Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Production
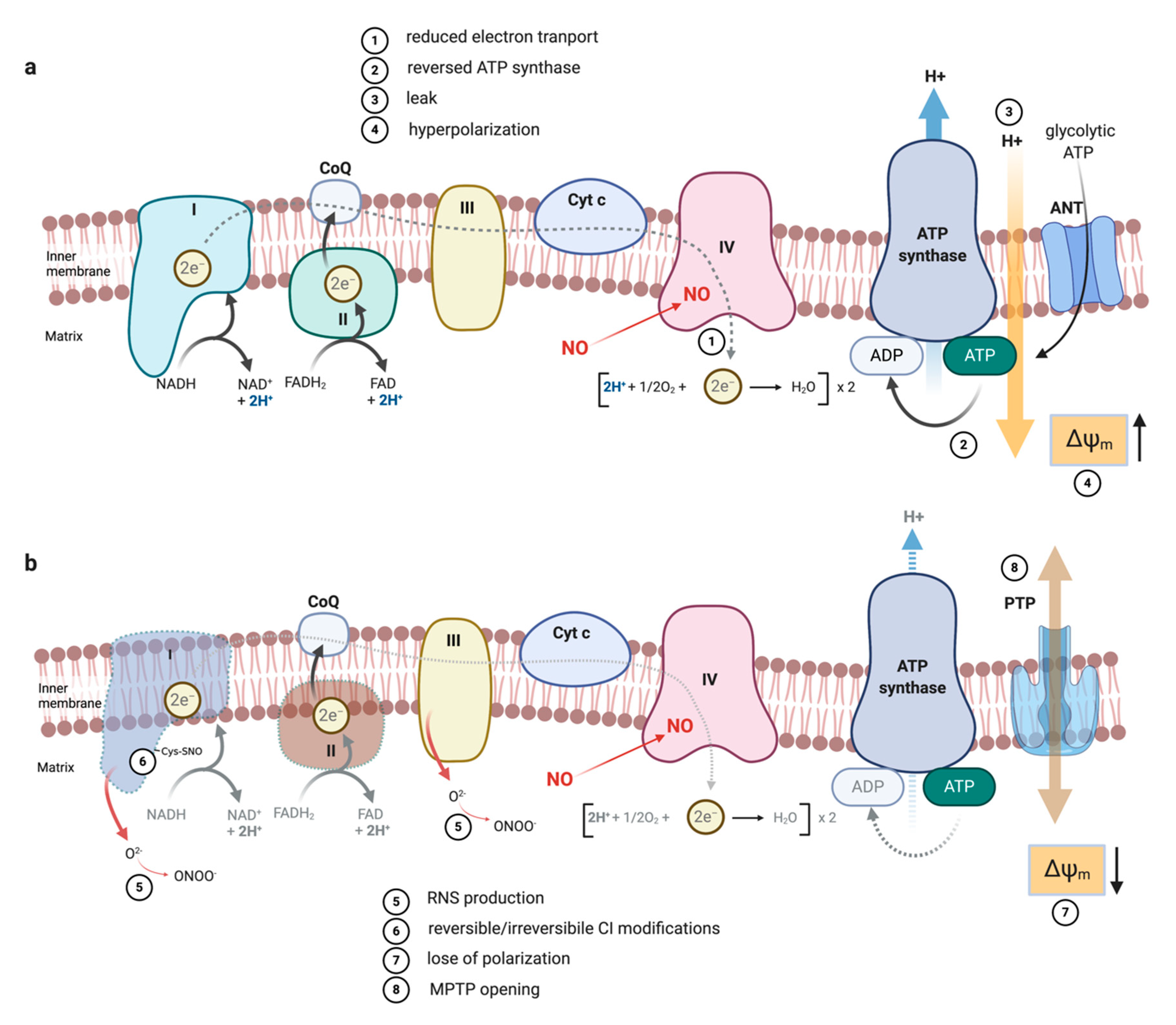
3. NO as Competitive Inhibitor of Complex IV
4. Inhibition of Non-Heme Enzymes
5. Macrophage Immunometabolism
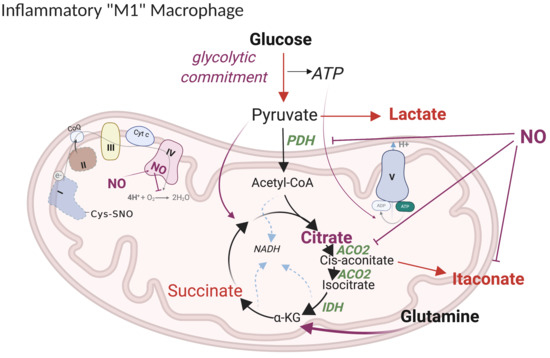
5.1. Current Model of M1 Macrophage Polarization
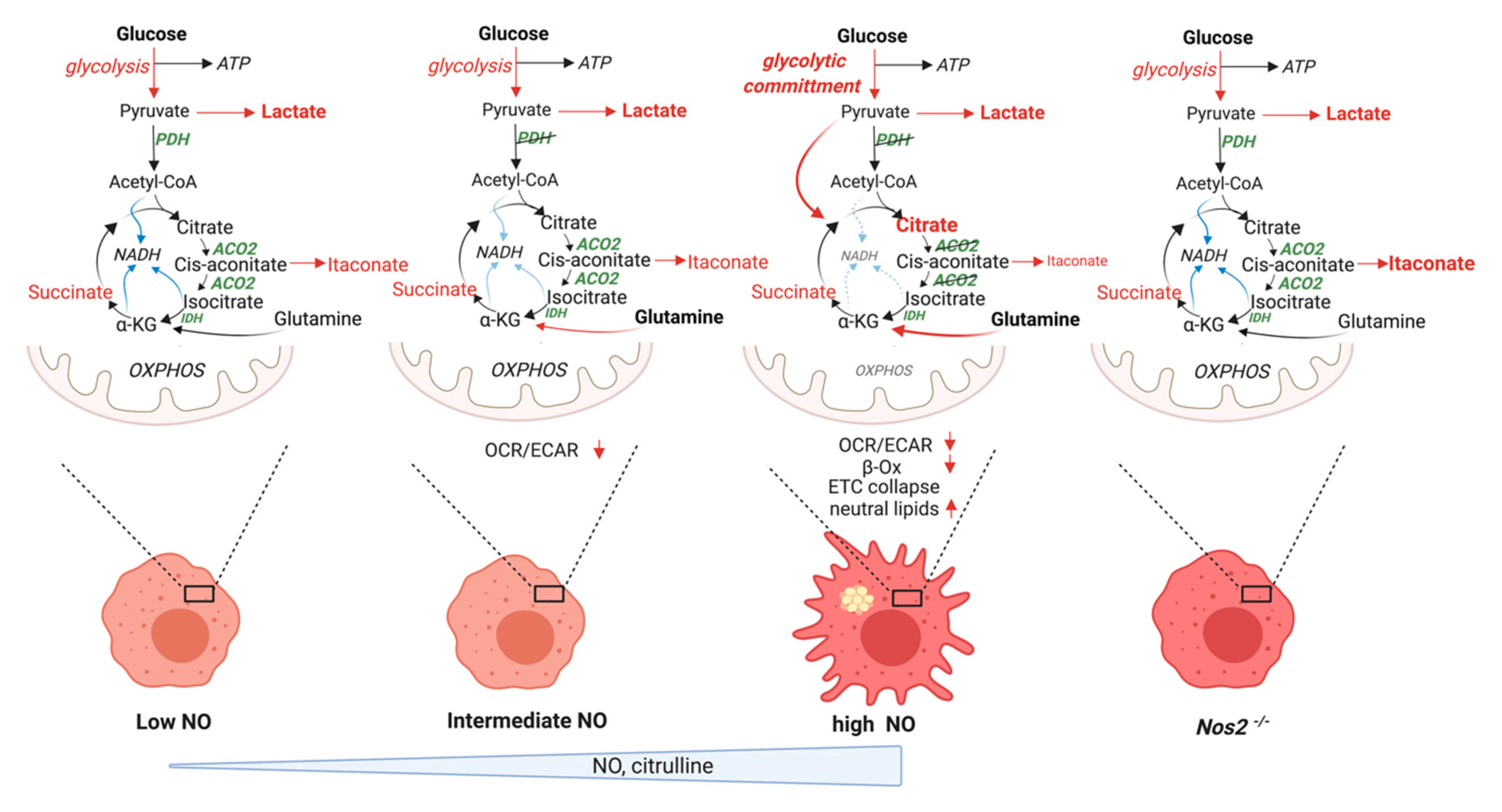
5.2. Revised Model
5.2.1. “Glycolytic Commitment”
5.2.2. HIF1α -PDK Axis
5.2.3. TCA Break at Citrate
5.2.4. Lipid Accumulation
5.2.5. Itaconate
5.2.6. Anaplerotic Pathways
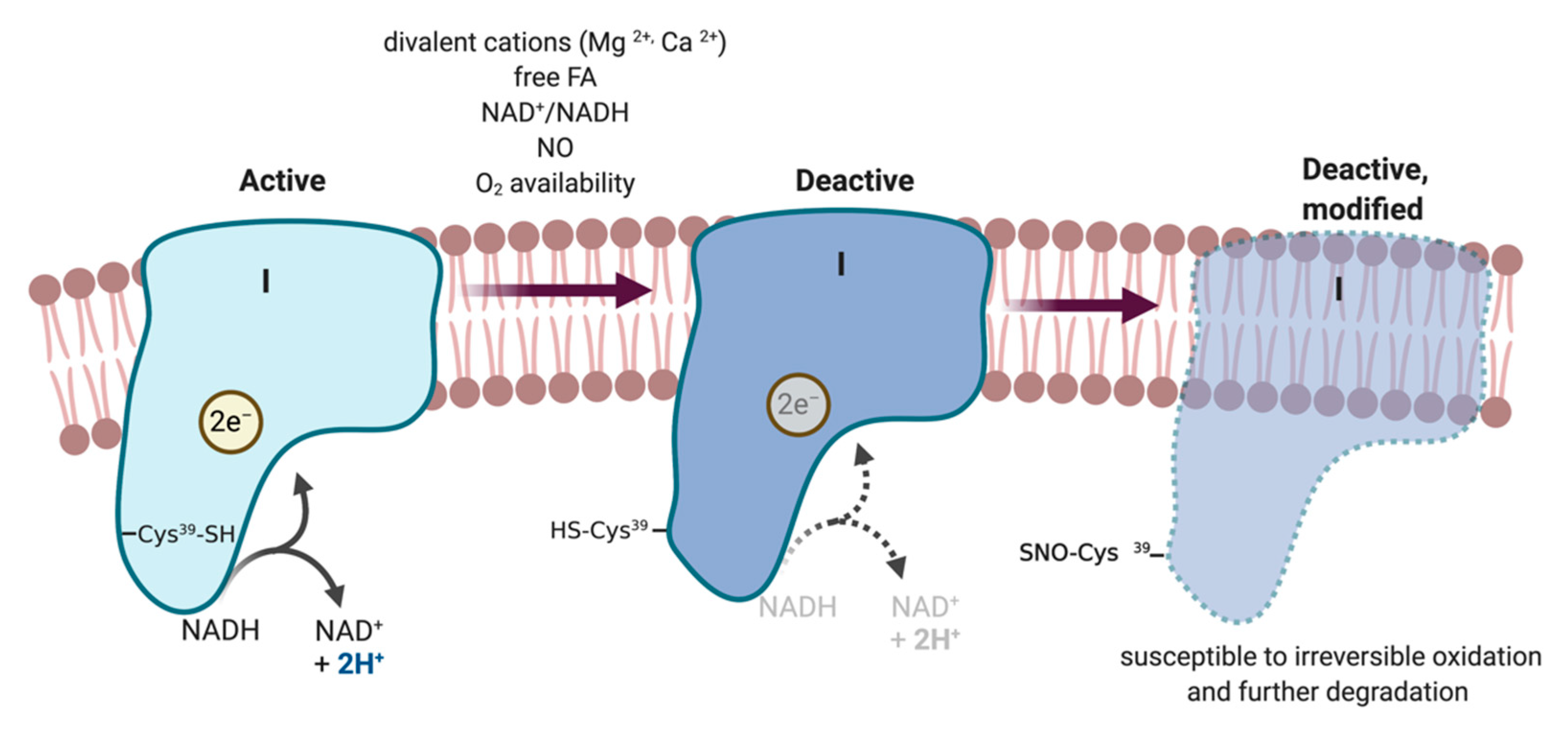
5.2.7. ETC Complexes
5.2.8. Metabolic Targets of NO Fluxes
5.3. The Impact of NO-Mediated TCA Rewiring in Macrophage Function
5.3.1. Inflammatory Cytokine Production
5.3.2. NRF2 Activation
5.3.3. Lipid Mediators of the Inflammatory Response
5.4. NO and the Human Monocyte/Macrophage Dilemma
6. Microenvironments and Niche Specificity
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Acetyl-CoA | acetyl-coenzyme A |
| ACO2 | mitochondrial aconitase |
| α-KG | α-ketoglutarate |
| ANT | adenosine nucleotide translocator |
| ADP | adenosine diphosphate |
| ATP | adenosine triphosphate |
| BCKDH | branched chain alpha keto acid dehydrogenase |
| BH4 | tetrahydrobiopterin |
| BMDM | bone marrow derived macrophage |
| CAT | catalase |
| cGMP | cyclic guanosine monophosphate |
| CM | conditional medium |
| CO | carbon monoxide |
| CPT | carnitine palmitoyltransferase |
| Cyt c | cytochrome c |
| DC | dendritic cell |
| DETA/NO | diethylenetriamine NONOate |
| DGAT | diglyceride acyltransferase |
| DLD | dihydrolipoamide dehydrogenase |
| ECAR | extracellular acidification rate |
| EDHF | endothelial-derived hyperpolarizing factor |
| eNOS | endothelial nitric oxide synthase (aka NOS3) |
| ER | endoplasmic reticulum |
| ETC | mitochondrial electron transport chain |
| FADH2 | flavin-adenine dinucleotide reduced |
| GABA | gamma-Aminobutyric acid |
| GSH | glutathione |
| GPAT | glycerol-3-Phosphate Acyltransferase |
| GSNO | s-nitrosoglutathione |
| HIF | hypoxia-inducible factor |
| HNO | nitroxyl |
| IDH | isocitrate dehydrogenase |
| IFNγ | interferon gamma |
| IL | interleukin |
| iNOS | inducible nitric oxide synthase (aka NOS2) |
| IRG | Immune-Responsive Gene |
| KEAP | Kelch-like ECH-associated protein |
| LPS | lipopolysaccharides |
| MRC | maximal respiratory capacity |
| mTOR | mammalian target of rapamycin |
| N2O3 | dinitrogen trioxide |
| NADH | nicotinamide adenine dinucleotide reduced |
| NADPH | nicotinamide adenine dinucleotide phosphate reduced |
| NDUFV | NADH dehydrogenase [ubiquinone] flavoprotein |
| NMMA | NG-monomethyl-l-arginine |
| nNOS | neuronal nitric oxide synthase (aka NOS1) |
| NO | nitric oxide |
| NO2− | nitrite |
| NO3− | nitrate |
| NOHA | n-hydroxy-l-arginine |
| NOX | NADPH oxidase |
| NRF | nuclear factor erythroid-derived |
| O2 | molecular oxygen |
| O2− | superoxide |
| OCR | oxygen consumption rate |
| OGDH | oxoglutarate dehydrogenase |
| ONOO− | peroxynitrite |
| OXPHOS | oxidative phosphorylation |
| PC | phosphatidylcholine |
| PDH | pyruvate dehydrogenase |
| PDK | pyruvate dehydrogenase kinase |
| PE | phosphatidylethanolamine |
| PGE2 | prostaglandin E2 |
| PI | phosphatidylinositol |
| Q | ubiquinone |
| PTP | permeability transition pore |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| RSH | thiol |
| RSNO | s-nitrosothiol |
| RS(O)NH2 | sulfinamide |
| SDH | succinate dehydrogenase |
| sGC | soluble guanylyl cyclase |
| SOD | superoxide dismutase |
| TCA | tricarboxylic acid cycle |
| TLR | toll like receptor |
| TG | triglycerides |
| TME | tumor microenvironment |
| TNFα | tumor necrosis factor alpha |
References
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nat. Cell Biol. 1980, 288, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Villani, G.M.; Jothianandan, D.; Furchgott, R.F. Selective blockade of endothelium-dependent and glyceryl trinitrate-induced relaxation by hemoglobin and by methylene blue in the rabbit aorta. J. Pharmacol. Exp. Ther. 1985, 232, 708–716. [Google Scholar] [PubMed]
- Ignarro, L.J.; Byrns, R.E.; Buga, G.M.; Wood, K.S. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circ. Res. 1987, 61, 866–879. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef]
- Ignarro, L.J. Nitric oxide is not just blowing in the wind. Br. J. Pharmacol. 2018, 176, 131–134. [Google Scholar] [CrossRef]
- Weinberg, J.B.; Hibbs, J.B. Endocytosis of red blood cells or haemoglobin by activated macrophages inhibits their tumoricidal effect. Nat. Cell Biol. 1977, 269, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Stuehr, D.J.; Marletta, M.A. Mammalian nitrate biosynthesis: Mouse macrophages produce nitrite and nitrate in response to Escherichia coli lipopolysaccharide. Proc. Natl. Acad. Sci. USA 1985, 82, 7738–7742. [Google Scholar] [CrossRef]
- Hibbs, J.B.; Taintor, R.R.; Vavrin, Z.; Rachlin, E.M. Nitric oxide: A cytotoxic activated macrophage effector molecule. Biochem. Biophys. Res. Commun. 1988, 157, 87–94. [Google Scholar] [CrossRef]
- Stuehr, D.J.; Nathan, C.F. Nitric oxide. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J. Exp. Med. 1989, 169, 1543–1555. [Google Scholar] [CrossRef]
- Thomas, D.D.; Wink, D.A. NOS2 as an Emergent Player in Progression of Cancer. Antioxid Redox Sign. 2017, 26, 963–965. [Google Scholar] [CrossRef]
- Wink, D.A.; Ridnour, L.A.; Hussain, S.P.; Harris, C.C. The reemergence of nitric oxide and cancer. Nitric Oxide 2008, 19, 65–67. [Google Scholar] [CrossRef]
- Green, L.E.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.S.; Tannenbaum, S.R. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal. Biochem. 1982, 126, 131–138. [Google Scholar] [CrossRef]
- Hibbs, J.B.; Taintor, R.R.; Vavrin, Z. Macrophage cytotoxicity: Role for l-arginine deiminase and imino nitrogen oxidation to nitrite. Science 1987, 235, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Tannenbaum, S.R.; Sinskey, A.J.; Weisman, M.; Bishop, W. Nitrite in human saliva. Its possible relationship to nitrosamine formation. J. Natl. Cancer I. 1974, 53, 79–84. [Google Scholar]
- Coleman, J.W. Nitric oxide in immunity and inflammation. Int. Immunopharmacol. 2001, 1, 1397–1406. [Google Scholar] [CrossRef]
- Wink, D.; Osawa, Y.; Darbyshire, J.; Jones, C.; Eshenaur, S.; Nims, R.W. Inhibition of Cytochromes P450 by Nitric Oxide and a Nitric Oxide-Releasing Agent. Arch. Biochem. Biophys. 1993, 300, 115–123. [Google Scholar] [CrossRef]
- Abu-Soud, H.M.; Wang, J.; Rousseau, D.L.; Fukuto, J.M.; Ignarro, L.J.; Stuehr, D.J. Neuronal Nitric Oxide Synthase Self-inactivates by Forming a Ferrous-Nitrosyl Complex during Aerobic Catalysis. J. Biol. Chem. 1995, 270, 22997–23006. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar] [PubMed]
- James, S.L. Nitric Oxide in Health and Disease. Parasitol. Today 1998, 14, 504. [Google Scholar] [CrossRef]
- Ford, P.C.; Lorkovic, I.M. Mechanistic Aspects of the Reactions of Nitric Oxide with Transition-Metal Complexes. Chem. Rev. 2002, 102, 993–1018. [Google Scholar] [CrossRef]
