. Neurology 2007, 68, 2003–2007. [Google Scholar] [CrossRef] [PubMed]
- Kirkinezos, I.G.; Hernandez, D.; Bradley, W.G.; Moraes, C.T. Regular exercise is beneficial to a mouse model of amyotrophic lateral sclerosis. Ann. Neurol. 2003, 53, 804–807. [Google Scholar] [CrossRef]
- Gerber, Y.N.; Sabourin, J.C.; Hugnot, J.P.; Perrin, F.E. Unlike Physical Exercise, Modified Environment Increases the Lifespan of SOD1G93A Mice However Both Conditions Induce Cellular Changes. PLoS ONE 2012, 7, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Hombrados, L.; Molina-Torres, G.; Galán-Mercant, A.; Sánchez-Guerrero, E.; González-Sánchez, M.; Ruiz-Muñoz, M. Systematic Review of Therapeutic Physical Exercise in Patients with Amyotrophic Lateral Sclerosis over Time. Int. J. Environ. Res. Public Health 2021, 18, 1074. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Porporato, P.E. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 2016, 5, 200. [Google Scholar] [CrossRef] [PubMed]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 330. [Google Scholar] [CrossRef]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. DMM Dis. Model. Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef]
- Cohen, S.; Nathan, J.A.; Goldberg, A.L. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2014, 14, 58–74. [Google Scholar] [CrossRef]
- Costelli, P.; Muscaritoli, M.; Bossola, M.; Penna, F.; Reffo, P.; Bonetto, A.; Busquets, S.; Bonelli, G.; Lopez-Soriano, F.J.; Doglietto, G.B.; et al. IGF-1 is downregulated in experimental cancer cachexia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R674–R683. [Google Scholar] [CrossRef]
- Asp, M.L.; Tian, M.; Wendel, A.A.; Belury, M.A. Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice. Int. J. Cancer 2010, 126, 756–763. [Google Scholar] [CrossRef]
- Rofe, A.M.; Bourgeois, C.S.; Coyle, P.; Taylor, A.; Abdi, E.A. Altered insulin response to glucose in weight-losing cancer patients. Anticancer Res. 1994, 14, 647–650. [Google Scholar] [PubMed]
- Mantovani, G.; Macciò, A.; Mura, L.; Massa, E.; Mudu, M.C.; Mulas, C.; Lusso, M.R.; Madeddu, C.; Dessì, A. Serum levels of leptin and proinflammatory cytokines in patients with advanced-stage cancer at different sites. J. Mol. Med. 2000, 78, 554–561. [Google Scholar] [CrossRef] [PubMed]
- He, W.A.; Calore, F.; Londhe, P.; Canella, A.; Guttridge, D.C.; Croce, C.M. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc. Natl. Acad. Sci. USA 2014, 111, 4525–4529. [Google Scholar] [CrossRef] [PubMed]
- Qiao, F.; Pan, P.; Yan, J.; Sun, J.; Zong, Y.; Wu, Z.; Lu, X.; Chen, N.; Mi, R.; Ma, Y.; et al. Role of tumor-derived extracellular vesicles in cancer progression and their clinical applications (Review). Int. J. Oncol. 2019, 54, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Daou, N.; Hassani, M.; Matos, E.; De Castro, G.S.; Costa, R.G.F.; Seelaender, M.; Moresi, V.; Rocchi, M.; Adamo, S.; Li, Z.; et al. Displaced myonuclei in cancer cachexia suggest altered innervation. Int. J. Mol. Sci. 2020, 21, 1092. [Google Scholar] [CrossRef] [PubMed]
- Boehm, I.; Miller, J.; Wishart, T.M.; Wigmore, S.J.; Skipworth, R.J.E.; Jones, R.A.; Gillingwater, T.H. Neuromuscular junctions are stable in patients with cancer cachexia. J. Clin. Investig. 2020, 130, 1461–1465. [Google Scholar] [CrossRef]
- Fermoselle, C.; García-Arumí, E.; Puig-Vilanova, E.; Andreu, A.L.; Urtreger, A.J.; de Kier Joffé, E.D.B.; Tejedor, A.; Puente-Maestu, L.; Barreiro, E. Mitochondrial dysfunction and therapeutic approaches in respiratory and limb muscles of cancer cachectic mice. Exp. Physiol. 2013, 98, 1349–1365. [Google Scholar] [CrossRef]
- Aria Tzika, A.; Fontes-Oliveira, C.C.; Shestov, A.A.; Constantinou, C.; Psychogios, N.; Righi, V.; Mintzopoulos, D.; Busquets, S.; Lopez-Soriano, F.J.; Milot, S.; et al. Skeletal muscle mitochondrial uncoupling in a murine cancer cachexia model. Int. J. Oncol. 2013, 43, 886–894. [Google Scholar] [CrossRef]
- Antunes, D.; Padrão, A.I.; Maciel, E.; Santinha, D.; Oliveira, P.; Vitorino, R.; Moreira-Gonçalves, D.; Colaço, B.; Pires, M.J.; Nunes, C.; et al. Molecular insights into mitochondrial dysfunction in cancer-related muscle wasting. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Neyroud, D.; Nosacka, R.L.; Judge, A.R.; Hepple, R.T. Colon 26 adenocarcinoma (C26)-induced cancer cachexia impairs skeletal muscle mitochondrial function and content. J. Muscle Res. Cell Motil. 2019, 40, 59–65. [Google Scholar] [CrossRef]
- Barreiro, E.; De La Puente, B.; Busquets, S.; López-Soriano, F.J.; Gea, J.; Argilés, J.M. Both oxidative and nitrosative stress are associated with muscle wasting in tumour-bearing rats. FEBS Lett. 2005, 579, 1646–1652. [Google Scholar] [CrossRef]
- Sullivan-Gunn, M.J.; Campbell-O’Sullivan, S.P.; Tisdale, M.J.; Lewandowski, P.A. Decreased NADPH oxidase expression and antioxidant activity in cachectic skeletal muscle. J. Cachexia Sarcopenia Muscle 2011, 2, 181–188. [Google Scholar] [CrossRef]
- McLean, J.B.; Moylan, J.S.; Andrade, F.H. Mitochondria dysfunction in lung cancer-induced muscle wasting in C2C12 myotubes. Front. Physiol. 2014, 5, 503. [Google Scholar] [CrossRef]
- Cancer Research. Differential Reconstitution of Mitochondrial Respiratory Chain Activity and Plasma Redox State by Cysteine and Ornithine in a Model of Cancer Cachexia. Available online: https://cancerres.aacrjournals.org/content/59/14/3527.long (accessed on 6 May 2021).
- Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Blackwell, T.A.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Hardee, J.P.; Carson, J.A.; Wiggs, M.P.; et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Vanderveen, B.N.; Fix, D.K.; Carson, J.A. Disrupted Skeletal Muscle Mitochondrial Dynamics, Mitophagy, and Biogenesis during Cancer Cachexia: A Role for Inflammation. Oxid. Med. Cell. Longev. 2017, 2017, 3292087. [Google Scholar] [CrossRef]
- Argilés, J.M.; Fontes-Oliveira, C.C.; Toledo, M.; López-Soriano, F.J.; Busquets, S. Cachexia: A problem of energetic inefficiency. J. Cachexia Sarcopenia Muscle 2014, 5, 279–286. [Google Scholar] [CrossRef]
- Sanchís, D.; Busquets, S.; Alvarez, B.; Ricquier, D.; López-Soriano, F.J.; Argilés, J.M. Skeletal muscle UCP2 and UCP3 gene expression in a rat cancer cachexia model. FEBS Lett. 1998, 436, 415–418. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef]
- Unger, R.H. Minireview: Weapons of Lean Body Mass Destruction: The Role of Ectopic Lipids in the Metabolic Syndrome. Endocrinology 2003, 144, 5159–5165. [Google Scholar] [CrossRef]
- Koopman, R.; Van Loon, L.J.C. Aging, exercise, and muscle protein metabolism. J. Appl. Physiol. 2009, 106, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Johns, N.; Hatakeyama, S.; Stephens, N.A.; Degen, M.; Degen, S.; Frieauff, W.; Lambert, C.; Ross, J.A.; Roubenoff, R.; Glass, D.J.; et al. Clinical classification of cancer cachexia: Phenotypic correlates in human skeletal muscle. PLoS ONE 2014, 9, e83618. [Google Scholar] [CrossRef]
- Taskin, S.; Stumpf, V.I.; Bachmann, J.; Weber, C.; Martignoni, M.E.; Friedrich, O. Motor protein function in skeletal abdominal muscle of cachectic cancer patients. J. Cell. Mol. Med. 2014, 18, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Op den Kamp, C.M.; Gosker, H.R.; Lagarde, S.; Tan, D.Y.; Snepvangers, F.J.; Dingemans, A.M.C.; Langen, R.C.J.; Schols, A.M.W.J. Preserved muscle oxidative metabolic phenotype in newly diagnosed non-small cell lung cancer cachexia. J. Cachexia Sarcopenia Muscle 2015, 6, 164–173. [Google Scholar] [CrossRef]
- Aulino, P.; Berardi, E.; Cardillo, V.M.; Rizzuto, E.; Perniconi, B.; Ramina, C.; Padula, F.; Spugnini, E.P.; Baldi, A.; Faiola, F.; et al. Molecular, cellular and physiological characterization of the cancer cachexia-inducing C26 colon carcinoma in mouse. BMC Cancer 2010, 10, 363. [Google Scholar] [CrossRef]
- Stephens, N.A.; Skipworth, R.J.E.; MacDonald, A.J.; Greig, C.A.; Ross, J.A.; Fearon, K.C.H. Intramyocellular lipid droplets increase with progression of cachexia in cancer patients. J. Cachexia Sarcopenia Muscle 2011, 2, 111–117. [Google Scholar] [CrossRef]
- Almasud, A.A.; Giles, K.H.; Miklavcic, J.J.; Martins, K.J.B.; Baracos, V.E.; Putman, C.T.; Guan, L.L.; Mazurak, V.C. Fish oil mitigates myosteatosis and improves chemotherapy efficacy in a preclinical model of colon cancer. PLoS ONE 2017, 12, e0183576. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, D.P.J.; Bakens, M.J.A.M.; Coolsen, M.M.E.; Rensen, S.S.; van Dam, R.M.; Bours, M.J.L.; Weijenberg, M.P.; Dejong, C.H.C.; Olde Damink, S.W.M. Low skeletal muscle radiation attenuation and visceral adiposity are associated with overall survival and surgical site infections in patients with pancreatic cancer. J. Cachexia Sarcopenia Muscle 2017, 8, 317–326. [Google Scholar] [CrossRef]
- Narasimhan, A.; Ghosh, S.; Stretch, C.; Greiner, R.; Bathe, O.F.; Baracos, V.; Damaraju, S. Small RNAome profiling from human skeletal muscle: Novel miRNAs and their targets associated with cancer cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Johns, N.; Stretch, C.; Tan, B.H.L.; Solheim, T.S.; Sørhaug, S.; Stephens, N.A.; Gioulbasanis, I.; Skipworth, R.J.E.; Deans, D.A.C.; Vigano, A.; et al. New genetic signatures associated with cancer cachexia as defined by low skeletal muscle index and weight loss. J. Cachexia Sarcopenia Muscle 2017, 8, 122–130. [Google Scholar] [CrossRef]
- Narasimhan, A.; Greiner, R.; Bathe, O.F.; Baracos, V.; Damaraju, S. Differentially expressed alternatively spliced genes in skeletal muscle from cancer patients with cachexia. J. Cachexia Sarcopenia Muscle 2018, 9, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Von Haehling, S.; Anker, S.D. Cachexia as a major underestimated and unmet medical need: Facts and numbers. J. Cachexia Sarcopenia Muscle 2010, 1, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef]
- Pigna, E.; Berardi, E.; Aulino, P.; Rizzuto, E.; Zampieri, S.; Carraro, U.; Kern, H.; Merigliano, S.; Gruppo, M.; Mericskay, M.; et al. Aerobic Exercise and Pharmacological Treatments Counteract Cachexia by Modulating Autophagy in Colon Cancer. Sci. Rep. 2016, 6, 26991. [Google Scholar] [CrossRef]
- Solheim, T.S.; Laird, B.J.A. Evidence base for multimodal therapy in cachexia. Curr. Opin. Support. Palliat. Care 2012, 6, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, C.; Weekes, C.E. Dietary advice with or without oral nutritional supplements for disease-related malnutrition in adults. Cochrane Database Syst. Rev. 2011. [Google Scholar] [CrossRef] [PubMed]
- Wallengren, O.; Lundholm, K.; Bosaeus, I. Diet energy density and energy intake in palliative care cancer patients. Clin. Nutr. 2005, 24, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Engelen, M.P.K.J.; Safar, A.M.; Bartter, T.; Koeman, F.; Deutz, N.E.P. High anabolic potential of essential amino acid mixtures in advanced nonsmall cell lung cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 1960–1966. [Google Scholar] [CrossRef]
