Electrophile Modulation of Inflammation: A Two-Hit Approach


Abstract
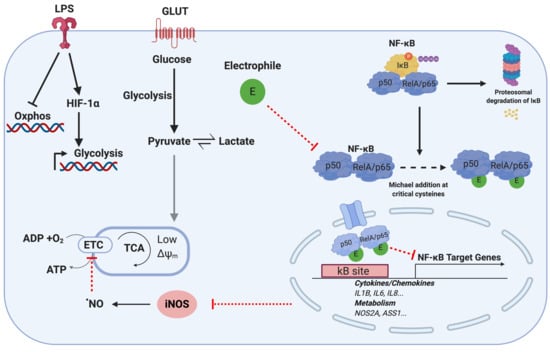
1. Introduction
2. Electrophile Overview, Structure and Function
3. Metabolism and Inflammation
3.1. NRF2
3.2. NF-κB
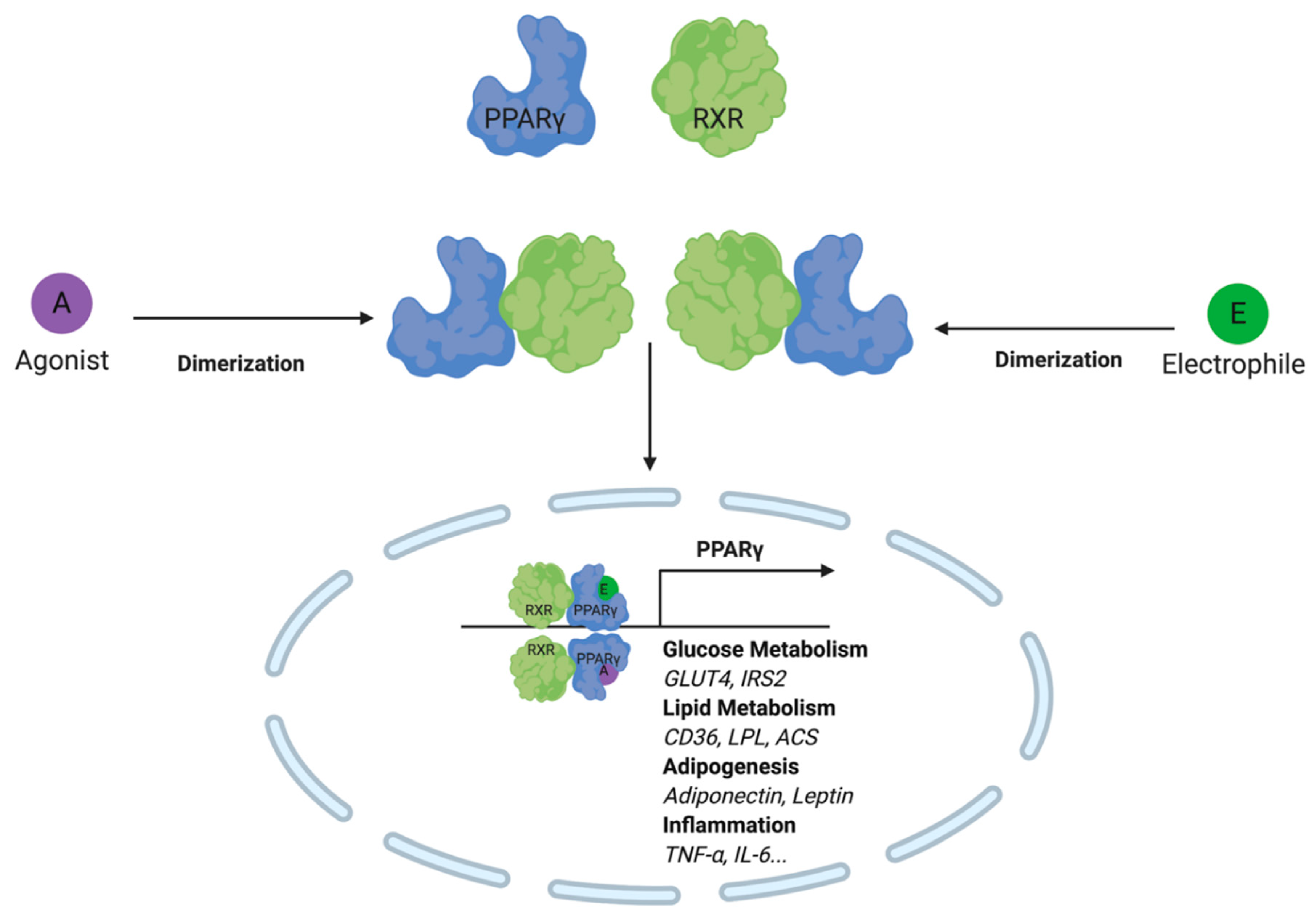
3.3. PPARγ
4. Electrophiles as Potential Therapeutics
4.1. Lipid Electrophiles
4.2. Dimethyl Fumarate
4.3. Bardoxolone
4.4. Sulforaphane
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Weynad, C.; Jörg, G. Immunometabolism in early and late stages of rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory Mechanisms in Obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed]
- Sorci-Thomas, M.G.; Thomas, M.J. Microdomains, Inflammation, and Atherosclerosis. Circ. Res. 2016, 118, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.; Gilroy, D.; Serhan, C. Pro-Resolving lipid mediators and Mechanisms in the resolution of acute inflammation. Immunity 2014, 40, 315–327, Pro-Resolving. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Novel Pro-Resolving Lipid Mediators in Inflammation Are Leads for Resolution Physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef]
- Sugimoto, M.A.; Sousa, L.P.; Pinho, V.; Perretti, M.; Teixeira, M.M. Resolution of inflammation: What controls its onset? Front. Immunol. 2016, 7, 160. [Google Scholar] [CrossRef]
- McGarry, T.; Biniecka, M.; Veale, D.J.; Fearon, U. Hypoxia, oxidative stress and inflammation. Free. Radic. Biol. Med. 2018, 125, 15–24. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2008, 417, 1–13. [Google Scholar] [CrossRef]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Kettle, A.J. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid. Redox Signal. 2013, 18, 642–660. [Google Scholar] [CrossRef] [PubMed]
- Franchina, D.G.; Dostert, C.; Brenner, D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol. 2018, 39, 489–502. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, I.; Mullen, L.; Bekeschus, S.; Hanschmann, E.-M. Redox Regulation of Inflammatory Processes Is Enzymatically Controlled. Oxid. Med. Cell Longev. 2017, 2017, 8459402. [Google Scholar] [CrossRef]
- Brüne, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; Von Knethen, A.; Weigert, A. Redox control of inflammation in macrophages. Antioxid. Redox Signal. 2013, 19, 595–637. [Google Scholar] [CrossRef] [PubMed]
- Schopfer, F.J.; Cipollina, C.; Freeman, B.A. Formation and signaling actions of electrophilic lipids. Chem. Rev. 2011, 111, 5997–6021. [Google Scholar] [CrossRef]
- Groeger, A.L.; Freeman, B.A. Signaling Actions of Electrophiles. Mol. Interv. 2010, 10, 39–50. [Google Scholar] [CrossRef]
- Lopachin, R.M.; Barber, D.S.; Gavin, T. Molecular mechanisms of the conjugated α,β-unsaturated carbonyl derivatives: Relevance to neurotoxicity and neurodegenerative diseases. Toxicol. Sci. 2008, 104, 235–249. [Google Scholar] [CrossRef]
- Farmer, E.E.; Davoine, C. Reactive electrophile species. Curr. Opin. Plant. Biol. 2007, 10, 380–386. [Google Scholar] [CrossRef]
- Dick, R.A.; Kwak, M.K.; Sutter, T.R.; Kensler, T.W. Antioxidative function and substrate specificity of NAD(P)H-dependent alkenal/one oxidoreductase. A new role for leukotriene B4 12-hydroxydehydrogenase/15-oxoprostaglandin 13-reductase. J. Biol. Chem. 2001, 276, 40803–40810. [Google Scholar] [CrossRef]
- Erlemann, K.-R.; Cossette, C.; Grant, G.E.; Lee, G.-J.; Patel, P.; Rokach, J.; Powell, W.S. Regulation of 5-hydroxyeicosanoid dehydrogenase activity in monocytic cells. Biochem. J. 2007, 403, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Gebicki, J.; Puhl, H.; Jürgens, G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free. Radic. Biol. Med. 1992, 13, 341–390. [Google Scholar] [CrossRef]
- Yamada, S.; Kumazawa, S.; Ishii, T.; Nakayama, T.; Itakura, K.; Shibata, N.; Kobayashi, M.; Sakai, K.; Osawa, T.; Uchida, K. Immunochemical detection of a lipofuscin-like fluorophore derived from malondialdehyde and lysine. J. Lipid Res. 2001, 42, 1187–1196. [Google Scholar] [PubMed]
- Koenitzer, J.R.; Bonacci, G.; Woodcock, S.R.; Chen, C.-S.; Cantu-Medellin, N.; Kelley, E.E.; Schopfer, F.J. Fatty acid nitroalkenes induce resistance to ischemic cardiac injury by modulating mitochondrial respiration at complex II. Redox Biol. 2016, 8, 1–10. [Google Scholar] [CrossRef]
- Esterbauer, H.; Cheeseman, K.H.; Dianzani, M.U.; Poli, G.; Slater, T.F. Separation and characterization of the aldehydic products of lipid peroxidation stimulated by ADP-Fe2+ in rat liver microsomes. Biochem. J. 1982, 208, 129–140. [Google Scholar] [CrossRef]
- Esterbauer, H.; Cheeseman, K.H. Determination of aldehydic lipid peroxidation products: Malonaldehyde and 4-hydroxynonenal. Methods Enzymol. 1990, 186, 407–421. [Google Scholar] [CrossRef]
- Doorn, J.A.; Petersen, D.R. Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxy-2-nonenal and 4-oxo-2-nonenal. Chem. Res. Toxicol. 2002, 15, 1445–1450. [Google Scholar] [CrossRef]