- Thomas, D.D.; Heinecke, J.L.; Ridnour, L.A.; Cheng, R.Y.; Kesarwala, A.H.; Switzer, C.H.; McVicar, D.W.; Roberts, D.D.; Glynn, S.A.; Fukuto, J.M.; et al. Signaling and stress: The redox landscape in NOS2 biology. Free. Radic. Biol. Med. 2015, 87, 204–225. [Google Scholar] [CrossRef]
- Liew, F.Y.; Li, Y.; Moss, D.; Parkinson, C.; Rogers, M.V.; Moncada, S. Resistance toLeishmania major infection correlates with the induction of nitric oxide synthase in murine macrophages. Eur. J. Immunol. 1991, 21, 3009–3014. [Google Scholar] [CrossRef]
- Brunet, L.R. Nitric oxide in parasitic infections. Int. Immunopharmacol. 2001, 1, 1457–1467. [Google Scholar] [CrossRef]
- Kröncke, K.-D.; Fehsel, K.; Suschek, C.; Kolb-Bachofen, V. Inducible nitric oxide synthase-derived nitric oxide in gene regulation, cell death and cell survival. Int. Immunopharmacol. 2001, 1, 1407–1420. [Google Scholar] [CrossRef]
- Langrehr, J.M.; Hoffman, R.A.; Lancaster, J.R.; Simmons, R.L. Nitric Oxide—A New Endogenous Immunomodulator. Transplantation 1993, 55, 1205–1212. [Google Scholar] [CrossRef]
- Bogdan, C.; Röllinghoff, M.; Diefenbach, A. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr. Opin. Immunol. 2000, 12, 64–76. [Google Scholar] [CrossRef]
- Thomassen, M.J.; Kavuru, M.S. Human alveolar macrophages and monocytes as a source and target for nitric oxide. Int. Immunopharmacol. 2001, 1, 1479–1490. [Google Scholar] [CrossRef]
- Van Der Veen, R.C. Nitric oxide and T helper cell immunity. Int. Immunopharmacol. 2001, 1, 1491–1500. [Google Scholar] [CrossRef]
- Taylor-Robinson, A.W.; Liew, F.Y.; Severn, A.; Xu, D.; McSorley, S.J.; Garside, P.; Padrón, J.; Phillips, R.S. Regulation of the immune response by nitric oxide differentially produced by T helper type 1 and T helper type 2 cells. Eur. J. Immunol. 1994, 24, 980–984. [Google Scholar] [CrossRef]
- Wei, X.-Q.; Charles, I.G.; Smith, A.; Ure, J.; Feng, G.-J.; Huang, F.-P.; Xu, D.; Mullers, W.; Moncada, S.; Liew, F.Y. Altered immune responses in mice lacking inducible nitric oxide synthase. Nat. Cell Biol. 1995, 375, 408–411. [Google Scholar] [CrossRef]
- Barnes, P.J.; Liew, F. Nitric oxide and asthmatic inflammation. Immunol. Today 1995, 16, 128–130. [Google Scholar] [CrossRef]
- Somasundaram, V.; Gilmore, A.C.; Basudhar, D.; Palmieri, E.M.; Scheiblin, D.A.; Heinz, W.F.; Cheng, R.; Ridnour, L.A.; Altan-Bonnet, G.; Lockett, S.J.; et al. Inducible nitric oxide synthase-derived extracellular nitric oxide flux regulates proinflammatory responses at the single cell level. Redox Biol. 2020, 28, 101354. [Google Scholar] [CrossRef] [PubMed]
- Espey, M.G.; Miranda, K.M.; Pluta, R.M.; Wink, D.A. Nitrosative Capacity of Macrophages Is Dependent on Nitric-oxide Synthase Induction Signals. J. Biol. Chem. 2000, 275, 11341–11347. [Google Scholar] [CrossRef]
- Somasundaram, V.; Basudhar, D.; Bharadwaj, G.; No, J.H.; Ridnour, L.A.; Cheng, R.Y.; Fujita, M.; Thomas, U.D.; Anderson, S.K.; McVicar, D.W.; et al. Molecular Mechanisms of Nitric Oxide in Cancer Progression, Signal Transduction, and Metabolism. Antioxid. Redox Signal. 2019, 30, 1124–1143. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, T.A.; Da Silva, R.S.; Miranda, K.M.; Switzer, C.H.; Wink, D.A.; Fukuto, J.M. Biological nitric oxide signalling: Chemistry and terminology. Br. J. Pharmacol. 2013, 169, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Wainio, W.W. Reactions of cytochrome oxidase. J. Biol. Chem. 1955, 212, 723–733. [Google Scholar] [PubMed]
- Cleeter, M.; Cooper, J.; Darley-Usmar, V.; Moncada, S.; Schapira, A. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide: Implications for neurodegenerative diseases. FEBS Lett. 1994, 345, 50–54. [Google Scholar] [CrossRef]
- Brown, G.C.; Cooper, C. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994, 356, 295–298. [Google Scholar] [CrossRef]
- Schweizer, M.; Richter, C. Nitric Oxide Potently and Reversibly Deenergizes Mitochondria at Low Oxygen Tension. Biochem. Biophys. Res. Commun. 1994, 204, 169–175. [Google Scholar] [CrossRef]
- Torres, J.; Darley-Usmar, V.; Wilson, M.T. Inhibition of cytochrome c oxidase in turnover by nitric oxide: Mechanism and implications for control of respiration. Biochem. J. 1995, 312, 169–173. [Google Scholar] [CrossRef]
- Giuffrè, A.; Sarti, P.; D’Itri, E.; Buse, G.; Soulimane, T.; Brunori, M. On the Mechanism of Inhibition of CytochromecOxidase by Nitric Oxide. J. Biol. Chem. 1996, 271, 33404–33408. [Google Scholar] [CrossRef]
- Tamura, M.; Hazeki, O.; Nioka, S.; Chance, B. In Vivo Study of Tissue Oxygen Metabolism using Optical and Nuclear Magnetic Resonance Spectroscopies. Annu. Rev. Physiol. 1989, 51, 813–834. [Google Scholar] [CrossRef] [PubMed]
- Shibuki, K.; Okada, D. Endogenous nitric oxide release required for long-term synaptic depression in the cerebellum. Nat. Cell Biol. 1991, 349, 326–328. [Google Scholar] [CrossRef]
- Malinski, T.; Taha, Z.; Grunfeld, S.; Patton, S.; Kapturczak, M.; Tomboulian, P. Diffusion of Nitric Oxide in the Aorta Wall Monitored in Situ by Porphyrinic Microsensors. Biochem. Biophys. Res. Commun. 1993, 193, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Erusalimsky, J.D. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat. Rev. Mol. Cell Biol. 2002, 3, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Poderoso, J.J.; Carreras, M.C.; Lisderoa, C.; Riobóa, N.; Schöpfera, F.; Boverisb, A. Nitric Oxide Inhibits Electron Transfer and Increases Superoxide Radical Production in Rat Heart Mitochondria and Submitochondrial Particles. Arch. Biochem. Biophys. 1996, 328, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Thomas, U.D.; Liu, X.; Kantrow, S.P.; Lancaster, J.R. The biological lifetime of nitric oxide: Implications for the perivascular dynamics of NO and O. Proc. Natl. Acad. Sci. USA 2000, 98, 355–360. [Google Scholar] [CrossRef]
- Pohl, U.; Busse, R. Hypoxia stimulates release of endothelium-derived relaxant factor. Am. J. Physiol. Circ. Physiol. 1989, 256, H1595–H1600. [Google Scholar] [CrossRef]
- Rees, D.D.; Monkhouse, J.E.; Cambridge, D.; Moncada, S. Nitric oxide and the haemodynamic profile of endotoxin shock in the conscious mouse. Br. J. Pharmacol. 1998, 124, 540–546. [Google Scholar] [CrossRef]
- Brown, G.C.; Foxwell, N.; Moncada, S. Transcellular regulation of cell respiration by nitric oxide generated by activated macrophages. FEBS Lett. 1998, 439, 321–324. [Google Scholar] [CrossRef]
- Granger, D.L.; Taintor, R.R.; Cook, J.L.; Hibbs, J.B. Injury of neoplastic cells by murine macrophages leads to inhibition of mitochondrial respiration. J. Clin. Investig. 1980, 65, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.L.; Lehninger, A.L. Sites of inhibition of mitochondrial electron transport in macrophage-injured neoplastic cells. J. Cell Biol. 1982, 95, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Drapier, J.C.; Hibbs, J.B. Murine cytotoxic activated macrophages inhibit aconitase in tumor cells. Inhibition involves the iron-sulfur prosthetic group and is reversible. J. Clin. Investig. 1986, 78, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.C.; Emptage, M.H.; Dreyer, J.L.; Beinert, H. The role of iron in the activation-inactivation of aconitase. J. Biol. Chem. 1983, 258, 11098–11105. [Google Scholar]