- Fujita, S.; Dreyer, H.C.; Drummond, M.J.; Glynn, E.L.; Cadenas, J.G.; Yoshizawa, F.; Volpi, E.; Rasmussen, B.B. Nutrient signalling in the regulation of human muscle protein synthesis. J. Physiol. 2007, 582, 813–823. [Google Scholar] [CrossRef]
- Den Kamp, C.M.O.; Langen, R.C.; Haegens, A.; Schols, A.M. Muscle atrophy in cachexia: Can dietary protein tip the balance? Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 611–616. [Google Scholar] [CrossRef]
- Arends, J.; Bachmann, P.; Baracos, V.; Barthelemy, N.; Bertz, H.; Bozzetti, F.; Fearon, K.; Hütterer, E.; Isenring, E.; Kaasa, S.; et al. ESPEN guidelines on nutrition in cancer patients. Clin. Nutr. 2017, 36, 11–48. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Cao, D.; Chen, Z.; Chen, B.; Li, J.; Guo, J.; Dong, Q.; Liu, L.; Wei, Q. Red and processed meat consumption and cancer outcomes: Umbrella review. Food Chem. 2021, 356, 129697. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J.; Brehm, N.; Sweeney, R.A. Ketogenic diets in medical oncology: A systematic review with focus on clinical outcomes. Med. Oncol. 2020, 37, 14. [Google Scholar] [CrossRef]
- Ruiz-García, V.; López-Briz, E.; Carbonell-Sanchis, R.; Bort-Martí, S.; Gonzálvez-Perales, J.L. Megestrol acetate for cachexia–anorexia syndrome. A systematic review. J. Cachexia Sarcopenia Muscle 2018, 9, 444–452. [Google Scholar] [CrossRef]
- Esfahani, M.; Sahafi, S.; Derakhshandeh, A.; Moghaddas, A. The anti-wasting effects of L-carnitine supplementation on cancer: Experimental data and clinical studies. Asia Pac. J. Clin. Nutr. 2018, 27, 503–511. [Google Scholar] [PubMed]
- Bar-Sela, G.; Zalman, D.; Semenysty, V.; Ballan, E. The Effects of Dosage-Controlled Cannabis Capsules on Cancer-Related Cachexia and Anorexia Syndrome in Advanced Cancer Patients: Pilot Study. Integr. Cancer Ther. 2019, 18. [Google Scholar] [CrossRef]
- Khatib, M.N.; Gaidhane, A.; Gaidhane, S.; Quazi, Z.S. Ghrelin as a promising therapeutic option for cancer cachexia. Cell. Physiol. Biochem. 2018, 48, 2172–2188. [Google Scholar] [CrossRef] [PubMed]
- Siff, T.; Parajuli, P.; Razzaque, M.S.; Atfi, A. Cancer-Mediated Muscle Cachexia: Etiology and Clinical Management. Trends Endocrinol. Metab. 2021, 32, 382–402. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Ohmori, H.; Luo, Y.; Mori, S.; Miyagawa, Y.; Nukaga, S.; Goto, K.; Fujiwara-Tani, R.; Kishi, S.; Sasaki, T.; et al. Giving combined medium-chain fatty acids and glucose protects against cancer-associated skeletal muscle atrophy. Cancer Sci. 2019, 110, 3391–3399. [Google Scholar] [CrossRef]
- Gorjao, R.; dos Santos, C.M.M.; Serdan, T.D.A.; Diniz, V.L.S.; Alba-Loureiro, T.C.; Cury-Boaventura, M.F.; Hatanaka, E.; Levada-Pires, A.C.; Sato, F.T.; Pithon-Curi, T.C.; et al. New insights on the regulation of cancer cachexia by N-3 polyunsaturated fatty acids. Pharmacol. Ther. 2019, 196, 117–134. [Google Scholar] [CrossRef]
- Yan, Z.; Zhong, Y.; Duan, Y.; Chen, Q.; Li, F. Antioxidant mechanism of tea polyphenols and its impact on health benefits. Anim. Nutr. 2020, 6, 115–123. [Google Scholar] [CrossRef]
- Penna, F.; Camperi, A.; Muscaritoli, M.; Filigheddu, N.; Costelli, P. The role of Vitamin D in cancer cachexia. Curr. Opin. Support. Palliat. Care 2017, 11, 287–292. [Google Scholar] [CrossRef]
- Tisdale, M.J. Mechanism of lipid mobilization associated with cancer cachexia: Interaction between the polyunsaturated fatty acid, eicosapentaenoic acid, and inhibitory guanine nucleotide-regulatory protein. Prostaglandins Leukot. Essent. Fat. Acids 1993, 48, 105–109. [Google Scholar] [CrossRef]
- Anti, M.; Marra, G.; Armelao, F.; Bartoli, G.M.; Ficarelli, R.; Percesepe, A.; De Vitis, I.; Maria, G.; Sofo, L.; Rapaccini, G.L.; et al. Effect of ω-3 fatty acids on rectal mucosal cell proliferation in subjects at risk for colon cancer. Gastroenterology 1992, 103, 883–891. [Google Scholar] [CrossRef]
- Rose, D.P.; Connolly, J.M. Effects of dietary omega-3 fatty acids on human breast cancer growth and metastases in nude mice. J. Natl. Cancer Inst. 1993, 85, 1743–1747. [Google Scholar] [CrossRef]
- La Guardia, M.; Giammanco, S.; Di Majo, D.; Tabacchi, G.; Tripoli, E.; Giammanco, M. Omega 3 Fatty Acids: Biological Activity and Effects on Human Health. Panminerva Med. 2005, 47, 245–257. Available online: https://pubmed.ncbi.nlm.nih.gov/16489323/ (accessed on 9 May 2021). [PubMed]
- Dunbar, B.S.; Bosire, R.V.; Deckelbaum, R.J. Omega 3 and omega 6 fatty acids in human and animal health: An African perspective. Mol. Cell. Endocrinol. 2014, 398, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.D.; Ross, J.A.; Voss, A.C.; Tisdale, M.J.; Fearon, K.C.H. The effect of an oral nutritional supplement enriched with fish oil on weight-loss in patients with pancreatic cancer. Br. J. Cancer 1999, 81, 80–86. [Google Scholar] [CrossRef]
- Read, J.A.; Beale, P.J.; Volker, D.H.; Smith, N.; Childs, A.; Clarke, S.J. Nutrition intervention using an eicosapentaenoic acid (EPA)-containing supplement in patients with advanced colorectal cancer. Effects on nutritional and inflammatory status: A phase II trial. Support. Care Cancer 2007, 15, 301–307. [Google Scholar] [CrossRef]
- Song, M.; Zhang, X.; Meyerhardt, J.A.; Giovannucci, E.L.; Ogino, S.; Fuchs, C.S.; Chan, A.T. Marine ω-3 polyunsaturated fatty acid intake and survival after colorectal cancer diagnosis. Gut 2017, 66, 1790–1796. [Google Scholar] [CrossRef]