- Uchida, K.; Kanematsu, M.; Morimitsu, Y.; Osawa, T.; Noguchi, N.; Niki, E. Acrolein is a product of lipid peroxidation reaction. Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J. Biol. Chem. 1998, 273, 16058–16066. [Google Scholar] [CrossRef]
- Musiek, E.S.; Brooks, J.D.; Joo, M.; Brunoldi, E.; Porta, A.; Zanoni, G.; Vidari, G.; Blackwell, T.S.; Montine, T.J.; Milne, G.L.; et al. Electrophilic cyclopentenone neuroprostanes are anti-inflammatory mediators formed from the peroxidation of the omega-3 polyunsaturated fatty acid docosahexaenoic acid. J. Biol. Chem. 2008, 283, 19927–19935. [Google Scholar] [CrossRef]
- Liebler, D. Protein Damage by Reactive Electrophiles: Targets and Consequences. Chem. Res. Toxicol. 2008, 21, 117–128. [Google Scholar] [CrossRef]
- Hong, F.; Sekhar, K.E.; Freeman, M.L.; Liebler, D.C. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J. Biol. Chem. 2005, 280, 31768–31775. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Saleh, S.; Liebler, D.C. Reversibility of covalent electrophile-protein adducts and chemical toxicity. Chem. Res. Toxicol. 2008, 21, 2361–2369. [Google Scholar] [CrossRef] [PubMed]
- Davoine, C.; Douki, T.; Iacazio, G.; Montillet, J.L.; Triantaphylidès, C. Conjugation of keto fatty acids to glutathione in plant tissues. Characterization and quantification by HPLC-tandem mass spectrometry. Anal. Chem. 2005, 77, 7366–7372. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Oe, T.; Blair, I. Determination of cellular redox status by stable isotope dilution liquid chromatography/mass spectrometry analysis of glutathione and glutathione disulfide. Rapid Commun. Mass Spectrom. 2008, 22, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Jobbagy, S.; Vitturi, D.A.; Salvatore, S.R.; Turell, L.; Pires, M.F.; Kansanen, E.; Batthyany, C.; Lancaster, J.R.; Freeman, B.A.; Schopfer, F.J. Electrophiles modulate glutathione reductase activity via alkylation and upregulation of glutathione biosynthesis. Redox Biol. 2019, 21, 101050. [Google Scholar] [CrossRef] [PubMed]
- Nerland, D. The antioxidant/electrophile response element motif. Drug Metab. Rev. 2007, 39, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Tong, K.I.; Yamamoto, M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free. Radic. Biol. Med. 2004, 36, 1208–1213. [Google Scholar] [CrossRef]
- Lingappan, K. NF-κB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Khoo, N.K.H.; Li, L.; Salvatore, S.R.; Schopfer, F.J.; Freeman, B.A. Electrophilic fatty acid nitroalkenes regulate Nrf2 and NF-κB signaling:A medicinal chemistry investigation of structure-function relationships. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Groeger, A.L.; Cipollina, C.; Cole, M.P.; Woodcock, S.R.; Bonacci, G.; Rudolph, T.K.; Rudolph, V.; Freeman, B.A.; Schopfer, F.J. Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids NIH Public Access Author Manuscript. Nat. Chem. Biol. 2010, 6, 433–441. [Google Scholar] [CrossRef]
- Li, Y.H.; Yan, Z.Q.; Brauner, A.; Tullus, K. Activation of macrophage nuclear factor-κB and induct ion of inducible nitric oxide synthase by LPS. Respir Res. 2002, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, W.; Shi, B.; Klein, S.; Su, X. Peroxisomal regulation of redox homeostasis and adipocyte metabolism. Redox Biol. 2019, 24, 101167. [Google Scholar] [CrossRef] [PubMed]
- Muralikumar, S.; Vetrivel, U.; Narayanasamy, A.; Das, U.N. Probing the intermolecular interactions of PPARγ-LBD with polyunsaturated fatty acids and their anti-inflammatory metabolites to infer most potential binding moieties. Lipids Health Dis. 2017, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kuri-Harcuch, W.; Velez-delValle, C.; Vazquez-Sandoval, A.; Hernández-Mosqueira, C.; Fernandez-Sanchez, V. A cellular perspective of adipogenesis transcriptional regulation. J. Cell Physiol. 2019, 234, 1111–1129. [Google Scholar] [CrossRef]
- Levonen, A.L.; Dickinson, D.A.; Moellering, D.R.; Timothy Mulcahy, R.; Forman, H.J.; Darley-Usmar, V.M. Biphasic effects of 15-deoxy-Δ12,14-prostaglandin J2 on glutathione induction and apoptosis in human endothelial cells. Arter. Thromb. Vasc. Biol. 2001, 21, 1846–1851. [Google Scholar] [CrossRef]
- Rudolph, V.; Rudolph, T.K.; Schopfer, F.J.; Bonacci, G.; Woodcock, S.R.; Cole, M.P.; Baker, P.R.; Ramani, R.; Freeman, B.A. Endogenous generation and protective effects of nitro-fatty acids in a murine model of focal cardiac ischaemia and reperfusion. Cardiovasc. Res. 2010, 85, 155–166. [Google Scholar] [CrossRef]
- Falletti, O.; Douli, T. Low glutathione level favors formation of DNA adducts to 4-hydroxy-2(E)-nonenal, a major lipid peroxidation product. Chem. Res. Toxicol. 2008, 21, 2097–2105. [Google Scholar] [CrossRef]
- Perluigi, M.; Sultana, R.; Cenini, G.; Di Domenico, F.; Memo, M.; Pierce, W.M.; Coccia, R.; Butterfield, D.A. Redox proteomics identification of 4-hydroxynonenal-modified brain proteins in Alzheimer’s disease: Role of lipid peroxidation in Alzheimer’s disease pathogenesis. Proteom. Clin. Appl. 2010, 3, 682–693. [Google Scholar] [CrossRef]