- Kent, T.A.; Emptage, M.H.; Merkle, H.; Kennedy, M.C.; Beinert, H.; Münck, E. Mössbauer studies of aconitase. Substrate and inhibitor binding, reaction intermediates, and hyperfine interactions of reduced 3Fe and 4Fe clusters. J. Biol. Chem. 1985, 260, 6871–6881. [Google Scholar]
- Wink, D.A.; Laval, J. The Fpg protein, a DNA repair enzyme, is inhibited by the biomediator nitric oxide in vitro and in vivo. Carcinogenesis 1994, 15, 2125–2129. [Google Scholar] [CrossRef]
- Hatefi, Y. The Mitochondrial Electron Transport and Oxidative Phosphorylation System. Annu. Rev. Biochem. 1985, 54, 1015–1069. [Google Scholar] [CrossRef]
- Ackrell, B.A.; Kearney, E.B.; Mims, W.B.; Peisach, J.; Beinert, H. Iron-sulfur cluster 3 of beef heart succinate-ubiquinone oxidoreductase is a 3-iron cluster. J. Biol. Chem. 1984, 259, 4015–4018. [Google Scholar]
- Reddy, D.; Lancaster, J.R.; Cornforth, D.P. Nitrite inhibition of Clostridium botulinum: Electron spin resonance detection of iron-nitric oxide complexes. Science 1983, 221, 769–770. [Google Scholar] [CrossRef]
- Lushchak, O.; Piroddi, M.; Galli, F.; Lushchak, V.I. Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species. Redox Rep. 2013, 19, 8–15. [Google Scholar] [CrossRef]
- Liu, Q.; Simpson, D.C.; Gronert, S. Carbonylation of mitochondrial aconitase with 4-hydroxy-2-(E)-nonenal: Localization and relative reactivity of addition sites. Biochim. et Biophys. Acta (BBA) Proteins Proteom. 2013, 1834, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Miller, M.J.S.; Joshi, M.S.; Thomas, U.D.; Lancaster, J.R. Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc. Natl. Acad. Sci. USA 1998, 95, 2175–2179. [Google Scholar] [CrossRef] [PubMed]
- López-Figueroa, M.O.; Caamaño, C.; Morano, M.; Rønn, L.C.; Akil, H.; Watson, S.J. Direct Evidence of Nitric Oxide Presence within Mitochondria. Biochem. Biophys. Res. Commun. 2000, 272, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Mason, M.G.; Nicholls, P.; Wilson, M.T.; Cooper, C.E. Nitric oxide inhibition of respiration involves both competitive (heme) and noncompetitive (copper) binding to cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2006, 103, 708–713. [Google Scholar] [CrossRef]
- Stuehr, D.J.; Marletta, M.A. Induction of nitrite/nitrate synthesis in murine macrophages by BCG infection, lymphokines, or interferon-gamma. J. Immunol. 1987, 139, 518. [Google Scholar]
- Hibbs, J.B.; Vavrin, Z.; Taintor, R.R. l-arginine is required for expression of the activated macrophage effector mechanism causing selective metabolic inhibition in target cells. J. Immunol. 1987, 138, 550–565. [Google Scholar]
- Drapier, J.-C.; Wietzerbin, J.; Hibbs, J.B. Interferon-γ and tumor necrosis factor induce the l-arginine-dependent cytotoxic effector mechanism in murine macrophages. Eur. J. Immunol. 1988, 18, 1587–1592. [Google Scholar] [CrossRef]
- Drapier, J.C.; Hibbs, J.B. Differentiation of murine macrophages to express nonspecific cytotoxicity for tumor cells results in l-arginine-dependent inhibition of mitochondrial iron-sulfur enzymes in the macrophage effector cells. J. Immunol. 1988, 140, 2829–2838. [Google Scholar]
- Stadler, J.; Billiar, T.R.; Curran, R.D.; Stuehr, D.J.; Ochoa, J.B.; Simmons, R.L. Effect of exogenous and endogenous nitric oxide on mitochondrial respiration of rat hepatocytes. Am. J. Physiol. 1991, 260, C910–C916. [Google Scholar] [CrossRef]
- Pellat, C.; Henry, Y.; Drapier, J.-C. IFN-γ-activated macrophages: Detection by electron paramagnetic resonance of complexes between l-Arginine-derived nitric oxide and non-heme iron proteins. Biochem. Biophys. Res. Commun. 1990, 166, 119–125. [Google Scholar] [CrossRef]
- Bolaños, J.P.; Peuchen, S.; Heales, S.J.R.; Land, J.M.; Clark, J.B. Nitric Oxide-Mediated Inhibition of the Mitochondrial Respiratory Chain in Cultured Astrocytes. J. Neurochem. 2002, 63, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Lizasoain, I.; Moro, M.A.; Knowles, R.G.; Darley-Usmar, V.; Moncada, S. Nitric oxide and peroxynitrite exert distinct effects on mitochondrial respiration which are differentially blocked by glutathione or glucose. Biochem. J. 1996, 314, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.A.; Porteous, C.M.; Murphy, M.P. Superoxide production by mitochondria in the presence of nitric oxide forms peroxynitrite. IUBMB Life 1996, 40, 527–534. [Google Scholar] [CrossRef]
- Maragos, C.M.; Morley, D.; Wink, D.A.; Dunams, T.M.; Saavedra, J.E.; Hoffman, A.; Bove, A.A.; Isaac, L.; Hrabie, J.A.; Keefer, L.K. Complexes of.NO with nucleophiles as agents for the controlled biological release of nitric oxide. Vasorelaxant effects. J. Med. Chem. 1991, 34, 3242–3247. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzellie, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free. Radic. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Radi, R.; Rodríguez, M.; Castro, L.; Telleri, R. Inhibition of Mitochondrial Electron Transport by Peroxynitrite. Arch. Biochem. Biophys. 1994, 308, 89–95. [Google Scholar] [CrossRef]
- Thomas, D.D.; Ridnour, L.A.; Espey, M.G.; Donzelli, S.; Ambs, S.; Hussain, S.P.; Harris, C.C.; DeGraff, W.; Roberts, D.D.; Mitchell, J.B.; et al. Superoxide Fluxes Limit Nitric Oxide-induced Signaling. J. Biol. Chem. 2006, 281, 25984–25993. [Google Scholar] [CrossRef]
- Jourd’Heuil, D.; Jourd’Heuil, F.L.; Kutchukian, P.S.; Musah, R.A.; Wink, D.A.; Grisham, M.B. Reaction of Superoxide and Nitric Oxide with Peroxynitrite. J. Biol. Chem. 2001, 276, 28799–28805. [Google Scholar] [CrossRef]
- Cleeter, M.W.J.; Cooper, J.M.; Schapira, A.H. Irreversible Inhibition of Mitochondrial Complex I by 1-Methyl-4-Phenylpyridinium: Evidence for Free Radical Involvement. J. Neurochem. 1992, 58, 786–789. [Google Scholar] [CrossRef]
- Beltrán, B.; Mathur, A.; Duchen, M.R.; Erusalimsky, J.D.; Moncada, S. The effect of nitric oxide on cell respiration: A key to understanding its role in cell survival or death. Proc. Natl. Acad. Sci. USA 2000, 97, 14602–14607. [Google Scholar] [CrossRef]
- Radi, R.; Beckman, J.S.; Bush, K.M.; Freeman, B.A. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J. Biol. Chem. 1991, 266, 4244–4250. [Google Scholar] [PubMed]
- Clementi, E.; Brown, G.C.; Feelisch, M.; Moncada, S. Persistent inhibition of cell respiration by nitric oxide: Crucial role of s-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc. Natl. Acad. Sci. USA 1998, 95, 7631–7636. [Google Scholar] [CrossRef] [PubMed]
- Bolaños, J.P.; Heales, S.J.; Peuchen, S.; Barker, J.E.; Land, J.M.; Clark, J.B. Nitric oxide-mediated mitochondrial damage: A potential neuroprotective role for glutathione. Free. Radic. Biol. Med. 1996, 21, 995–1001. [Google Scholar] [CrossRef]
- Moro, M.A.; Darley-Usmar, V.M.; Goodwin, D.A.; Read, N.G.; Zamora-Pino, R.; Feelisch, M.; Radomski, M.W.; Moncada, S. Paradoxical fate and biological action of peroxynitrite on human platelets. Proc. Natl. Acad. Sci. USA 1994, 91, 6702–6706. [Google Scholar] [CrossRef] [PubMed]