- Gogos, C.A.; Ginopoulos, P.; Salsa, B.; Apostolidou, E.; Zoumbos, N.C.; Kalfarentzos, F. Dietary omega-3 polyunsaturated fatty acids plus vitamin E restore immunodeficiency and prolong survival for severely ill patients with generalized malignancy: A randomized control trial. Cancer 1998, 82, 395–402. Available online: https://pubmed.ncbi.nlm.nih.gov/9445198/ (accessed on 9 May 2021). [CrossRef]
- Lipina, C.; Hundal, H.S. Lipid modulation of skeletal muscle mass and function. J. Cachexia Sarcopenia Muscle 2017, 8, 190–201. [Google Scholar] [CrossRef]
- Pinel, A.; Rigaudière, J.P.; Laillet, B.; Pouyet, C.; Malpuech-Brugère, C.; Prip-Buus, C.; Morio, B.; Capel, F. N—3PUFA differentially modulate palmitate-induced lipotoxicity through alterations of its metabolism in C2C12 muscle cells. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2016, 1861, 12–20. [Google Scholar] [CrossRef]
- Smith, G.I.; Julliand, S.; Reeds, D.N.; Sinacore, D.R.; Klein, S.; Mittendorfer, B. Fish oil-derived n-3 PUFA therapy increases muscle mass and function in healthy older adults. Am. J. Clin. Nutr. 2015, 102, 115–122. [Google Scholar] [CrossRef]
- Smith, G.I.; Atherton, P.; Reeds, D.N.; Mohammed, B.S.; Rankin, D.; Rennie, M.J.; Mittendorfer, B. Omega-3 polyunsaturated fatty acids augment the muscle protein anabolic response to hyperinsulinaemia-hyperaminoacidaemia in healthy young and middle-aged men and women. Clin. Sci. 2011, 121, 267–278. [Google Scholar] [CrossRef]
- Baker, E.J.; Miles, E.A.; Burdge, G.C.; Yaqoob, P.; Calder, P.C. Metabolism and functional effects of plant-derived omega-3 fatty acids in humans. Prog. Lipid Res. 2016, 64, 30–56. [Google Scholar] [CrossRef]
- Smith, G.I.; Atherton, P.; Reeds, D.N.; Mohammed, B.S.; Rankin, D.; Rennie, M.J.; Mittendorfer, B. Dietary omega-3 fatty acid supplementation increases the rate of muscle protein synthesis in older adults: A randomized controlled trial. Am. J. Clin. Nutr. 2011, 93, 402–412. [Google Scholar] [CrossRef]
- Hussey, H.J.; Tisdale, M.J. Effect of a cachectic factor on carbohydrate metabolism and attenuation by eicosapentaenoic acid. Br. J. Cancer 1999, 80, 1231–1235. [Google Scholar] [CrossRef][Green Version]
- Wang, H.; Lai, Y.J.; Chan, Y.L.; Li, T.L.; Wu, C.J. Epigallocatechin-3-gallate effectively attenuates skeletal muscle atrophy caused by cancer cachexia. Cancer Lett. 2011, 305, 40–49. [Google Scholar] [CrossRef]
- Davis, J.M.; Murphy, E.A.; Carmichael, M.D.; Davis, B. Quercetin increases brain and muscle mitochondrial biogenesis and exercise tolerance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R1071–R1077. [Google Scholar] [CrossRef] [PubMed]
- Alway, S.E.; McCrory, J.L.; Kearcher, K.; Vickers, A.; Frear, B.; Gilleland, D.L.; Bonner, D.E.; Thomas, J.M.; Donley, D.A.; Lively, M.W.; et al. Resveratrol Enhances Exercise-Induced Cellular and Functional Adaptations of Skeletal Muscle in Older Men and Women. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2017, 72, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Murphy, E.A.; Carmichael, M.D. Effects of the dietary flavonoid quercetin upon performance and health. Curr. Sports Med. Rep. 2009, 8, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Aquila, G.; Re Cecconi, A.D.; Brault, J.J.; Corli, O.; Piccirillo, R. Nutraceuticals and Exercise against Muscle Wasting during Cancer Cachexia. Cells 2020, 9, 2536. [Google Scholar] [CrossRef] [PubMed]
- Antoun, S.; Raynard, B. Muscle protein anabolism in advanced cancer patients: Response to protein and amino acids support, and to physical activity. Ann. Oncol. 2018, 29, ii10–ii17. [Google Scholar] [CrossRef]
- Kawamura, I.; Sato, H.; Ogoshi, S.; Blackburn, G.L. Use of an intravenous branched chain amino acid enriched diet in the tumor-bearing rat. Jpn. J. Surg. 1985, 15, 471–476. [Google Scholar] [CrossRef]
- Crosby, L.E.; Bistrian, B.R.; Ling, P.R.; Istfan, N.W.; Blackburn, G.L.; Hoffman, S.B. Effects of branched chain amino acid-enriched total parenteral nutrition on amino acid utilization in rats bearing Yoshida sarcoma. Cancer Res. 1988, 48, 2698–2702. Available online: https://pubmed.ncbi.nlm.nih.gov/3129186/ (accessed on 9 May 2021). [PubMed]
- Eley, H.L.; Russell, S.T.; Tisdale, M.J. Effect of branched-chain amino acids on muscle atrophy in cancer cachexia. Biochem. J. 2007, 407, 113–120. [Google Scholar] [CrossRef]
- Penna, F.; Ballarò, R.; Beltrá, M.; De Lucia, S.; Costelli, P. Modulating metabolism to improve cancer-induced muscle wasting. Oxid. Med. Cell. Longev. 2018, 2018, 7153610. [Google Scholar] [CrossRef] [PubMed]
- Girgis, C.M.; Clifton-Bligh, R.J.; Hamrick, M.W.; Holick, M.F.; Gunton, J.E. The roles of vitamin D in skeletal muscle: Form, function, and metabolism. Endocr. Rev. 2013, 34, 33–83. [Google Scholar] [CrossRef]
- Owens, D.J.; Sharples, A.P.; Polydorou, I.; Alwan, N.; Donovan, T.; Tang, J.; Fraser, W.D.; Cooper, R.G.; Morton, J.P.; Stewart, C.; et al. A systems-based investigation into vitamin D and skeletal muscle repair, regeneration, and hypertrophy. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E1019–E1031. [Google Scholar] [CrossRef]
- Von Hurst, P.R.; Stonehouse, W.; Coad, J. Vitamin D supplementation reduces insulin resistance in South Asian women living in New Zealand who are insulin resistant and vitamin D deficient-a randomised, placebo-controlled trial. Br. J. Nutr. 2010, 103, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, P.; John Weisnagel, S.; Caron, A.Z.; Julien, A.S.; Morisset, A.S.; Carreau, A.M.; Poirier, J.; Tchernof, A.; Robitaille, J.; Bergeron, J.; et al. Effects of 6-month Vitamin D supplementation on insulin sensitivity and secretion: A randomised, placebo-controlled trial. Eur. J. Endocrinol. 2019, 181, 287–299. [Google Scholar] [CrossRef]