- Codreanu, S.G.; Ullery, J.C.; Zhu, J.; Tallman, K.A.; Beavers, W.N.; Porter, N.A.; Marnett, L.J.; Zhang, B.; Liebler, D.C. Alkylation damage by lipid electrophiles targets functional protein systems. Mol. Cell Proteomics. 2014, 13, 849–859. [Google Scholar] [CrossRef]
- West, J.D.; Marnett, L.J. Endogenous reactive intermediates as modulators of cell signaling and cell death. Chem Res. Toxicol. 2006, 19, 173–194. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Andrejeva, G.; Rathmell, J.C. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Hannink, M. Distinct Cysteine Residues in Keap1 Are Required for Keap1-Dependent Ubiquitination of Nrf2 and for Stabilization of Nrf2 by Chemopreventive Agents and Oxidative Stress. Mol. Cell Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; Von Knethen, A. Nrf2, the master regulator of anti-oxidative responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2015, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Bonacci, G.; Schopfer, F.J.; Kuosmanen, S.M.; Tong, K.I.; Leinonen, H.; Woodcock, S.R.; Yamamoto, M.; Carlberg, C.; Ylä-Herttuala, S.; et al. Electrophilic nitro-fatty acids activate Nrf2 by a Keap1 cysteine 151-independent mechanism. J. Biol. Chem. 2011, 286, 14019–14027. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Eggler, A.L.; Mesecar, A.D.; van Breemen, R.B. Modification of keap1 cysteine residues by sulforaphane. Chem. Res. Toxicol. 2011, 24, 515–521. [Google Scholar] [CrossRef]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial Role of Nrf2 in Regulating NADPH Generation and Consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef]
- Emitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell. 2012, 22, 66–79. [Google Scholar] [CrossRef]
- Lee, J.-M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar] [PubMed]
- Singh, A.; Happel, C.; Manna, S.K.; Acquaah-Mensah, G.K.; Carrerero, J.; Kumar, S.; Nasipuri, P.; Krausz, K.W.; Wakabayashi, N.; Dewi, R.; et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Investig. 2013, 123, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Chartoumpekis, D.V.; Ziros, P.G.; Psyrogiannis, A.I.; Papavassiliou, A.G.; Kyriazopoulou, V.E.; Sykiotis, G.P.; Habeos, I.G. Nrf2 represses FGF21 during long-term high-fat—Induced obesity in mice. Diabetes 2011, 60, 2465–2473. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Y.; Li, Y.; Yu, Q.; Jin, X.; Wang, X.; Jia, A.; Hu, Y.; Han, L.; Wang, J.; et al. HIF1α-dependent glycolysis promotes macrophage functional activities in protecting against bacterial and fungal infection. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Köhler, T.; Reizis, B.; Johnson, R.S.; Weighardt, H.; Förster, I. Influence of hypoxia-inducible factor 1α on dendritic cell differentiation and migration. Eur. J. Immunol. 2012, 42, 1226–1236. [Google Scholar] [CrossRef]
- Cho, S.H.; Raybuck, A.L.; Blagih, J.; Kemboi, E.; Haase, V.H.; Jones, R.G.; Boothby, M.R. Hypoxia-inducible factors in CD4+ T cells promote metabolism, switch cytokine secretion, and T cell help in humoral immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 8975–8984. [Google Scholar] [CrossRef]
- Holyoak, T.; Zhang, B.; Deng, J.; Tang, Q.; Prasannan, C.; Fenton, A. Energetic coupling between an oxidizable cysteine and the phosphorylatable N-terminus of human liver pyruvate kinase. Biochemistry 2013, 52, 466–476. [Google Scholar] [CrossRef]
- Anastasiou, D. Inhibition of Pyruvate Kinase M2 by Reactive Oxygen Species Contributes to Cellular Antioxidant Responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef]
- Yates, M.S.; Tran, Q.T.; Dolan, P.M.; Osburn, W.O.; Shin, S.; McCulloch, C.C.; Silkworth, J.B.; Taguchi, K.; Yamamoto, M.; Williams, C.R.; et al. Genetic versus chemoprotective activation of Nrf2 signaling: Overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 2009, 30, 1024–1031. [Google Scholar] [CrossRef]
- Humphries, K.M.; Szweda, L.I. Selective inactivation of α-ketoglutarate dehydrogenase and pyruvate dehydrogenase: Reaction of lipoic acid with 4-hydroxy-2-nonenal. Biochemistry 1998, 37, 15835–15841. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T.R.; Collins, Y.; Abakumova, I.; Chouchani, E.T.; Baranowski, B.; Fearnley, I.M.; Prime, T.A.; Murphy, M.P.; James, A.M. Inactivation of pyruvate dehydrogenase kinase 2 by mitochondrial reactive oxygen species. J. Biol. Chem. 2012, 287, 35153–35160. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T.R.; Prime, T.A.; Harbour, M.E.; Lilley, K.S.; Murphy, M.P. Detection of reactive oxygen species-sensitive thiol proteins by redox difference gel electrophoresis: Implications for mitochondrial redox signaling. J. Biol. Chem. 2007, 282, 22040–22051. [Google Scholar] [CrossRef] [PubMed]
- Cardaci, S.; Filomeni, G.; Ciriolo, M.R. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J. Cell Sci. 2012, 125, 2115–2125. [Google Scholar] [CrossRef]