- Beltrán, B.; Orsi, A.; Clementi, E.; Moncada, S. Oxidative stress and s-nitrosylation of proteins in cells. Br. J. Pharmacol. 2000, 129, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Babot, M.; Birch, A.; Labarbuta, P.; Galkin, A. Characterisation of the active/de-active transition of mitochondrial complex I. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1837, 1083–1092. [Google Scholar] [CrossRef]
- Vinogradov, A.D. Catalytic properties of the mitochondrial NADH–ubiquinone oxidoreductase (Complex I) and the pseudo-reversible active/inactive enzyme transition. Biochim. Biophys. Acta (BBA) Bioenerg. 1998, 1364, 169–185. [Google Scholar] [CrossRef]
- Galkin, A.; Abramov, A.Y.; Frakich, N.; Duchen, M.R.; Moncada, S. Lack of Oxygen Deactivates Mitochondrial Complex, I. J. Biol. Chem. 2009, 284, 36055–36061. [Google Scholar] [CrossRef]
- Gavrikova, E.V.; Vinogradov, A.D. Active/de-active state transition of the mitochondrial complex I as revealed by specific sulfhydryl group labeling. FEBS Lett. 1999, 455, 36–40. [Google Scholar] [CrossRef]
- Galkin, A.; Meyer, B.; Wittig, I.; Karas, M.; Schägger, H.; Vinogradov, A.; Brandt, U. Identification of the Mitochondrial ND3 Subunit as a Structural Component Involved in the Active/Deactive Enzyme Transition of Respiratory Complex, I. J. Biol. Chem. 2008, 283, 20907–20913. [Google Scholar] [CrossRef]
- Babot, M.; Labarbuta, P.; Birch, A.; Kee, S.; Fuszard, M.; Botting, C.H.; Wittig, I.; Heide, H.; Galkin, A. ND3, ND1 and 39kDa subunits are more exposed in the de-active form of bovine mitochondrial complex I. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1837, 929–939. [Google Scholar] [CrossRef]
- Galkin, A.; Moncada, S. s-Nitrosation of Mitochondrial Complex I Depends on Its Structural Conformation. J. Biol. Chem. 2007, 282, 37448–37453. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cochemé, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by s-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef]
- Russell, D.G.; Huang, L.; VanderVen, B.C. Immunometabolism at the interface between macrophages and pathogens. Nat. Rev. Immunol. 2019, 19, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Kato, M. Site of Action of Lipid A on Mitochondria. J. Bacteriol. 1972, 112, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Gawehn, K.; Geissler, A.W. [Metabolism of leukocytes]. Z. fur Naturforschung. Teil B Chemie Biochem. Biophys. Biol. Verwandte- Geb. 1958, 13, 515–516. [Google Scholar]
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; DeBerardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A roadmap for interpreting 13 C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015, 34, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Divakaruni, A.S.; Paradyse, A.; Ferrick, D.A.; Murphy, A.N.; Jastroch, M. Analysis and Interpretation of Microplate-Based Oxygen Consumption and pH Data. Methods Enzymol. 2014, 547, 309–354. [Google Scholar] [CrossRef]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor–induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef]
- Jones, A.E.; Divakaruni, A.S. Macrophage activation as an archetype of mitochondrial repurposing. Mol. Asp. Med. 2020, 71, 100838. [Google Scholar] [CrossRef]
- Rodríguez-Prados, J.-C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate Fate in Activated Macrophages: A Comparison between Innate, Classic, and Alternative Activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.T.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nat. Cell Biol. 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Haschemi, A.; Kosma, P.; Gille, L.; Evans, C.R.; Burant, C.F.; Starkl, P.; Knapp, B.; Haas, R.; Schmid, J.A.; Jandl, C.; et al. The Sedoheptulose Kinase CARKL Directs Macrophage Polarization through Control of Glucose Metabolism. Cell Metab. 2012, 15, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Kellett, D.N. 2-Deoxyglucose and inflammation. J. Pharm. Pharmacol. 1966, 18, 199–200. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Li, Y.; Yu, Q.; Jin, X.; Wang, X.; Jia, A.; Hu, Y.; Han, L.; Wang, J.; et al. HIF1α-dependent glycolysis promotes macrophage functional activities in protecting against bacterial and fungal infection. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules that Regulate Macrophage Polarization. Immunology 2015, 42, 419–430. [Google Scholar] [CrossRef]
- Michelucci, A.; Cordes, T.; Ghelfi, J.; Pailot, A.; Reiling, N.; Goldmann, O.; Binz, T.; Wegner, A.; Tallam, A.; Rausell, A.; et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. USA 2013, 110, 7820–7825. [Google Scholar] [CrossRef]
- Bailey, J.D.; Diotallevi, M.; Nicol, T.; McNeill, E.; Shaw, A.; Chuaiphichai, S.; Hale, A.; Starr, A.; Nandi, M.; Stylianou, E.; et al. Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep. 2019, 28, 218–230.e7. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- De Souza, D.P.; Achuthan, A.; Lee, M.K.; Binger, K.J.; Lee, M.-C.; Davidson, S.; Tull, D.L.; McConville, M.J.; Cook, A.D.; Murphy, A.J.; et al. Autocrine IFN-I inhibits isocitrate dehydrogenase in the TCA cycle of LPS-stimulated macrophages. J. Clin. Investig. 2019, 129, 4239–4244. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.-C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Cordes, T.; Wallace, M.; Michelucci, A.; Divakaruni, A.S.; Sapcariu, S.C.; Sousa, C.; Koseki, H.; Cabrales, P.; Murphy, A.N.; Hiller, K.; et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J. Biol. Chem. 2016, 291, 14274–14284. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.-C.; Chou, C.-H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2015, 213, 15–23. [Google Scholar] [CrossRef]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef]
- MacKenzie, E.D.; Selak, M.A.; Tennant, D.A.; Payne, L.J.; Crosby, S.; Frederiksen, C.M.; Watson, D.G.; Gottlieb, E. Cell-Permeating α-Ketoglutarate Derivatives Alleviate Pseudohypoxia in Succinate Dehydrogenase-Deficient Cells. Mol. Cell. Biol. 2007, 27, 3282–3289. [Google Scholar] [CrossRef]
- Everts, B.; Amiel, E.; Huang, S.C.-C.; Smith, S.; Chang, C.-H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; Van Der Windt, G.J.W.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; Di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436. [Google Scholar] [CrossRef]
- Strelko, C.L.; Lu, W.; Dufort, F.J.; Seyfried, T.N.; Chiles, T.C.; Rabinowitz, J.D.; Mary, F.R. Itaconic Acid Is a Mammalian Metabolite Induced during Macrophage Activation. J. Am. Chem. Soc. 2011, 133, 16386–16389. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.R.; McFadden, B.A. Caenorhabditis elegans and Ascaris suum: Inhibition of isocitrate lyase by itaconate. Exp. Parasitol. 1978, 44, 262–268. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Huynh, J.P.; Lampropoulou, V.; Loginicheva, E.; Esaulova, E.; Gounder, A.P.; Boon, A.C.; Schwarzkopf, E.A.; Bradstreet, T.R.; Edelson, B.T.; et al. Irg1 expression in myeloid cells prevents immunopathology during M. tuberculosis infection. J. Exp. Med. 2018, 215, 1035–1045. [Google Scholar] [CrossRef]