- Mager, D.R.; Carroll, M.W.; Wine, E.; Siminoski, K.; MacDonald, K.; Kluthe, C.L.; Medvedev, P.; Chen, M.; Wu, J.; Turner, J.M.; et al. Vitamin D status and risk for sarcopenia in youth with inflammatory bowel diseases. Eur. J. Clin. Nutr. 2018, 72, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Snijder, M.B.; Van Schoor, N.M.; Pluijm, S.M.F.; Van Dam, R.M.; Visser, M.; Lips, P. Vitamin D Status in Relation to One-Year Risk of Recurrent Falling in Older Men and Women. J. Clin. Endocrinol. Metab. 2006, 91, 2980–2985. [Google Scholar] [CrossRef]
- Garcia, M.; Seelaender, M.; Sotiropoulos, A.; Coletti, D.; Lancha, A.H. Vitamin D, muscle recovery, sarcopenia, cachexia, and muscle atrophy. Nutrition 2019, 60, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Dev, R.; Del Fabbro, E.; Schwartz, G.G.; Hui, D.; Palla, S.L.; Gutierrez, N.; Bruera, E. Preliminary Report: Vitamin D Deficiency in Advanced Cancer Patients with Symptoms of Fatigue or Anorexia. Oncologist 2011, 16, 1637–1641. [Google Scholar] [CrossRef]
- Sustova, H.; De Feudis, M.; Reano, S.; Alves Teixeira, M.; Valle, I.; Zaggia, I.; Agosti, E.; Prodam, F.; Filigheddu, N. Opposing effects of 25-hydroxy- and 1α,25-dihydroxy-vitamin D3 on pro-cachectic cytokine-and cancer conditioned medium-induced atrophy in C2C12 myotubes. Acta Physiol. 2019, 226, e13269. [Google Scholar] [CrossRef] [PubMed]
- Camperi, A.; Pin, F.; Costamagna, D.; Penna, F.; Menduina, M.L.; Aversa, Z.; Zimmers, T.; Verzaro, R.; Fittipaldi, R.; Caretti, G.; et al. Vitamin D and VDR in cancer cachexia and muscle regeneration. Oncotarget 2017, 8, 21778–21793. [Google Scholar] [CrossRef]
- Ryan, Z.C.; Craig, T.A.; Wang, X.; Delmotte, P.; Salisbury, J.L.; Lanza, I.R.; Sieck, G.C.; Kumar, R. 1α,25-dihydroxyvitamin D3 mitigates cancer cell mediated mitochondrial dysfunction in human skeletal muscle cells. Biochem. Biophys. Res. Commun. 2018, 496, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Padrão, A.I.; Figueira, A.C.C.; Faustino-Rocha, A.I.; Gama, A.; Loureiro, M.M.; Neuparth, M.J.; Moreira-Gonçalves, D.; Vitorino, R.; Amado, F.; Santos, L.L.; et al. Long-term exercise training prevents mammary tumorigenesis-induced muscle wasting in rats through the regulation of TWEAK signalling. Acta Physiol. 2017, 219, 803–813. [Google Scholar] [CrossRef]
- Battaglini, C.L.; Hackney, A.C.; Goodwin, M.L. Cancer cachexia: Muscle physiology and exercise training. Cancers 2012, 4, 1247–1251. [Google Scholar] [CrossRef]
- Eschke, R.C.K.R.; Lampit, A.; Schenk, A.; Javelle, F.; Steindorf, K.; Diel, P.; Bloch, W.; Zimmer, P. Impact of physical exercise on growth and progression of cancer in rodents-a systematic review and meta-analysis. Front. Oncol. 2019, 9, 35. [Google Scholar] [CrossRef]
- Bacuau, R.F.P.; Belmonte, M.A.; Seelaender, M.C.L.; Costa Rosa, L.F.B.P. Effect of a moderate intensity exercise training protocol on the metabolism of macrophages and lymphocytes of tumour-bearing rats. Cell Biochem. Funct. 2000, 18, 249–258. [Google Scholar] [CrossRef]
- Bacurau, A.V.N.; Belmonte, M.A.; Navarro, F.; Moraes, M.R.; Pontes, F.L.; Pesquero, J.L.; Araújo, R.C.; Bacurau, R.F.P. Effect of a high-intensity exercise training on the metabolism and function of macrophages and lymphocytes of walker 256 tumor-bearing rats. Exp. Biol. Med. 2007, 232, 1289–1299. [Google Scholar] [CrossRef]
- Ranjbar, K.I.A.; Ballarò, R.; Bover, Q.; Pin, F.; Beltrà, M.; Penna, F.; Costelli, P. Combined Exercise Training Positively Affects Muscle Wasting in Tumor-Bearing Mice. Med. Sci. Sports Exerc. 2019, 51, 1387–1395. [Google Scholar] [CrossRef]
- Ballarò, R.; Beltrà, M.; De Lucia, S.; Pin, F.; Ranjbar, K.; Hulmi, J.J.; Costelli, P.; Penna, F. Moderate exercise in mice improves cancer plus chemotherapy-induced muscle wasting and mitochondrial alterations. FASEB J. 2019, 33, 5482–5494. [Google Scholar] [CrossRef]
- Aversa, Z.; Pin, F.; Lucia, S.; Penna, F.; Verzaro, R.; Fazi, M.; Colasante, G.; Tirone, A.; Fanelli, F.R.; Ramaccini, C.; et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci. Rep. 2016, 6, 30340. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Narkar, V.A.; Downes, M.; Yu, R.T.; Embler, E.; Wang, Y.X.; Banayo, E.; Mihaylova, M.M.; Nelson, M.C.; Zou, Y.; Juguilon, H.; et al. AMPK and PPARδ Agonists Are Exercise Mimetics. Cell 2008, 134, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C. Caloric restriction and exercise “mimetics” Ready for prime time? Pharmacol. Res. 2016, 103, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Ballarò, R.; Martinez-Cristobal, P.; Sala, D.; Sebastian, D.; Busquets, S.; Muscaritoli, M.; Argilés, J.M.; Costelli, P.; Zorzano, A. Autophagy Exacerbates Muscle Wasting in Cancer Cachexia and Impairs Mitochondrial Function. J. Mol. Biol. 2019, 431, 2674–2686. [Google Scholar] [CrossRef] [PubMed]
- Montalvo, R.N.; Hardee, J.P.; Vanderveen, B.N.; Carson, J.A. Resistance Exercise’s Ability to Reverse Cancer-Induced Anabolic Resistance. Exerc. Sport Sci. Rev. 2018, 46, 247–253. [Google Scholar] [CrossRef]
- Lira, F.S.; Antunes, B.D.M.M.; Seelaender, M.; Neto, J.C.R. The therapeutic potential of exercise to treat cachexia. Curr. Opin. Support. Palliat. Care 2015, 9, 317–324. [Google Scholar] [CrossRef]
- Puppa, M.J.; Murphy, E.A.; Fayad, R.; Hand, G.A.; Carson, J.A. Cachectic skeletal muscle response to a novel bout of low-frequency stimulation. J. Appl. Physiol. 2014, 116, 1078–1087. [Google Scholar] [CrossRef]