- Aldini, G.; Carini, M.; Vistoli, G.; Shibata, T.; Kusano, Y.; Gamberoni, L.; Dalle-Donne, I.; Milzani, A.D.G.; Uchida, K. Identification of actin as a 15-deoxy-Delta12,14-prostaglandin J2 target in neuroblastoma cells: Mass spectrometric, computational, and functional approaches to investigate the effect on cytoskeletal derangement. Biochemistry 2007, 46, 2707–2718. [Google Scholar] [CrossRef]
- Cui, T.; Schopfer, F.J.; Zhang, J.; Chen, K.; Ichikawa, T.; Baker, P.R.S.; Batthyany, C.; Chacko, B.K.; Feng, X.; Patel, R.P.; et al. Nitrated fatty acids: Endogenous anti-inflammatory signaling mediators. J. Biol. Chem. 2006, 281, 35686–35698. [Google Scholar] [CrossRef]
- Straus, D.S.; Pascual, G.; Li, M.; Welch, J.S.; Ricote, M.; Hsiang, C.-H.; Sengchanthalangsy, L.L.; Ghosh, G.; Glass, C.K. 15-Deoxy-Δ12,14-prostaglandin J2 inhibits multiple steps in the NF-κB signaling pathway. Proc. Natl. Acad. Sci. USA 2000, 97, 4844–4849. [Google Scholar] [CrossRef]
- Snyder, N.W.; Golin-Bisello, F.; Gao, Y.; Blair, I.A.; Freeman, B.A.; Wendell, S.G. 15-Oxoeicosatetraenoic acid is a 15-hydroxyprostaglandin dehydrogenase-derived electrophilic mediator of inflammatory signaling pathways. Chem. Biol. Interact. 2015, 234, 144–153. [Google Scholar] [CrossRef]
- Villacorta, L.; Chang, L.; Salvatore, S.R.; Ichikawa, T.; Zhang, J.; Petrovic-Djergovic, D.; Jia, L.; Carlsen, H.; Schopfer, F.J.; Freeman, B.A.; et al. Electrophilic nitro-fatty acids inhibit vascular inflammation by disrupting LPS-dependent TLR4 signalling in lipid rafts. Cardiovasc. Res. 2013, 98, 116–124. [Google Scholar] [CrossRef]
- Woodcock, C.-S.C.; Huang, Y.; Woodcock, S.R.; Salvatore, S.R.; Singh, B.; Golin-Bisello, F.; Davidson, N.E.; Neumann, C.A.; Freeman, B.A.; Wendell, S.G. Nitro-fatty acid inhibition of triple-negative breast cancer cell viability, migration, invasion, and tumor growth. J. Biol Chem. 2018, 293, 1120–1137. [Google Scholar] [CrossRef]
- Ambrozova, G.; Fidlerova, T.; Verescakova, H.; Koudelka, A.; Rudolph, T.K.; Woodcock, S.R.; Freeman, B.A.; Kubala, L.; Pekarova, M. Nitro-oleic acid inhibits vascular endothelial inflammatory responses and the endothelial-mesenchymal transition. Biochim. Biophys. Acta Bioenerg. 2016, 1860, 2428–2437. [Google Scholar] [CrossRef] [PubMed]
- Wendell, S.G.; Golin-Bisello, F.; Wenzel, S.; Sobol, R.W.; Holguin, F.; Freeman, B.A. 15-Hydroxyprostaglandin dehydrogenase generation of electrophilic lipid signaling mediators from hydroxy ω-3 fatty acids. J. Biol. Chem. 2015, 290, 5868–5880. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.A. The role of dendritic cells in asthma. J. Allergy Clin. Immunol. 2012, 129, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Morelli, A.E.; Thomson, A.W. Dendritic cells under the spell of prostaglandins. Trends Immunol. 2003, 24, 108–111. [Google Scholar] [CrossRef]
- Delmastro-Greenwood, M.; Freeman, B.A.; Wendell, S.G. Redox-dependent anti-inflammatory signaling actions of unsaturated fatty acids. Annu. Rev. Physiol. 2014, 76, 79–105. [Google Scholar] [CrossRef] [PubMed]
- Cipollina, C.; Di Vincenzo, S.; Gerbino, S.; Siena, L.; Gjomarkaj, M.; Pace, E. Dual anti-oxidant and anti-inflammatory actions of the electrophilic cyclooxygenase-2-derived 17-oxo-DHA in lipopolysaccharide- and cigarette smoke-induced inflammation. Biochim. Biophys Acta. Gen. Subj. 2014, 1840, 2299–2309. [Google Scholar] [CrossRef] [PubMed]
- Cipollina, C.; Di Vincenzo, S.; Siena, L.; Di Sano, C.; Gjomarkaj, M.; Pace, E. 17-oxo-DHA displays additive anti-inflammatory effects with fluticasone propionate and inhibits the NLRP3 inflammasome. Sci. Rep. 2016, 6, 37625. [Google Scholar] [CrossRef]
- Bailey, J.D.; Diotallevi, M.; Nicol, T.; McNeill, E.; Shaw, A.; Chuaiphichai, S.; Hale, A.; Starr, A.; Nandi, M.; Stylianou, E.; et al. Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep. 2019, 28, 218–230.e7. [Google Scholar] [CrossRef]
- Evans, A. PPAR gamma, the good, the bad & the future. Nat. Med. 2013, 19. [Google Scholar] [CrossRef]
- Lefterova, M.I.; Haakonsson, A.K.; Lazar, M.A.; Mandrup, S. PPARγ and the global map of adipogenesis and beyond. Trends Endocrinol. Metab. 2014, 25, 293–302. [Google Scholar] [CrossRef]
- Le Menn, G.; Neels, J.G. Regulation of immune cell function by PPARs and the connection with metabolic and neurodegenerative diseases. Int. J. Mol. Sci. 2018, 19, 1575. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Ricote, M.; Akiyama, T.E.; Gonzalez, F.J.; Glass, C.K. PPARγ and PPARδ negatively regulate specific subsets of lipopolysaccharide and IFN-γ target genes in macrophages. Proc. Natl. Acad. Sci. USA 2003, 100, 6712–6717. [Google Scholar] [CrossRef] [PubMed]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, H.E.; Kulkarni, A.; Lehmann, G.M.; Garcia-Bates, T.M.; Thatcher, T.H.; Huxlin, K.R.; Phipps, R.P.; Sime, P.J. Electrophilic peroxisome proliferator-activated receptor-γ ligands have potent antifibrotic effects in human lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 2009, 41, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Bunney, P.; Zink, A.; Holm, A.; Billington, C.; Kotz, C. Molecular recognition of nitrated fatty acids by PPARγ. Nat. Struct. Mol. Biol. 2008, 15, 865–867. [Google Scholar] [CrossRef]