- Daniels, B.P.; Kofman, S.B.; Smith, J.R.; Norris, G.T.; Snyder, A.G.; Kolb, J.P.; Gao, X.; Locasale, J.W.; Martinez, J.; Gale, M.; et al. The Nucleotide Sensor ZBP1 and Kinase RIPK3 Induce the Enzyme IRG1 to Promote an Antiviral Metabolic State in Neurons. Immunology 2019, 50, 64–76.e4. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Deng, M.; Scott, M.J.; Fu, G.; Loughran, P.A.; Lei, Z.; Li, S.; Sun, P.; Yang, C.; Li, W.; et al. IRG1/Itaconate Activates Nrf2 in Hepatocytes to Protect Against Liver Ischemia-Reperfusion Injury. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.M.; Davies, L.C.; Karwan, M.; Ileva, L.; Ozaki, M.K.; Cheng, R.Y.; Ridnour, L.A.; Annunziata, C.M.; Wink, D.A.; McVicar, D.W. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J. Clin. Investig. 2018, 128, 3794–3805. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Artyomov, M.N. Itaconate: The poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 2019, 19, 273–281. [Google Scholar] [CrossRef]
- Hooftman, A.; O’Neill, L.A. The Immunomodulatory Potential of the Metabolite Itaconate. Trends Immunol. 2019, 40, 687–698. [Google Scholar] [CrossRef]
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwärzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008, 9, 1261–1269. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef]
- Jung, F.; Haendeler, J.; Hoffmann, J.; Reissner, A.; Dernbach, E.; Zeiher, A.M.; Dimmeler, S. Hypoxic Induction of the Hypoxia-Inducible Factor Is Mediated via the Adaptor Protein Shc in Endothelial Cells. Circ. Res. 2002, 91, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed]
- VanderVen, B.C.; Yates, R.M.; Russell, D.G. Intraphagosomal Measurement of the Magnitude and Duration of the Oxidative Burst. Traffic 2009, 10, 372–378. [Google Scholar] [CrossRef]
- MacMicking, J.; Xie, Q.-W.; Nathan, C. NITRIC OXIDE AND MACROPHAGE FUNCTION. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef]
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef]
- Cyster, J.G.; Dang, E.V.; Reboldi, A.; Yi, T. 25-Hydroxycholesterols in innate and adaptive immunity. Nat. Rev. Immunol. 2014, 14, 731–743. [Google Scholar] [CrossRef]
- Qualls, J.E.; Subramanian, C.; Rafi, W.; Smith, A.M.; Balouzian, L.; DeFreitas, A.A.; Shirey, K.A.; Reutterer, B.; Kernbauer, E.; Stockinger, S.; et al. Sustained Generation of Nitric Oxide and Control of Mycobacterial Infection Requires Argininosuccinate Synthase 1. Cell Host Microbe 2012, 12, 313–323. [Google Scholar] [CrossRef]
- Buga, G.M.; Wei, L.H.; Bauer, P.M.; Fukuto, J.M.; Ignarro, L.J. N G-hydroxy-l-arginine and nitric oxide inhibit Caco-2 tumor cell proliferation by distinct mechanisms. Am. J. Physiol. Integr. Comp. Physiol. 1998, 275, R1256–R1264. [Google Scholar] [CrossRef] [PubMed]
- Iniesta, V.; Gómez-Nieto, L.C.; Corraliza, I. The Inhibition of Arginase by Nω-Hydroxy-l-Arginine Controls the Growth of Leishmania Inside Macrophages. J. Exp. Med. 2001, 193, 777–784. [Google Scholar] [CrossRef]
- O’Neill, L.A. A Broken Krebs Cycle in Macrophages. Immunology 2015, 42, 393–394. [Google Scholar] [CrossRef]
- Everts, B.; Amiel, E.; Van Der Windt, G.J.W.; Freitas, T.C.; Chott, R.; Yarasheski, K.E.; Pearce, E.L.; Pearce, E.J. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 2012, 120, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Doulias, P.-T.; Tenopoulou, M.; Greene, J.L.; Raju, K.; Ischiropoulos, H. Nitric Oxide Regulates Mitochondrial Fatty Acid Metabolism Through Reversible Protein s-Nitrosylation. Sci. Signal. 2013, 6, rs1. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O’Neill, L.A.J. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Thwe, P.M.; Amiel, E. The role of nitric oxide in metabolic regulation of Dendritic cell immune function. Cancer Lett. 2018, 412, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Amiel, E.; Everts, B.; Fritz, D.; Beauchamp, S.; Ge, B.; Pearce, E.L.; Pearce, E.J. Mechanistic Target of Rapamycin Inhibition Extends Cellular Lifespan in Dendritic Cells by Preserving Mitochondrial Function. J. Immunol. 2014, 193, 2821–2830. [Google Scholar] [CrossRef]
- Kam, Y.; Swain, P.M.; Dranka, B.P. Abstract A67: Bi-phasic metabolic responses to in situ macrophage activation. Antitumor Immune Responses 2018, 6, A67. [Google Scholar] [CrossRef]
- Forkink, M.; Manjeri, G.R.; Liemburg-Apers, D.C.; Nibbeling, E.; Blanchard, M.; Wojtala, A.; Smeitink, J.A.; Wieckowski, M.R.; Willems, P.H.; Koopman, W.J. Mitochondrial hyperpolarization during chronic complex I inhibition is sustained by low activity of complex II, III, IV and V. Biochim. et Biophys. Acta (BBA) Bioenerg. 2014, 1837, 1247–1256. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Almeida, A.; Almeida, J.; Bolaños, J.P.; Moncada, S. Different responses of astrocytes and neurons to nitric oxide: The role of glycolytically generated ATP in astrocyte protection. Proc. Natl. Acad. Sci. USA 2001, 98, 15294–15299. [Google Scholar] [CrossRef]
- Baseler, W.A.; Davies, L.C.; Quigley, L.; Ridnour, L.A.; Weiss, J.M.; Hussain, S.P.; Wink, D.A.; McVicar, D.W. Autocrine IL-10 functions as a rheostat for M1 macrophage glycolytic commitment by tuning nitric oxide production. Redox Biol. 2016, 10, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, M.; Yang, M.; Yu, Y.; Zhu, S.; Hou, W.; Kang, R.; Lotze, M.T.; Billiar, T.R.; Wang, H.; et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Seim, G.L.; Britt, E.C.; John, S.V.; Yeo, F.J.; Johnson, A.R.; Eisenstein, R.S.; Pagliarini, D.J.; Fan, J. Two-stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon-γ stimulation. Nat. Metab. 2019, 1, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.-J.; Liu, L.; Forster, M.J. Reversible inactivation of dihydrolipoamide dehydrogenase by Angeli’s salt. Acta Biophys. Sin. 2012, 28, 341–350. [Google Scholar]
- Piantadosi, C.A. Regulation of mitochondrial processes by protein s-nitrosylation. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 2012, 1820, 712–721. [Google Scholar] [CrossRef]
- Feingold, K.R.; Shigenaga, J.K.; Kazemi, M.R.; McDonald, C.M.; Patzek, S.M.; Cross, A.S.; Moser, A.; Grunfeld, C. Mechanisms of triglyceride accumulation in activated macrophages. J. Leukoc. Biol. 2012, 92, 829–839. [Google Scholar] [CrossRef]
- Rosas-Ballina, M.; Guan, X.L.; Schmidt, A.; Bumann, D. Classical Activation of Macrophages Leads to Lipid Droplet Formation Without de novo Fatty Acid Synthesis. Front. Immunol. 2020, 11, 131. [Google Scholar] [CrossRef]
- Zirath, H.; Frenzel, A.; Oliynyk, G.; Segerström, L.; Westermark, U.K.; Larsson, K.; Persson, M.M.; Hultenby, K.; Lehtiö, J.; Einvik, C.; et al. MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10258–10263. [Google Scholar] [CrossRef]
- Dobbins, R.L.; Szczepaniak, L.S.; Bentley, B.; Esser, V.; Myhill, J.; McGarry, J.D. Prolonged Inhibition of Muscle Carnitine Palmitoyltransferase-1 Promotes Intramyocellular Lipid Accumulation and Insulin Resistance in Rats. Diabetes 2001, 50, 123–130. [Google Scholar] [CrossRef]
- Boren, J.J.; Brindle, K.M. Apoptosis-induced mitochondrial dysfunction causes cytoplasmic lipid droplet formation. Cell Death Differ. 2012, 19, 1561–1570. [Google Scholar] [CrossRef]