- Van der Ende, M.; Grefte, S.; Plas, R.; Meijerink, J.; Witkamp, R.F.; Keijer, J.; van Norren, K. Mitochondrial dynamics in cancer-induced cachexia. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 137–150. [Google Scholar] [CrossRef]
- Halle, J.L.; Counts, B.R.; Carson, J.A. Exercise as a therapy for cancer-induced muscle wasting. Sport. Med. Health Sci. 2020, 2, 186–194. [Google Scholar] [CrossRef]
- Carson, J.A.; Hardee, J.P.; VanderVeen, B.N. The emerging role of skeletal muscle oxidative metabolism as a biological target and cellular regulator of cancer-induced muscle wasting. Semin. Cell Dev. Biol. 2016, 54, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Pin, F.; Busquets, S.; Toledo, M.; Camperi, A.; Lopez-Soriano, F.J.; Costelli, P.; Argilés, J.M.; Penna, F. Combination of exercise training and erythropoietin prevents cancer-induced muscle alterations. Oncotarget 2015, 6, 43202–43215. [Google Scholar] [CrossRef]
- Wang, X.; Pickrell, A.M.; Zimmers, T.A.; Moraes, C.T. Increase in muscle mitochondrial biogenesis does not prevent muscle loss but increased tumor size in a mouse model of acute cancer-induced cachexia. PLoS ONE 2012, 7, 33426. [Google Scholar] [CrossRef]
- Ballarò, R.; Penna, F.; Ferraro, E.; Costelli, P. Muscle mitochondria and oxidative metabolism as targets against cancer cachexia. J. Cancer Metastasis Treat. 2019, 5, 61. [Google Scholar] [CrossRef]
- Kido, K.; Ato, S.; Yokokawa, T.; Makanae, Y.; Sato, K.; Fujita, S. Acute resistance exercise-induced IGF1 expression and subsequent GLUT4 translocation. Physiol. Rep. 2016, 4, e12907. [Google Scholar] [CrossRef]
- Egan, B.; Zierath, J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [PubMed]
- Honors, M.A.; Kinzig, K.P. The role of insulin resistance in the development of muscle wasting during cancer cachexia. J. Cachexia Sarcopenia Muscle 2012, 3, 5–11. [Google Scholar] [CrossRef]
- Buffart, L.M.; Galvão, D.A.; Brug, J.; Chinapaw, M.J.M.; Newton, R.U. Evidence-based physical activity guidelines for cancer survivors: Current guidelines, knowledge gaps and future research directions. Cancer Treat. Rev. 2014, 40, 327–340. [Google Scholar] [CrossRef]
- Fuller, J.T.; Hartland, M.C.; Maloney, L.T.; Davison, K. Therapeutic effects of aerobic and resistance exercises for cancer survivors: A systematic review of meta-analyses of clinical trials. Br. J. Sports Med. 2018, 52, 1311. [Google Scholar] [CrossRef]
- Mustian, K.M.; Sprod, L.K.; Janelsins, M.; Peppone, L.J.; Mohile, S. Exercise Recommendations for Cancer-Related Fatigue, Cognitive Impairment, Sleep problems, Depression, Pain, Anxiety, and Physical Dysfunction: A Review. Oncol. Hematol. Rev. 2012, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Bushman, B.A. Complete Guide to Fitness & Health, 2nd ed.; The American College of Sports Medicine: Indianapolis, IN, USA, 2017. [Google Scholar]
- Schmitz, K.H.; Courneya, K.S.; Matthews, C.; Demark-Wahnefried, W.; Galvão, D.A.; Pinto, B.M.; Irwin, M.L.; Wolin, K.Y.; Segal, R.J.; Lucia, A.; et al. American College of Sports Medicine roundtable on exercise guidelines for cancer survivors. Med. Sci. Sports Exerc. 2010, 42, 1409–1426. [Google Scholar] [CrossRef] [PubMed]
- Wolin, K.Y.; Schwartz, A.L.; Matthews, C.E.; Courneya, K.S.; Schmitz, K.H. Implementing the exercise guidelines for cancer survivors. J. Support. Oncol. 2012, 10, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Pin, F.; Ballarò, R.; Baccino, F.M.; Costelli, P. Novel investigational drugs mimicking exercise for the treatment of cachexia. Expert Opin. Investig. Drugs 2016, 25, 63–72. [Google Scholar] [CrossRef]
- Gatta, L.; Vitiello, L.; Gorini, S.; Chiandotto, S.; Costelli, P.; Giammarioli, A.M.; Malorni, W.; Rosano, G.; Ferraro, E. Modulating the metabolism by trimetazidine enhances myoblast differentiation and promotes myogenesis in cachectic tumor-bearing c26 mice. Oncotarget 2017, 8, 113938–113956. [Google Scholar] [CrossRef]
- De Lima, E.A.; Teixeira, A.A.d.S.; Biondo, L.A.; Diniz, T.A.; Silveira, L.S.; Coletti, D.; Rius, S.B.; Neto, J.C.R. Exercise reduces the resumption of tumor growth and proteolytic pathways in the skeletal muscle of mice following chemotherapy. Cancers 2020, 12, 3466. [Google Scholar] [CrossRef]
- Falcão-Tebas, F.; Bento-Santos, A.; Antônio Fidalgo, M.; De Almeida, M.B.; Dos Santos, J.A.; De Souza, S.L.; Manhães-De-Castro, R.; Leandro, C.G. Maternal low-protein diet-induced delayed reflex ontogeny is attenuated by moderate physical training during gestation in rats. Br. J. Nutr. 2012, 107, 372–377. [Google Scholar] [CrossRef]
- Fidalgo, M.; Falcão-Tebas, F.; Bento-Santos, A.; De Oliveira, E.; Nogueira-Neto, J.F.; De Moura, E.G.; Lisboa, P.C.; De Castro, R.M.; Leandro, C.G. Programmed changes in the adult rat offspring caused by maternal protein restriction during gestation and lactation are attenuated by maternal moderate-low physical training. Br. J. Nutr. 2013, 109, 449–456. [Google Scholar] [CrossRef]
- Bayol, S.A.; Simbi, B.H.; Stickland, N.C. A maternal cafeteria diet during gestation and lactation promotes adiposity and impairs skeletal muscle development and metabolism in rat offspring at weaning. J. Physiol. 2005, 567, 951–961. [Google Scholar] [CrossRef]
- Stanford, K.I.; Lee, M.Y.; Getchell, K.M.; So, K.; Hirshman, M.F.; Goodyear, L.J. Exercise before and during pregnancy prevents the deleterious effects of maternal high-fat feeding on metabolic health of male offspring. Diabetes 2015, 64, 427–433. [Google Scholar] [CrossRef]
- Masuyama, H.; Hiramatsu, Y. Effects of a high-fat diet exposure in utero on the metabolic syndrome-like phenomenon in mouse offspring through epigenetic changes in adipocytokine gene expression. Endocrinology 2012, 153, 2823–2830. [Google Scholar] [CrossRef]