- Villacorta, L.; Schopfer, F.J.; Zhang, J.; Freeman, B.A.; Eugene, Y. PPARγ and its ligands: Therapeutic implications in cardiovascular disease. Clin. Sci. 2009, 116, 205–218. [Google Scholar] [CrossRef]
- Itoh, T.; Fairall, L.; Amin, K.; Inaba, Y.; Szanto, A.; Bálint, B.L.; Nagy, L.; Yamamoto, K.; Schwabe, J.W.R. Structural basis for the activation of PPARγ by oxidized fatty acids. Nat. Struct. Mol. Biol. 2008, 15, 924–931. [Google Scholar] [CrossRef]
- Waku, T.; Shiraki, T.; Oyama, T.; Fujimoto, Y.; Maebara, K.; Kamiya, N.; Jingami, H.; Morikawa, K. Structural Insight into PPARγ Activation Through Covalent Modification with Endogenous Fatty Acids. J. Mol. Biol. 2009, 385, 188–199. [Google Scholar] [CrossRef]
- Egawa, D.; Itoh, T.; Akiyama, Y.; Saito, T.; Yamamoto, K. 17-OxoDHA Is a PPARα/γ Dual Covalent Modifier and Agonist. ACS Chem. Biol. 2016, 11, 2447–2455. [Google Scholar] [CrossRef]
- Kiss, M.; Czimmerer, Z.; Nagy, L. The role of lipid-activated nuclear receptors in shaping macrophage and dendritic cell function: From physiology to pathology. J. Allergy Clin. Immunol. 2013, 132, 264–286. [Google Scholar] [CrossRef]
- Szatmari, I.; Töröcsik, D.; Agostini, M.; Nagy, T.; Gurnell, M.; Barta, E.; Chatterjee, K.; Nagy, L. PPARγ regulates the function of human dendritic cells primarily by altering lipid metabolism. Blood 2007, 110, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Szatmari, I.; Pap, A.; Rühl, R.; Ma, J.-X.; Illarionov, P.A.; Besra, G.S.; Rajnavolgyi, E.; Dezso, B.; Nagy, L. PPARγ controls CD1d expression by turning on retinoic acid synthesis in developing human dendritic cells. J. Exp. Med. 2006, 203, 2351–2362. [Google Scholar] [CrossRef] [PubMed]
- Szatmari, I.; Gogolak, P.; Im, J.S.; Dezso, B.; Rajnavolgyi, E.; Nagy, L. Activation of PPARγ specifies a dendritic cell subtype capable of enhanced induction of iNKT cell expansion. Immunity 2004, 21, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1beta attenuate macrophage mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef]
- Park, H.-J.; Kim, D.-H.; Choi, J.-Y.; Kim, W.-J.; Kim, J.Y.; Senejani, A.G.; Hwang, S.S.; Kim, L.K.; Tobiasova, Z.; Lee, G.R.; et al. PPARγ negatively regulates T cell activation to prevent follicular helper T cells and germinal center formation. PLoS ONE 2014, 9, e99127. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Eagle, A.R.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef]
- Kanaoka, Y.; Boyce, J.A. Cysteinyl leukotrienes and their receptors; emerging concepts. Allergy Asthma Immunol. Res. 2014, 6, 288–295. [Google Scholar] [CrossRef]
- Kanaoka, Y.; Austen, K.F. Roles of cysteinyl leukotrienes and their receptors in immune cell-related functions. Adv. Immunol. 2019, 142, 65–84. [Google Scholar] [CrossRef]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Ching-Cheng Huang, S.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2017, 24, 158–166. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Hooftman, A.; Angiari, S.; Hester, S.; Corcoran, S.E.; Runtsch, M.C.; Ling, C.; Ruzek, M.C.; Slivka, P.F.; McGettrick, A.F.; Banahan, K.; et al. The Immunomodulatory Metabolite Itaconate Modifies NLRP3 and Inhibits Inflammasome Activation. Cell Metab. 2020, 32, 468–478.e7. [Google Scholar] [CrossRef] [PubMed]
- Bollong, M.J.; Lee, G.; Coukos, J.S.; Yun, H.; Zambaldo, C.; Chang, J.W.; Chin, E.N.; Ahmad, I.; Chatterjee, A.K.; Lairson, L.L.; et al. A metabolite-derived protein modification integrates glycolysis with KEAP1–NRF2 signalling. Nature 2018, 562, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, D.O.; Jennings, E.Q.; Anderson, C.C.; Marentette, J.O.; Shi, T.; Oxvig, A.-M.S.; Streeter, M.D.; Johannsen, M.; Spiegel, D.A.; Chapman, E.; et al. Non-enzymatic Lysine Lactoylation of Glycolytic Enzymes. Cell Chem. Biol. 2020, 27, 206–213.e6. [Google Scholar] [CrossRef]
- Ichikawa, T.; Zhang, J.; Chen, K.; Liu, Y.; Schopfer, F.J.; Baker, P.R.S.; Freeman, B.A.; Chen, Y.E.; Cui, T. Nitroalkenes suppress lipopolysaccharide-induced signal transducer and activator of transcription signaling in macrophages: A critical role of mitogen-activated protein kinase phosphatase 1. Endocrinology 2008, 149, 4086–4094. [Google Scholar] [CrossRef]
- Gorczynski, M.J.; Smitherman, P.K.; Akiyama, T.E.; Wood, H.B.; Berger, J.P.; King, S.B.; Morrow, C.S. Activation of peroxisome proliferator-activated receptor γ (PPARγ) by nitroalkene fatty acids: Importance of nitration position and degree of unsaturation. J. Med. Chem. 2009, 52, 4631–4639. [Google Scholar] [CrossRef]
- Kansanen, E.; Jyrkkänen, H.-K.; Volger, O.L.; Leinonen, H.; Kivelä, A.M.; Häkkinen, S.-K.; Woodcock, S.R.; Schopfer, F.J.; Horrevoets, A.J.; Ylä-Herttuala, S.; et al. Nrf2-dependent and -independent responses to nitro-fatty acids in human endothelial cells: Identification of heat shock response as the major pathway activated by nitro-oleic acid. J. Biol. Chem. 2009, 284, 33233–33241. [Google Scholar] [CrossRef]