- Tong, W.-H.; Maio, N.; Zhang, D.-L.; Palmieri, E.M.; Ollivierre, H.; Ghosh, M.C.; McVicar, D.W.; Rouault, T.A. TLR-activated repression of Fe-S cluster biogenesis drives a metabolic shift and alters histone and tubulin acetylation. Blood Adv. 2018, 2, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Moncada, S. Nitric Oxide, Cytochrome C Oxidase, and the Cellular Response to Hypoxia. Arter. Thromb. Vasc. Biol. 2010, 30, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Nystrom, T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005, 24, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Timón-Gómez, A.; Sanfeliu-Redondo, D.; Pascual-Ahuir, A.; Proft, M. Regulation of the Stress-Activated Degradation of Mitochondrial Respiratory Complexes in Yeast. Front. Microbiol. 2018, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Tomin, T.; Schittmayer, M.; Honeder, S.; Heininger, C.; Birner-Gruenberger, R. Irreversible oxidative post-translational modifications in heart disease. Expert Rev. Proteom. 2019, 16, 681–693. [Google Scholar] [CrossRef]
- Ugarte, N.; Petropoulos, I.; Friguet, B. Oxidized Mitochondrial Protein Degradation and Repair in Aging and Oxidative Stress. Antioxidants Redox Signal. 2010, 13, 539–549. [Google Scholar] [CrossRef]
- Diers, A.R.; Broniowska, K.A.; Darley-Usmar, V.M.; Hogg, N. Differential regulation of metabolism by nitric oxide and s-nitrosothiols in endothelial cells. Am. J. Physiol. Circ. Physiol. 2011, 301, H803–H812. [Google Scholar] [CrossRef]
- Lachmandas, E.; Boutens, L.; Ratter, J.M.; Hijmans, A.; Hooiveld, G.J.E.J.; Joosten, L.A.B.; Rodenburg, R.J.; Fransen, J.; Houtkooper, R.H.; Van Crevel, R.; et al. Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat. Microbiol. 2016, 2, 16246. [Google Scholar] [CrossRef]
- Netea, M.; Van Der Meer, J.W.M. Trained Immunity: An Ancient Way of Remembering. Cell Host Microbe 2017, 21, 297–300. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, S.; Jeon, R.; Vuckovic, I.; Jiang, X.; Lerman, A.; Folmes, C.D.; Dzeja, P.D.; Herrmann, J. Interferon Gamma Induces Reversible Metabolic Reprogramming of M1 Macrophages to Sustain Cell Viability and Pro-Inflammatory Activity. EBioMedicine 2018, 30, 303–316. [Google Scholar] [CrossRef]
- Fensterheim, B.A.; Young, J.D.; Luan, L.; Kleinbard, R.R.; Stothers, C.L.; Patil, N.K.; McAtee-Pereira, A.G.; Guo, Y.; Trenary, I.; Hernandez, A.; et al. The TLR4 Agonist Monophosphoryl Lipid A Drives Broad Resistance to Infection via Dynamic Reprogramming of Macrophage Metabolism. J. Immunol. 2018, 200, 3777–3789. [Google Scholar] [CrossRef] [PubMed]
- Garaude, J.; Acín-Pérez, R.; Martínez-Cano, S.; Enamorado, M.; Ugolini, M.; Nistal-Villán, E.; Hervás-Stubbs, E.N.-V.S.; Pelegrín, P.; Sander, M.U.L.E.; Enríquez, J.A.; et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 2016, 17, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Giordano, D.; Li, C.; Suthar, M.S.; Draves, K.E.; Ma, D.Y.; Gale, M.; Clark, E.A. Nitric oxide controls an inflammatory-like Ly6C(hi)PDCA1+ DC subset that regulates Th1 immune responses. J. Leukoc. Biol. 2010, 89, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.B.; Lovewell, R.R.; Olive, A.J.; Zhang, G.; Wang, W.; Eugenin, E.; Smith, C.M.; Phuah, J.Y.; Long, J.E.; Dubuke, M.L.; et al. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat. Microbiol. 2017, 2, 1–11. [Google Scholar] [CrossRef]
- Simioni, P.U.; Fernandes, L.G.; Tamashiro, W.M.D.S.C. Downregulation of l-arginine metabolism in dendritic cells induces tolerance to exogenous antigen. Int. J. Immunopathol. Pharmacol. 2017, 30, 44–57. [Google Scholar] [CrossRef]
- Noe, J.T.; Mitchell, R.A. Tricarboxylic acid cycle metabolites in the control of macrophage activation and effector phenotypes. J. Leukoc. Biol. 2019, 106, 359–367. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Bossche, J.V.D.; Baardman, J.; Otto, N.A.; Van Der Velden, S.; Neele, A.E.; Berg, S.M.V.D.; Luque-Martin, R.; Chen, H.-J.; Boshuizen, M.C.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef]
- Hernandez-Cuellar, E.; Tsuchiya, K.; Hara, H.; Fang, R.; Sakai, S.; Kawamura, I.; Akira, S.; Mitsuyama, M. Cutting Edge: Nitric Oxide Inhibits the NLRP3 Inflammasome. J. Immunol. 2012, 189, 5113–5117. [Google Scholar] [CrossRef]
- Mao, K.; Chen, S.; Chen, M.; Ma, Y.; Wang, Y.; Huang, B.; He, Z.; Zeng, Y.; Hu, Y.; Sun, S.; et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013, 23, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.B.; Rathinam, V.A.K.; Martens, G.W.; Martinot, A.J.; Kornfeld, H.; Fitzgerald, K.A.; Sassetti, C.M. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome–dependent processing of IL-1β. Nat. Immunol. 2012, 14, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.; Hale, A.B.; Patel, J.; Chuaiphichai, S.; Zen, A.A.H.; Rashbrook, V.S.; Trelfa, L.; Crabtree, M.J.; McNeill, E.; Channon, K.M. Roles for endothelial cell and macrophage Gch1 and tetrahydrobiopterin in atherosclerosis progression. Cardiovasc. Res. 2018, 114, 1385–1399. [Google Scholar] [CrossRef] [PubMed]
- McNeill, E.; Crabtree, M.J.; Sahgal, N.; Patel, J.; Chuaiphichai, S.; Iqbal, A.J.; Hale, A.B.; Greaves, D.R.; Channon, K.M. Regulation of iNOS function and cellular redox state by macrophage Gch1 reveals specific requirements for tetrahydrobiopterin in NRF2 activation. Free. Radic. Biol. Med. 2015, 79, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Krepel, S.A.; Gonzalez-Cotto, M.; Palmieri, E.M.; McVicar, D.W. Elucidation of the respective roles of itaconate and nitric oxide in the nuclear accumulation and activation of Nrf2 in activated macrophages. J. Immunol. 2020, 204, 128–149. [Google Scholar]
- Sun, K.A.; Li, Y.; Meliton, A.Y.; Woods, P.S.; Kimmig, L.M.; Cetin-Atalay, R.; Hamanaka, R.B.; Mutlu, G.M. Endogenous itaconate is not required for particulate matter-induced NRF2 expression or inflammatory response. eLife 2020, 9. [Google Scholar] [CrossRef]
- Swain, A.; Bambouskova, M.; Kim, H.; Andhey, P.S.; Duncan, D.; Auclair, K.; Chubukov, V.; Simons, D.M.; Roddy, T.P.; Stewart, K.M.; et al. Comparative evaluation of itaconate and its derivatives reveals divergent inflammasome and type I interferon regulation in macrophages. Nat. Metab. 2020, 2, 594–602. [Google Scholar] [CrossRef]
- Li, C.-Q.; Kim, M.Y.; Godoy, L.C.; Thiantanawat, A.; Trudel, L.J.; Wogan, G.N. Nitric oxide activation of Keap1/Nrf2 signaling in human colon carcinoma cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14547–14551. [Google Scholar] [CrossRef]
- Buckley, B.J.; Marshall, Z.M.; Whorton, A.R. Nitric oxide stimulates Nrf2 nuclear translocation in vascular endothelium. Biochem. Biophys. Res. Commun. 2003, 307, 973–979. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, S.; Porter, A.G. Nitric Oxide-induced Transcriptional Up-regulation of Protective Genes by Nrf2 via the Antioxidant Response Element Counteracts Apoptosis of Neuroblastoma Cells. J. Biol. Chem. 2004, 279, 20096–20107. [Google Scholar] [CrossRef]
- Liu, X.-M.; Peyton, K.J.; Ensenat, D.; Wang, H.; Hannink, M.; Alam, J.; Durante, W. Nitric oxide stimulates heme oxygenase-1 gene transcription via the Nrf2/ARE complex to promote vascular smooth muscle cell survival. Cardiovasc. Res. 2007, 75, 381–389. [Google Scholar] [CrossRef]