- Fernandez-Twinn, D.S.; Hjort, L.; Novakovic, B.; Ozanne, S.E.; Saffery, R. Intrauterine programming of obesity and type 2 diabetes. Diabetologia 2019, 62, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, K.M.; Reynolds, R.M.; Prescott, S.L.; Nyirenda, M.; Jaddoe, V.W.V.; Eriksson, J.G.; Broekman, B.F.P. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017, 5, 53–64. [Google Scholar] [CrossRef]
- Armitage, J.; Poston, L.; Taylor, P. Developmental origins of obesity and the metabolic syndrome: The role of maternal obesity. Front. Horm. Res. 2007, 36, 73–84. [Google Scholar]
- Prats-Puig, A.; García-Retortillo, S.; Puig-Parnau, M.; Vasileva, F.; Font-Lladó, R.; Xargay-Torrent, S.; Carreras-Badosa, G.; Mas-Parés, B.; Bassols, J.; López-Bermejo, A. DNA Methylation Reorganization of Skeletal Muscle-Specific Genes in Response to Gestational Obesity. Front. Physiol. 2020, 11, 938. [Google Scholar] [CrossRef] [PubMed]
- Houshmand-Oeregaard, A.; Schrölkamp, M.; Kelstrup, L.; Hansen, N.S.; Hjort, L.; Thuesen, A.C.B.; Broholm, C.; Mathiesen, E.R.; Clausen, T.D.; Vaag, A.; et al. Increased expression of microRNA-15a and microRNA-15b in skeletal muscle from adult offspring of women with diabetes in pregnancy. Hum. Mol. Genet. 2018, 27, 1763–1771. [Google Scholar] [CrossRef]
- Laker, R.C.; Wlodek, M.E.; Connelly, J.; Yan, Z. Epigenetic origins of metabolic disease: The impact of the maternal condition to the offspring epigenome and later health consequences. Food Sci. Hum. Wellness 2013, 2, 1–11. [Google Scholar] [CrossRef]
- Simar, D.; Chen, H.; Lambert, K.; Mercier, J.; Morris, M.J. Interaction between maternal obesity and post-natal over-nutrition on skeletal muscle metabolism. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 269–276. [Google Scholar] [CrossRef]
- Beleza, J.; Stevanović-Silva, J.; Coxito, P.; Costa, R.C.; Ascensão, A.; Torrella, J.R.; Magalhães, J. Building-up fit muscles for the future: Transgenerational programming of skeletal muscle through physical exercise. Eur. J. Clin. Investig. 2021, e13515. [Google Scholar] [CrossRef]
- Bayol, S.A.; MacHaria, R.; Farrington, S.J.; Simbi, B.H.; Stickland, N.C. Evidence that a maternal “junk food” diet during pregnancy and lactation can reduce muscle force in offspring. Eur. J. Nutr. 2009, 48, 62–65. [Google Scholar] [CrossRef]
- Walter, I.; Klaus, S. Maternal high-fat diet consumption impairs exercise performance in offspring. J. Nutr. Sci. 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Pileggi, C.A.; Hedges, C.P.; Segovia, S.A.; Markworth, J.F.; Durainayagam, B.R.; Gray, C.; Zhang, X.D.; Barnett, M.P.G.; Vickers, M.H.; Hickey, A.J.R.; et al. Maternal high fat diet alters skeletal muscle mitochondrial catalytic activity in adult male rat offspring. Front. Physiol. 2016, 7, 546. [Google Scholar] [CrossRef] [PubMed]
- Latouche, C.; Heywood, S.E.; Henry, S.L.; Ziemann, M.; Lazarus, R.; El-Osta, A.; Armitage, J.A.; Kingwell, B.A. Maternal overnutrition programs changes in the expression of skeletal muscle genes that are associated with insulin resistance and defects of oxidative phosphorylation in adult male rat offspring. J. Nutr. 2014, 144, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Bodden, C.; Hannan, A.J.; Reichelt, A.C. Diet-Induced Modification of the Sperm Epigenome Programs Metabolism and Behavior. Trends Endocrinol. Metab. 2020, 31, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Patti, M.E. Paternal Nongenetic Intergenerational Transmission of Metabolic Disease Risk. Curr. Diabetes Rep. 2019, 19, 38. [Google Scholar] [CrossRef]
- De Castro Barbosa, T.; Ingerslev, L.R.; Alm, P.S.; Versteyhe, S.; Massart, J.; Rasmussen, M.; Donkin, I.; Sjögren, R.; Mudry, J.M.; Vetterli, L.; et al. High-fat diet reprograms the epigenome of rat spermatozoa and transgenerationally affects metabolism of the offspring. Mol. Metab. 2016, 5, 184–197. [Google Scholar] [CrossRef]
- Kusuyama, J.; Alves-Wagner, A.B.; Makarewicz, N.S.; Goodyear, L.J. Effects of maternal and paternal exercise on offspring metabolism. Nat. Metab. 2020, 2, 858–872. [Google Scholar] [CrossRef]
- Raipuria, M.; Bahari, H.; Morris, M.J. Effects of maternal diet and exercise during pregnancy on glucose metabolism in skeletal muscle and fat of weanling rats. PLoS ONE 2015, 10, e0120980. [Google Scholar] [CrossRef]
- Harris, J.E.; Baer, L.A.; Stanford, K.I. Maternal Exercise Improves the Metabolic Health of Adult Offspring. Trends Endocrinol. Metab. 2018, 29, 164–177. [Google Scholar] [CrossRef]
- Laker, R.C.; Lillard, T.S.; Okutsu, M.; Zhang, M.; Hoehn, K.L.; Connelly, J.J.; Yan, Z. Exercise prevents maternal high-fat diet-induced hypermethylation of the Pgc-1α gene and age-dependent metabolic dysfunction in the offspring. Diabetes 2014, 63, 1605–1611. [Google Scholar] [CrossRef]
- Kasch, J.; Kanzleiter, I.; Saussenthaler, S.; Schürmann, A.; Keijer, J.; van Schothorst, E.; Klaus, S.; Schumann, S. Insulin sensitivity linked skeletal muscle Nr4a1 DNA methylation is programmed by the maternal diet and modulated by voluntary exercise in mice. J. Nutr. Biochem. 2018, 57, 86–92. [Google Scholar] [CrossRef]
- Falcão-Tebas, F.; Marin, E.C.; Kuang, J.; Bishop, D.J.; McConell, G.K. Maternal exercise attenuates the lower skeletal muscle glucose uptake and insulin secretion caused by paternal obesity in female adult rat offspring. J. Physiol. 2020, 598, 4251–4270. [Google Scholar] [CrossRef]