- Nadtochiy, S.M.; Baker, P.R.S.; Freeman, B.A.; Brookes, P.S. Mitochondrial nitroalkene formation and mild uncoupling in ischaemic preconditioning: Implications for cardioprotection. Cardiovasc. Res. 2009, 82, 333–340. [Google Scholar] [CrossRef]
- Hurd, T.R.; Costa, N.J.; Dahm, C.C.; Beer, S.M.; Brown, S.E.; Filipovska, A.; Murphy, M.P. Glutathionylation of Mitochondrial Proteins. Antioxid. Redox Signal. 2005, 7, 999–1010. [Google Scholar] [CrossRef]
- Humphries, K.M.; Yoo, Y.; Szweda, L.I. Inhibition of NADH-linked mitochondrial respiration by 4-hydroxy-2-nonenal. Biochemistry 1998, 37, 552–557. [Google Scholar] [CrossRef]
- Chen, J.; Schenker, S.; Frosto, T.A.; Henderson, G.I. Inhibition of cytochrome c oxidase activity by 4-hydroxynonenal (HNE). Role of HNE adduct formation with the enzyme subunits. Biochim. Biophys. Acta Gen. Subj. 1998, 1380, 336–344. [Google Scholar] [CrossRef]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta. Mol. Basis Dis. 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Di Cara, F.; Andreoletti, P.; Trompier, D.; Vejux, A.; Bülow, M.H.; Sellin, J.; Lizard, G.; Cherkaoui-Malki, M.; Savary, S. Peroxisomes in immune response and inflammation. Int. J. Mol. Sci. 2019, 20, 3887. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A.; Waterham, H.R. Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 2006, 75, 295–332. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef]
- Horie, T.; Ono, K.; Nagao, K.; Nishi, H.; Kinoshita, M.; Kawamura, T.; Wada, H.; Shimatsu, A.; Kita, T.; Hasegawa, K. Oxidative stress induces GLUT4 translocation by activation of PI3-K/Akt and dual AMPK kinase in cardiac myocytes. J. Cell Physiol. 2008, 215, 733–742. [Google Scholar] [CrossRef]
- Griffiths, H.R.; Gao, D.; Pararasa, C. Redox regulation in metabolic programming and inflammation. Redox Biol. 2017, 12, 50–57. [Google Scholar] [CrossRef]
- Reagan, L.P.; Magariños, A.M.; Yee, D.K.; Swzeda, L.I.; Van Bueren, A.; McCall, A.L.; McEwen, B.S. Oxidative stress and HNE conjugation of GLUT3 are increased in the hippocampus of diabetic rats subjected to stress. Brain Res. 2000, 862, 292–300. [Google Scholar] [CrossRef]
- Chavez, J.; Chung, W.-G.; Miranda, C.L.; Singhal, M.; Stevens, J.F.; Maier, C.S. Site-specific protein adducts of 4-hydroxy-2(E)-nonenal in human THP-1 monocytic cells: Protein carbonylation is diminished by ascorbic acid. Chem. Res. Toxicol. 2010, 23, 37–47. [Google Scholar] [CrossRef]
- Batthyany, C.; Schopfer, F.J.; Baker, P.R.S.; Durán, R.; Baker, L.M.S.; Huang, Y.; Cerveñansky, C.; Branchaud, B.P.; Freeman, B.A. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J. Biol. Chem. 2006, 281, 20450–20463. [Google Scholar] [CrossRef]
- Ishii, T.; Tatsuda, E.; Kumazawa, S.; Nakayama, T.; Uchida, K. Molecular basis of enzyme inactivation by an endogenous electrophile 4-hydroxy-2-nonenal: Identification of modification sites in glyceraldehyde-3-phosphate dehydrogenase. Biochemistry 2003, 42, 3474–3480. [Google Scholar] [CrossRef] [PubMed]
- Oliva, J.L.; Pérez-Sala, D.; Castrillo, A.; Martínez, N.; Cañada, F.J.; Boscá, L.; Rojas, J.M. The cyclopentenone 15-deoxy-delta 12,14-prostaglandin J2 binds to and activates H-Ras. Proc. Natl. Acad. Sci. USA 2003, 100, 4772–4777. [Google Scholar] [CrossRef] [PubMed]
- Sampey, B.P.; Carbone, D.L.; Doorn, J.A.; Drechsel, D.A.; Petersen, D.R. 4-Hydroxy-2-nonenal adduction of extracellular signal-regulated kinase (Erk) and the inhibition of hepatocyte Erk-Est-like protein-1-activating protein-1 signal transduction. Mol. Pharmacol. 2007, 71, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.T.; Han, C.; Xu, D.Q.; Fu, X.W.; Wang, J.S.; Kong, L.Y. 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to exert anti-inflammatory effects. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Balak, D.M. Fumaric acid esters in the management of psoriasis. Psoriasis 2015, 5, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Litjens, N.H.R.; Van Strijen, E.; Van Gulpen, C.; Mattie, H.; Van Dissel, J.T.; Thio, H.B.; Nibbering, P.H. In vitro pharmacokinetics of anti-psoriatic fumaric acid esters. BMC Pharmacol. 2004, 4, 22. [Google Scholar] [CrossRef]
- Mills, E.A.; Ogrodnik, M.A.; Plave, A.; Mao-Draayer, Y. Emerging Understanding of the Mechanism of Action for Dimethyl Fumarate in the Treatment of Multiple Sclerosis. Front. Neurol. 2018, 9, 5. [Google Scholar] [CrossRef]
- Bomprezzi, R. Dimethyl fumarate in the treatment of relapsing-remitting multiple sclerosis: An overview. Ther. Adv. Neurol. Disord. 2015, 8, 20–30. [Google Scholar] [CrossRef]
- Kornberg, M.D.; Bhargava, P.; Kim, P.M.; Putluri, V.; Snowman, A.M.; Putluri, N.; Calabresi, P.A.; Snyder, S.H. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 2018, 360, 449–453. [Google Scholar] [CrossRef]
- Blewett, M.M.; Xie, J.; Zaro, B.W.; Backus, K.M.; Altman, A.; Teijaro, J.R.; Cravatt, B.F. Chemical proteomic map of dimethyl fumarate-sensitive cysteines in primary human T cells. Sci. Signal. 2016, 9, 1–11. [Google Scholar] [CrossRef]