- Sunshine, S.B.; Sunshine, J.C.; Cano, M.; Green, J.; Handa, J.T. Nitric Oxide Induces Nrf2 Signaling In RPE Cells In Vitro. Investig. Ophth. Vis. Sci. 2011, 52, 2345. [Google Scholar]
- Fourquet, S.; Guerois, R.; Biard, D.; Toledano, M.B. Activation of NRF2 by Nitrosative Agents and H2O2 Involves KEAP1 Disulfide Formation. J. Biol. Chem. 2010, 285, 8463–8471. [Google Scholar] [CrossRef] [PubMed]
- Um, H.-C.; Jang, J.-H.; Kim, D.-H.; Lee, C.; Dong, Z. Nitric oxide activates Nrf2 through s-nitrosylation of Keap1 in PC12 cells. Nitric Oxide 2011, 25, 161–168. [Google Scholar] [CrossRef]
- Bozza, P.T.; Magalhães, K.G.; Weller, P.F. Leukocyte lipid bodies—Biogenesis and functions in inflammation. Biochim. et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2009, 1791, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Manderson, A.P.; Kay, J.G.; Hammond, L.A.; Brown, D.L.; Stow, J.L. Subcompartments of the macrophage recycling endosome direct the differential secretion of IL-6 and TNFα. J. Cell Biol. 2007, 178, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Murray, R.Z.; Stow, J.L. Cytokine Secretion in Macrophages: SNAREs, Rabs, and Membrane Trafficking. Front. Immunol. 2014, 5, 538. [Google Scholar] [CrossRef]
- Castoldi, A.; Monteiro, L.B.; Bakker, N.V.T.; Sanin, D.E.; Rana, N.; Corrado, M.; Cameron, A.M.; Hässler, F.; Matsushita, M.; Caputa, G.; et al. Triacylglycerol synthesis enhances macrophage inflammatory function. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Basudhar, D.; Glynn, S.A.; Greer, M.; Somasundaram, V.; No, J.H.; Scheiblin, D.A.; Garrido, P.; Heinz, W.F.; Ryan, A.E.; Weiss, J.M.; et al. Coexpression of NOS2 and COX2 accelerates tumor growth and reduces survival in estrogen receptor-negative breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 13030–13035. [Google Scholar] [CrossRef]
- Taylor, B.S.; Alarcon, L.H.; Billiar, T.R. Inducible nitric oxide synthase in the liver: Regulation and function. Biochemistry 1998, 63, 766–781. [Google Scholar]
- Gross, T.J.; Kremens, K.; Powers, L.S.; Brink, B.; Knutson, T.; Domann, F.E.; Philibert, R.A.; Milhem, M.M.; Monick, M.M. Epigenetic Silencing of the HumanNOS2Gene: Rethinking the Role of Nitric Oxide in Human Macrophage Inflammatory Responses. J. Immunol. 2014, 192, 2326–2338. [Google Scholar] [CrossRef]
- Albina, J.E. On the expression of nitric oxide synthase by human macrophages. Why no NO? J. Leukoc. Biol. 1995, 58, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-S.; Rai, P.R.; Chu, H.W.; Cool, C.; Chan, E.D. Analysis of Nitric Oxide Synthase and Nitrotyrosine Expression in Human Pulmonary Tuberculosis. Am. J. Respir. Crit. Care Med. 2002, 166, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, S.; Bonecini-Almeida, M.D.G.; Silva, J.R.L.E.; Nathan, C.; Xie, Q.W.; Mumford, R.; Weidner, J.R.; Calaycay, J.; Geng, J.; Boechat, N.; et al. Inducible nitric oxide synthase in pulmonary alveolar macrophages from patients with tuberculosis. J. Exp. Med. 1996, 183, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Kohsaka, H.; Liu, M.F.; Higashiyama, H.; Hirata, Y.; Kanno, K.; Saito, I.; Miyasaka, N. Nitric oxide production and inducible nitric oxide synthase expression in inflammatory arthritides. J. Clin. Investig. 1995, 96, 2357–2363. [Google Scholar] [CrossRef]
- McInnes, I.B.; Leung, B.P.; Field, M.D.; Wei, X.; Huang, F.; Sturrock, R.D.; Kinninmonth, A.W.G.; Weidner, J.R.; Mumford, R.A.; Liew, F. Production of nitric oxide in the synovial membrane of rheumatoid and osteoarthritis patients. J. Exp. Med. 1996, 184, 1519–1524. [Google Scholar] [CrossRef]
- Bagasra, O.; Michaels, F.H.; Zheng, Y.M.; Bobroski, L.E.; Spitsin, S.V.; Fu, Z.F.; Tawadros, R.; Koprowski, H. Activation of the inducible form of nitric oxide synthase in the brains of patients with multiple sclerosis. Proc. Natl. Acad. Sci. USA 1995, 92, 12041–12045. [Google Scholar] [CrossRef]
- De Groot, C.J.; Ruuls, S.R.; Theeuwes, J.W.M.; Dijkstra, C.D.; Van Der Valk, P. Immunocytochemical Characterization of the Expression of Inducible and Constitutive Isoforms of Nitric Oxide Synthase in Demyelinating Multiple Sclerosis Lesions. J. Neuropathol. Exp. Neurol. 1997, 56, 10–20. [Google Scholar] [CrossRef]
- Konttinen, Y.T.; Platts, L.A.M.; Tuominen, S.; Eklund, K.K.; Santavirta, N.; Törnwall, J.; Sorsa, T.; Hukkanen, M.; Polak, J.M. Role of nitric oxide in Sjögren’s syndrome. Arthritis Rheum. 1997, 40, 875–883. [Google Scholar] [CrossRef]
- Wang, C.-H.; Liu, C.-Y.; Lin, H.-C.; Yu, C.-T.; Chung, K.F.; Kuo, H.-P. Increased exhaled nitric oxide in active pulmonary tuberculosis due to inducible NO synthase upregulation in alveolar macrophages. Eur. Respir. J. 1998, 11, 809–815. [Google Scholar] [CrossRef]
- Mathrani, V.; Kenyon, N.; Zeki, A.; Last, J. Mouse models of asthma: Can they give us mechanistic insights into the role of nitric oxide? Curr. Med. Chem. 2007, 14, 2204–2213. [Google Scholar] [CrossRef]
- Martin, J.H.; Edwards, S.W. Interferon-gamma enhances monocyte cytotoxicity via enhanced reactive oxygen intermediate production. Absence of an effect on macrophage cytotoxicity is due to failure to enhance reactive nitrogen intermediate production. Immunology 1994, 81, 592–597. [Google Scholar] [PubMed]
- Padgett, E.L.; Pruett, S.B. Evaluation of nitrite production by human monocyte-derived macrophages. Biochem. Biophys. Res. Commun. 1992, 186, 775–781. [Google Scholar] [CrossRef]
- Schneemann, M.; Schoedon, G.; Hofer, S.; Blau, N.; Guerrero, L.; Schaffner, A. Nitric Oxide Synthase Is Not a Constituent of the Antimicrobial Armature of Human Mononuclear Phagocytes. J. Infect. Dis. 1993, 167, 1358–1363. [Google Scholar] [CrossRef]
- Bukrinsky, M.; Nottet, H.S.; Schmidtmayerova, H.; Dubrovsky, L.; Flanagan, C.R.; Mullins, M.E.; Lipton, S.A.; Gendelman, H.E. Regulation of nitric oxide synthase activity in human immunodeficiency virus type 1 (HIV-1)-infected monocytes: Implications for HIV-associated neurological disease. J. Exp. Med. 1995, 181, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.B.; Misukonis, M.A.; Shami, P.J.; Mason, S.N.; Sauls, D.L.; Dittman, W.A.; Wood, E.R.; Smith, G.K.; McDonald, B.; Bachus, K.E.; et al. Human mononuclear phagocyte inducible nitric oxide synthase (iNOS): Analysis of iNOS mRNA, iNOS protein, biopterin, and nitric oxide production by blood monocytes and peritoneal macrophages. Blood 1995, 86, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fernández, M.A.; Fernández, M.A.; Fresno, M. Activation of human macrophages for the killing of intracellular Trypanosoma cruzi by TNF-alpha and IFN-gamma through a nitric oxide-dependent mechanism. Immunol. Lett. 1992, 33, 35–40. [Google Scholar] [CrossRef]
- Denis, M. Tumor Necrosis Factor and Granulocyte Macrophage-Colony Stimulating Factor Stimulate Human Macrophages to Restrict Growth of Virulent Mycobacterium avium and to Kill Avirulent, M. avium: Killing Effector Mechanism Depends on the Generation of Reactive Nitr. J. Leukoc. Biol. 1991, 49, 380–387. [Google Scholar] [CrossRef]