- McPherson, N.O.; Owens, J.A.; Fullston, T.; Lane, M. Preconception diet or exercise intervention in obese fathers normalizes sperm microRNA profile and metabolic syndrome in female offspring. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E805–E821. [Google Scholar] [CrossRef]
- Hargreaves, M.; Spriet, L.L. Skeletal muscle energy metabolism during exercise. Nat. Metab. 2020, 2, 817–828. [Google Scholar] [CrossRef]
- Lundby, C.; Jacobs, R.A. Adaptations of skeletal muscle mitochondria to exercise training. Exp. Physiol. 2016, 101, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Hoppeler, H. Molecular networks in skeletal muscle plasticity. J. Exp. Biol. 2016, 219, 205–213. [Google Scholar] [CrossRef] [PubMed]
- McGee, S.L.; Fairlie, E.; Garnham, A.P.; Hargreaves, M. Exercise-induced histone modifications in human skeletal muscle. J. Physiol. 2009, 587, 5951–5958. [Google Scholar] [CrossRef] [PubMed]
- Ultimo, S.; Zauli, G.; Martelli, A.M.; Vitale, M.; McCubrey, J.A.; Capitani, S.; Neri, L.M. Influence of physical exercise on microRNAs in skeletal muscle regeneration, aging and diseases. Oncotarget 2018, 9, 17220–17237. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.M.; Watt, M.J.; Febbraio, M.A. Metabolic communication during exercise. Nat. Metab. 2020, 2, 805–816. [Google Scholar] [CrossRef]
- Stanford, K.I.; Rasmussen, M.; Baer, L.A.; Lehnig, A.C.; Rowland, L.A.; White, J.D.; So, K.; De Sousa-Coelho, A.L.; Hirshman, M.F.; Patti, M.E.; et al. Paternal exercise improves glucose metabolism in adult offspring. Diabetes 2018, 67, 2530–2540. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, N.; Bae-Gartz, I.; Bauer, C.; Janoschek, R.; Koxholt, I.; Mahabir, E.; Appel, S.; Alejandre Alcazar, M.A.; Grossmann, N.; Vohlen, C.; et al. Exercise during pregnancy and its impact on mothers and offspring in humans and mice. J. Dev. Orig. Health Dis. 2018, 9, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Koletzko, B.; Cremer, M.; Flothkötter, M.; Graf, C.; Hauner, H.; Hellmers, C.; Kersting, M.; Krawinkel, M.; Przyrembel, H.; Röbl-Mathieu, M.; et al. Diet and Lifestyle before and during Pregnancy—Practical Recommendations of the Germany-wide Healthy Start—Young Family Network. Geburtshilfe Frauenheilkd. 2018, 78, 1262–1282. [Google Scholar] [CrossRef] [PubMed]
- McMillan, A.G.; May, L.E.; Gaines, G.G.; Isler, C.; Kuehn, D. Effects of Aerobic Exercise during Pregnancy on 1-Month Infant Neuromotor Skills. Med. Sci. Sports Exerc. 2019, 51, 1671–1676. [Google Scholar] [CrossRef]
- Mourtakos, S.P.; Tambalis, K.D.; Panagiotakos, D.B.; Antonogeorgos, G.; Arnaoutis, G.; Karteroliotis, K.; Sidossis, L.S. Maternal lifestyle characteristics during pregnancy, and the risk of obesity in the offspring: A study of 5,125 children. BMC Pregnancy Childbirth 2015, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Parastesh, M.; Heidarianpour, A.; Sadegh, M. Investigating the effects of endurance, resistance and combined training on reproductive hormones and sperm parameters of streptozotocin–nicotinamide diabetic male rats. J. Diabetes Metab. Disord. 2019, 18, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Messerlian, C.; Sun, Z.H.; Duan, P.; Chen, H.G.; Chen, Y.J.; Wang, P.; Wang, L.; Meng, T.Q.; Wang, Q.; et al. Physical activity and sedentary time in relation to semen quality in healthy men screened as potential sperm donors. Hum. Reprod. 2019, 34, 2330–2339. [Google Scholar] [CrossRef]
- Guth, L.M.; Ludlow, A.T.; Witkowski, S.; Marshall, M.R.; Lima, L.C.J.; Venezia, A.C.; Xiao, T.; Ting Lee, M.L.; Spangenburg, E.E.; Roth, S.M. Sex-specific effects of exercise ancestry on metabolic, morphological and gene expression phenotypes in multiple generations of mouse offspring. Exp. Physiol. 2013, 98, 1469–1484. [Google Scholar] [CrossRef][Green Version]
- Murashov, A.K.; Pak, E.S.; Koury, M.; Ajmera, A.; Jeyakumar, M.; Parker, M.; Williams, O.; Ding, J.; Walters, D.; Neufer, P.D. Paternal long-term exercise programs offspring for low energy expenditure and increased risk for obesity in mice. FASEB J. 2016, 30, 775–784. [Google Scholar] [CrossRef]
- Cedola, A.; Mastrogiacomo, M.; Lagomarsino, S.; Cancedda, R.; Giannini, C.; Guagliardi, A.; Ladisa, M.; Burghammer, M.; Rustichelli, F.; Komlev, V. Orientation of mineral crystals by collagen fibers during in vivo bone engineering: An X-ray diffraction imaging study. Spectrochim. Acta Part B At. Spectrosc. 2007, 62, 642–647. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renzini, A.; Riera, C.S.; Minic, I.; D’Ercole, C.; Lozanoska-Ochser, B.; Cedola, A.; Gigli, G.; Moresi, V.; Madaro, L. Metabolic Remodeling in Skeletal Muscle Atrophy as a Therapeutic Target. Metabolites 2021, 11, 517. https://doi.org/10.3390/metabo11080517
Renzini A, Riera CS, Minic I, D’Ercole C, Lozanoska-Ochser B, Cedola A, Gigli G, Moresi V, Madaro L. Metabolic Remodeling in Skeletal Muscle Atrophy as a Therapeutic Target. Metabolites. 2021; 11(8):517. https://doi.org/10.3390/metabo11080517
Chicago/Turabian StyleRenzini, Alessandra, Carles Sánchez Riera, Isidora Minic, Chiara D’Ercole, Biliana Lozanoska-Ochser, Alessia Cedola, Giuseppe Gigli, Viviana Moresi, and Luca Madaro. 2021. "Metabolic Remodeling in Skeletal Muscle Atrophy as a Therapeutic Target" Metabolites 11, no. 8: 517. https://doi.org/10.3390/metabo11080517
APA StyleRenzini, A., Riera, C. S., Minic, I., D’Ercole, C., Lozanoska-Ochser, B., Cedola, A., Gigli, G., Moresi, V., & Madaro, L. (2021). Metabolic Remodeling in Skeletal Muscle Atrophy as a Therapeutic Target. Metabolites, 11(8), 517. https://doi.org/10.3390/metabo11080517