- Zaro, B.W.; Vinogradova, E.V.; Lazar, D.C.; Blewett, M.M.; Suciu, R.M.; Takaya, J.; Studer, S.; De La Torre, J.C.; Casanova, J.-L.; Cravatt, B.F.; et al. Dimethyl Fumarate Disrupts Human Innate Immune Signaling by Targeting the IRAK4–MyD88 Complex. J. Immunol. 2019, 202, 2737–2746. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Fitzgerald, K.C.; Venkata, S.L.V.; Smith, M.D.; Kornberg, M.D.; Mowry, E.M.; Haughey, N.J.; Calabresi, P.A. Dimethyl fumarate treatment induces lipid metabolism alterations that are linked to immunological changes. Ann. Clin. Transl. Neurol. 2019, 6, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Lee, D.-H.; Ryan, S.; Van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Ockenfels, H.M.; Schultewolter, T.; Ockenfels, G.; Funk, R.; Goos, M. The antipsoriatic agent dimethylfumarate immunomodulates T-cell cytokine secretion and inhibits cytokines of the psoriatic cytokine network. Br. J. Dermatol. 1998, 139, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Guerau-De-Arellano, M.; Mehta, V.B.; Yang, Y.; Huss, D.J.; Papenfuss, T.L.; Lovett-Racke, A.E.; Racke, M.K. Dimethyl fumarate inhibits dendritic cell maturation via nuclear factor κB (NF-κB) and extracellular signal-regulated kinase 1 and 2 (ERK1/2) and mitogen stress-activated kinase 1 (MSK1) signaling. J. Biol. Chem. 2012, 287, 28017–28026. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, Q.; Mao, G.; Dowling, C.A.; Lundy, S.K.; Mao-Draayer, Y. Dimethyl Fumarate Selectively Reduces Memory T Cells and Shifts the Balance between Th1/Th17 and Th2 in Multiple Sclerosis Patients. J. Immunol. 2017, 198, 3069–3080. [Google Scholar] [CrossRef]
- Montes Diaz, G.; Fraussen, J.; Van Wijmeersch, B.; Hupperts, R.; Somers, V. Dimethyl fumarate induces a persistent change in the composition of the innate and adaptive immune system in multiple sclerosis patients. Sci. Rep. 2018, 8, 8194. [Google Scholar] [CrossRef]
- Refaat, A.; Pararasa, C.; Arif, M.; Brown, J.E.P.; Carmichael, A.; Ali, S.S.; Sakurai, H.; Griffiths, H.R. Bardoxolone-methyl inhibits migration and metabolism in MCF7 cells. Free. Radic. Res. 2017, 51, 211–221. [Google Scholar] [CrossRef]
- Kulkarni, A.A.; Thatcher, T.H.; Hsiao, H.-M.; Olsen, K.C.; Kottmann, R.M.; Morrissette, J.; Wright, T.W.; Phipps, R.P.; Sime, P.J. The Triterpenoid CDDO-Me Inhibits Bleomycin-Induced Lung Inflammation and Fibrosis. PLoS ONE 2013, 8, e63798. [Google Scholar] [CrossRef]
- De Zeeuw, D.; Akizawa, T.; Agarwal, R.; Audhya, P.; Bakris, G.L.; Chin, M.; Krauth, M.; Heerspink, H.J.L.; Meyer, C.J.; McMurray, J.J.; et al. Rationale and trial design of bardoxolone methyl evaluation in patients with chronic kidney disease and type 2 diabetes: The occurrence of renal events (BEACON). Am. J. Nephrol. 2013, 37, 212–222. [Google Scholar] [CrossRef]
- Qin, D.-J.; Tang, C.-X.; Yang, L.; Lei, H.; Wei, W.; Wang, Y.-Y.; Ma, C.-M.; Gao, F.-H.; Xu, H.-Z.; Wu, Y.-L. Hsp90 Is a Novel Target Molecule of CDDO-Me in Inhibiting Proliferation of Ovarian Cancer Cells. PLoS ONE 2015, 10, e0132337. [Google Scholar] [CrossRef] [PubMed]
- Jansson, K.H.; Tucker, J.B.; Stahl, L.E.; Simmons, J.K.; Fuller, C.; Beshiri, M.L.; Agarwal, S.; Fang, L.; Hynes, P.G.; Alilin, A.N.; et al. High-throughput screens identify HSP90 inhibitors as potent therapeutics that target inter-related growth and survival pathways in advanced prostate cancer. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C.; Angelin, A.; Lisanti, S.; Kossenkov, A.V.; Speicher, K.D.; Wang, H.; Michalek, R.D. Landscape of the mitrochondrial Hsp90 metabolome in tumors. Nat. Commun. 2013, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.; Risingsong, R.; Royce, D.; Williams, C.R.; Sporn, M.B.; Liby, K. The synthetic triterpenoid CDDO-methyl ester delays estrogen receptor-negative mammary carcinogenesis in polyoma middle T mice. Cancer Prev. Res. 2012, 5, 726–734. [Google Scholar] [CrossRef]
- Ball, M.S.; Shipman, E.P.; Kim, H.; Liby, K.T.; Pioli, P.A. CDDO-Me redirects activation of breast tumor associated macrophages. PLoS ONE 2016, 11, e0149600. [Google Scholar] [CrossRef]
- Ball, M.S.; Bhandari, R.; Torres, G.M.; Martyanov, V.; Eltanbouly, M.A.; Archambault, K.; Whitfield, M.L.; Liby, K.T.; Pioli, P.A. CDDO-Me Alters the Tumor Microenvironment in Estrogen Receptor Negative Breast Cancer. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Zhao, Y.; Ge, X.; He, J.; Cheng, Y.; Wang, Z.; Wang, J.; Sun, L. The prognostic value of tumor-infiltrating lymphocytes in colorectal cancer differs by anatomical subsite: A systematic review and meta-analysis. World J. Surg. Oncol. 2019, 17, 1–11. [Google Scholar] [CrossRef]