- Cameron, M.L.; Granger, D.L.; Weinberg, J.B.; Kozumbo, W.J.; Koren, H.S. Human Alveolar and Peritoneal Macrophages Mediate Fungistasis Independently of l-Arginine Oxidation to Nitrite or Nitrate. Am. Rev. Respir. Dis. 1990, 142, 1313–1319. [Google Scholar] [CrossRef]
- Schoedon, G.; Troppmair, J.; Fontana, A.; Huber, C.; Curtius, H.-C.; Niederwieser, A. Biosynthesis and metabolism of pterins in peripheral blood mononuclear cells and leukemia lines of man and mouse. JBIC J. Biol. Inorg. Chem. 1987, 166, 303–310. [Google Scholar] [CrossRef]
- Sakai, N.; Milstien, S. Availability of Tetrahydrobiopterin Is Not a Factor in the Inability to Detect Nitric Oxide Production by Human Macrophages. Biochem. Biophys. Res. Commun. 1993, 193, 378–383. [Google Scholar] [CrossRef]
- Mautino, G.; Paul-Eugène, N.; Chanez, P.; Vignola, A.M.; Kolb, J.P.; Bousquet, J.; Dugas, B. Heterogeneous spontaneous and interleukin-4-induced nitric oxide production by human monocytes. J. Leukoc. Biol. 1994, 56, 15–20. [Google Scholar] [CrossRef]
- Kolb, J.-P.; Paul-Eugene, N.; Damais, C.; Yamaoka, K.; Drapier, J.C.; Dugas, B. Interleukin-4 stimulates cGMP production by IFN-gamma-activated human monocytes. Involvement of the nitric oxide synthase pathway. J. Biol. Chem. 1994, 269, 9811–9816. [Google Scholar] [PubMed]
- De Maria, R.; Cifone, M.G.; Trotta, R.; Rippo, M.R.; Festuccia, C.; Santoni, A.; Testi, R. Triggering of human monocyte activation through CD69, a member of the natural killer cell gene complex family of signal transducing receptors. J. Exp. Med. 1994, 180, 1999–2004. [Google Scholar] [CrossRef]
- Vouldoukis, I.; Riveros-Moreno, V.; Dugas, B.; Ouaaz, F.; Becherel, P.; Debre, P.; Moncada, S.; Mossalayi, M.D. The killing of Leishmania major by human macrophages is mediated by nitric oxide induced after ligation of the Fc epsilon RII/CD23 surface antigen. Proc. Natl. Acad. Sci. USA 1995, 92, 7804–7808. [Google Scholar] [CrossRef]
- Kobzik, L. Translating NO Biology into Clinical Advances. Am. J. Respir. Cell Mol. Biol. 2009, 41, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, V.; Pradhan, P.; Braud, L.; Fuchs, H.R.; Gueler, F.; Motterlini, R.; Foresti, R.; Immenschuh, S. Human and murine macrophages exhibit differential metabolic responses to lipopolysaccharide—A divergent role for glycolysis. Redox Biol. 2019, 22, 101147. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W.; et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef]
- Llufrio, E.M.; Wang, L.; Naser, F.J.; Patti, G.J. Sorting cells alters their redox state and cellular metabolome. Redox Biol. 2018, 16, 381–387. [Google Scholar] [CrossRef]
- Li, L.M.; Kilbourn, R.G.; Adams, J.; Fidler, I.J. Role of nitric oxide in lysis of tumor cells by cytokine-activated endothelial cells. Cancer Res. 1991, 51, 2531–2535. [Google Scholar]
- Jiang, H.; Stewart, C.A.; Fast, D.J.; Leu, R.W. Tumor target-derived soluble factor synergizes with IFN-γ and IL-2 to activate macrophages for tumor necrosis factor and nitric oxide production to mediate cytotoxicity of the same target. J. Immunol. 1992, 149, 2137–2146. [Google Scholar]
- Xiao, L.; Eneroth, P.H.E.; Qureshi, G.A. Nitric Oxide Synthase Pathway May Mediate Human Natural Killer Cell Cytotoxicity. Scand. J. Immunol. 1995, 42, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D. Role in nitric oxide in Kupffer cell-mediated hepatoma cell cytotoxicity in vitro and ex vivo. Hepatology 1996, 24, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Nath, N.; Kashfi, K. Tumor associated macrophages and ‘NO’. Biochem. Pharmacol. 2020, 176, 113899. [Google Scholar] [CrossRef]
- Lee, C.-T.; Mace, T.; Repasky, E.A. Hypoxia-driven immunosuppression: A new reason to use thermal therapy in the treatment of cancer? Int. J. Hyperth. 2010, 26, 232–246. [Google Scholar] [CrossRef]
- Wink, D.A.; Vodovotz, Y.; Laval, J.; Laval, F.; Dewhirst, M.W.; Mitchell, J.B. The multifaceted roles of nitric oxide in cancer. Carcinogenesis 1998, 19, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.F.; Sonveaux, P. Targeting Tumor Perfusion and Oxygenation to Improve the Outcome of Anticancer Therapy1. Front. Pharmacol. 2012, 3, 94. [Google Scholar] [CrossRef]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef]
- Perrotta, C.; Cervia, D.; Di Renzo, I.; Moscheni, C.; Bassi, M.T.; Campana, L.; Martelli, C.; Catalani, E.; Giovarelli, M.; Zecchini, S.; et al. Nitric Oxide Generated by Tumor-Associated Macrophages Is Responsible for Cancer Resistance to Cisplatin and Correlated With Syntaxin 4 and Acid Sphingomyelinase Inhibition. Front. Immunol. 2018, 9, 1186. [Google Scholar] [CrossRef]
- Newsholme, P. Why Is l-Glutamine Metabolism Important to Cells of the Immune System in Health, Postinjury, Surgery or Infection? J. Nutr. 2001, 131, 2515S–2522S. [Google Scholar] [CrossRef]
- Wong, P.S.-Y.; Hyun, J.; Fukuto, J.M.; Shirota, F.N.; DeMaster, E.G.; Shoeman, D.W.; Nagasawa, H.T. Reaction between s-Nitrosothiols and Thiols: Generation of Nitroxyl (HNO) and Subsequent Chemistry. Biochemistry 1998, 37, 5362–5371. [Google Scholar] [CrossRef]
- Arnelle, D.; Stamler, J. NO+, NO, and NO− Donation by s-Nitrosothiols: Implications for Regulation of Physiological Functions by s-Nitrosylation and Acceleration of Disulfide Formation. Arch. Biochem. Biophys. 1995, 318, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Miranda, K.M. The chemistry of nitroxyl (HNO) and implications in biology. Co-ord. Chem. Rev. 2005, 249, 433–455. [Google Scholar] [CrossRef]
- Miranda, K.M.; Paolocci, N.; Katori, T.; Thomas, D.D.; Ford, E.; Bartberger, M.D.; Espey, M.G.; Kass, D.A.; Feelisch, M.; Fukuto, J.M.; et al. A biochemical rationale for the discrete behavior of nitroxyl and nitric oxide in the cardiovascular system. Proc. Natl. Acad. Sci. USA 2003, 100, 9196–9201. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.L.; Toscano, J.P.; Bartberger, M.D.; Fukuto, J.M. The chemical biology of HNO signaling. Arch. Biochem. Biophys. 2016, 617, 129–136. [Google Scholar] [CrossRef]
- DeMaster, E.G.; Redfern, B.; Nagasawa, H.T. Mechanisms of Inhibition of Aldehyde Dehydrogenase by Nitroxyl, the Active Metabolite of the Alcohol Deterrent Agent Cyanamide. Biochem. Pharmacol. 1998, 55, 2007–2015. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palmieri, E.M.; McGinity, C.; Wink, D.A.; McVicar, D.W. Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight. Metabolites 2020, 10, 429. https://doi.org/10.3390/metabo10110429
Palmieri EM, McGinity C, Wink DA, McVicar DW. Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight. Metabolites. 2020; 10(11):429. https://doi.org/10.3390/metabo10110429
Chicago/Turabian StylePalmieri, Erika M., Christopher McGinity, David A. Wink, and Daniel W. McVicar. 2020. "Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight" Metabolites 10, no. 11: 429. https://doi.org/10.3390/metabo10110429
APA StylePalmieri, E. M., McGinity, C., Wink, D. A., & McVicar, D. W. (2020). Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight. Metabolites, 10(11), 429. https://doi.org/10.3390/metabo10110429