- Idos, G.E.; Kwok, J.; Bonthala, N.; Kysh, L.; Gruber, S.B.; Qu, C. The Prognostic Implications of Tumor Infiltrating Lymphocytes in Colorectal Cancer: A Systematic Review and Meta-Analysis. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The metabolic signature of macrophage responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Mehla, K.; Singh, P.K. Metabolic Regulation of Macrophage Polarization in Cancer. Trends Cancer 2019, 5, 822–834. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Talalay, P.; Cho, C.G.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef] [PubMed]
- Heber, D.; Bowerman, S. Research Conference on Diet, Nutrition and Cancer Applying Science to Changing Dietary Patterns 1. J. Nutr. 2001, 131, 3078–3081. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Axelsson, A.S.; Vial, G.; Wollheim, C.B.; Rieusset, J.; Rosengren, A.H. Sulforaphane improves disrupted ER-mitochondria interactions and suppresses exaggerated hepatic glucose production. Mol. Cell Endocrinol. 2018, 461, 205–214. [Google Scholar] [CrossRef]
- Chaudhuri, D.; Orsulic, S.; Ashok, B.T. Antiproliferative activity of sulforaphane in Akt-overexpressing ovarian cancer cells. Mol. Cacer Ther. 2007, 6, 334–345. [Google Scholar] [CrossRef]
- Liang, J.; Jahraus, B.; Balta, E.; Ziegler, J.D.; Hübner, K.; Blank, N.; Niesler, B.; Wabnitz, G.H.; Samstag, Y. Sulforaphane inhibits inflammatory responses of primary human T-cells by increasing ROS and depleting glutathione. Front. Immunol. 2018, 9, 2584. [Google Scholar] [CrossRef]
- Geisel, J.; Brück, J.; Glocova, I.; Dengler, K.; Sinnberg, T.; Rothfuss, O.; Walter, M.; Schulze-Osthoff, K.; Röcken, M.; Ghoreschi, K. Sulforaphane Protects from T Cell–Mediated Autoimmune Disease by Inhibition of IL-23 and IL-12 in Dendritic Cells. J. Immunol. 2014, 192, 3530–3539. [Google Scholar] [CrossRef]
- Teng, W.; Li, Y.; Du, M.; Lei, X.; Xie, S.; Ren, F. Sulforaphane Prevents Hepatic Insulin Resistance by Blocking Serine Palmitoyltransferase 3-Mediated Ceramide Biosynthesis. Nutrients 2019, 11, 1185. [Google Scholar] [CrossRef]
- Xu, D.; Xu, M.; Jeong, S.; Qian, Y.; Wu, H.; Xia, Q.; Kong, X. The role of Nrf2 in liver disease: Novel molecular mechanisms and therapeutic approaches. Front. Pharmacol. 2019, 9, 1428. [Google Scholar] [CrossRef]
- Singh, K.B.; Kim, S.H.; Hahm, E.R.; Pore, S.K.; Jacobs, B.L.; Singh, S.V. Prostate cancer chemoprevention by sulforaphane in a preclinical mouse model is associated with inhibition of fatty acid metabolism. Carcinogenesis 2018, 39, 826–837. [Google Scholar] [CrossRef]
- Lee, C.-H.; Jeong, S.-J.; Yun, S.-M.; Kim, J.-H.; Lee, H.-J.; Ahn, K.S.; Won, S.-H.; Kim, H.; Lee, H.-J.; Ahn, K.-S.; et al. Down-regulation of phosphoglucomutase 3 mediates sulforaphane-induced cell death in LNCaP prostate cancer cells. Proteome Sci. 2010, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; De Mooij, T.; Peterson, T.E.; Kaptzan, T.; Johnson, A.J.; Daniels, D.J.; Parney, I.F. Modulating glioma-mediated myeloid-derived suppressor cell development with sulforaphane. PLoS ONE 2017, 12, e0179012. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-Cell dysfunction in glioblastoma: Applying a new framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.; Chawla, A. The immune system as a sensor of the metabolic state. Immunity 2013, 38, 644–654. [Google Scholar] [CrossRef]
- Jantsch, J.; Chakravortty, D.; Turza, N.; Prechtel, A.T.; Buchholz, B.; Gerlach, R.G.; Volke, M.; Gläsner, J.; Warnecke, C.; Wiesener, M.S.; et al. Hypoxia and Hypoxia-Inducible Factor-1α Modulate Lipopolysaccharide-Induced Dendritic Cell Activation and Function. J. Immunol. 2008, 180, 4697–4705. [Google Scholar] [CrossRef]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef]
- Yan, D.; Adeshakin, A.O.; Xu, M.; Afolabi, L.O.; Zhang, G.; Chen, Y.H.; Wan, X. Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Front. Immunol. 2019, 10, 1399. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Electrophile | Residue | Ref |
|---|---|---|---|
| GAPDH | NO2-OA | C-153, C-244, H-108, H-134, H-327 | [130] |
| DMF | C-152, C-156, C-247 | [135] | |
| HNE | C-244, C-281, H-327, H-164, K-331 | [131] | |
| 4-OI | C-22 | [134] | |
| Aldolase | HNE | H-246 | [132] |
| Enolase | HNE | Not well-defined | [75,129] |
| GLUT3 | HNE | Not well-defined | [128] |
| LDH | 15-d-PGJ2 | Not well-defined | [75] |
| H-Ras | 15-d-PGJ2 | C-184 | [132] |
| Erk-2 | HNE | H-178 | [133] |
| α-KGDH | HNE | Lipoic acid sulfhydryl | [71] |
| PDH | HNE | Lipoic acid sulfhydryl | [71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Brien, J.; Wendell, S.G. Electrophile Modulation of Inflammation: A Two-Hit Approach. Metabolites 2020, 10, 453. https://doi.org/10.3390/metabo10110453
O’Brien J, Wendell SG. Electrophile Modulation of Inflammation: A Two-Hit Approach. Metabolites. 2020; 10(11):453. https://doi.org/10.3390/metabo10110453
Chicago/Turabian StyleO’Brien, James, and Stacy G. Wendell. 2020. "Electrophile Modulation of Inflammation: A Two-Hit Approach" Metabolites 10, no. 11: 453. https://doi.org/10.3390/metabo10110453
APA StyleO’Brien, J., & Wendell, S. G. (2020). Electrophile Modulation of Inflammation: A Two-Hit Approach. Metabolites, 10(11), 453. https://doi.org/10.3390/metabo10110453