
Development of New Extracts of Crocus sativus L. By-Product from Two Different Italian Regions as New Potential Active Ingredient in Cosmetic Formulations

 ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Plant Materials
2.3. Extraction Procedures
2.4. Total Phenol Content Determination
2.5. Evaluation of the Antioxidant Capacity (AC)
2.6. Ultra Performance Liquid Chromatography—Diode Array-Electrospray Ionization-Mass Spectrometry Analysis
2.7. Identification of Unknown Molecules by High-Resolution Mass Spectrometry (HRMS)
2.8. Determination of Cytotoxicity, Cytoprotection and Antioxidant Activity in Keratinocyte Cells
2.8.1. Cell Cultures
2.8.2. UVA and UVB Irradiation
2.8.3. Cytotoxicity
2.8.4. Cytoprotection
2.8.5. Intracellular Antioxidant Activity
2.9. Statistical Analysis
3. Results and Discussion
3.1. Determination of the Total Phenol Content and Antioxidant Capacity of Extracts
3.2. Identification of Flavonols and Anthocyanins
3.2.1. Ultra Performance Liquid Chromatography-Tandem Mass Spectrometry (UPLC-MS/MS)
3.2.2. Identification of Unknown Molecules by High-Resolution Mass Spectrometry (HRMS)
3.2.3. Quantitative Analysis by UPLC-DAD-ESI-MS
3.3. Antioxidant and Cytoprotective Activity of Sibillini Extracts in Human Keratinocytes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fontana, A.R.; Antoniolli, A.; Bottini, R. Grape pomace as a sustainable source of bioactive compounds: Extraction, characterization, and biotechnological applications of phenolics. J. Agric. Food Chem. 2013, 61, 8987–9003. [Google Scholar] [CrossRef]
- Zuin, V.G.; Ramin, L.Z. Green and Sustainable Separation of Natural Products from Agro-Industrial Waste: Challenges, Potentialities, and Perspectives on Emerging Approaches. Top. Cur. Chem. 2018, 376, 3. [Google Scholar] [CrossRef] [PubMed]
- Jadouali, S.M.; Atifi, H.; Bouzoubaa, Z.; Majourhat, K.; Gharby, S.; Achemchem, F.; Elmoslih, A.; Laknifli, A.; Mamouni, R. Chemical characterization, antioxidant and antibacterial activity of Moroccan Crocus sativus L petals and leaves. J. Mater. Environ. Sci. 2018, 9, 113–118. [Google Scholar] [CrossRef]
- Zeka, K.; Ruparelia, K.C.; Continenza, M.A.; Stagos, D.; Veglio, F.; Arroo, R.R.J. Petals of Crocus sativus L. as a potential source of the antioxidants crocin and kaempferol. Fitoterapia 2015, 107, 128–134. [Google Scholar] [CrossRef]
- Loriana, C.; Donato, C.; Michele, P.; Nunzia, C.; Vincenzo, C. Saffron (Crocus sativus L.), the king of spices: An overview. Sci. Hortic. 2020, 272, 109560. [Google Scholar] [CrossRef]
- Maggi, L.; Carmona, M.; Kelly, S.D.; Marigheto, N.; Alonso, G.L. Geographical origin differentiation of saffron spice (Crocus sativus L. stigmas)—Preliminary investigation using chemical and multi-element (H, C, N) stable isotope analysis. Food Chem. 2011, 128, 543–548. [Google Scholar] [CrossRef]
- Karimi, E.; Oskoueian, E.; Hendra, R.; Jaafar, H.Z.E. Evaluation of Crocus sativus L. stigma phenolic and flavonoid compounds and its antioxidant activity. Molecules 2010, 15, 6244–6256. [Google Scholar] [CrossRef] [PubMed]
- Assimopoulou, A.N.; Sinakos, Z.; Papageorgiou, V.P. Radical scavenging activity of Crocus sativus L. extract and its bioactive constituents. Phytother. Res. 2005, 19, 997–1000. [Google Scholar] [CrossRef] [PubMed]
- Mashmoul, M.; Azlan, A.; Khaza’ai, H.; Mohd Yusof, B.N.; Mohd Noor, S. Saffron: A Natural Potent Antioxidant as a Promising Anti-Obesity Drug. Antioxidants 2013, 2, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Tuberoso, C.I.G.; Rosa, A.; Montoro, P.; Fenu, M.A.; Pizza, C. Antioxidant activity, cytotoxic activity and metabolic profiling of juices obtained from saffron (Crocus sativus L.) floral by-product. Food Chem. 2016, 199, 18–27. [Google Scholar] [CrossRef]
- Zeka, K.; Ruparelia, K.C.; Sansone, C.; Macchiarelli, G.; Continenza, M.A.; Arroo, R.R.J. New Hydrogels Enriched with Antioxidants from Saffron Crocus Can Find Applications in Wound Treatment and/or Beautification. Skin Pharmacol. Physiol. 2018, 31, 95–98. [Google Scholar] [CrossRef]
- Moratalla-López, N.; José Bagur, M.; Lorenzo, C.; Martínez-Navarro, M.E.; Salinas, M.R.; Alonso, G.L. Bioactivity and Bioavailability of the Major Metabolites of Crocus sativus L. Flower. Molecules 2019, 24, 2827. [Google Scholar] [CrossRef]
- Li, C.-Y.; Lee, E.J.; Wu, T.-S. Antityrosinase Principles and Constituents of the Petals of Crocus sativus. J. Nat. Prod. 2004, 67, 437–440. [Google Scholar] [CrossRef]
- Serrano-Diaz, J.; Sanchez, A.M.; Martinez-Tome, M.; Winterhalter, P.; Alonso, G.L. Flavonoid Determination in the Quality Control of Floral Bioresidues from Crocus sativus L. J. Agric. Food Chem. 2014, 62, 3125–3133. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Diaz, J.; Estevan, C.; Sogorb, M.A.; Carmona, M.; Alonso, G.L.; Vilanova, E. Cytotoxic effect against 3T3 fibroblasts cells of saffron floral bio-residues extracts. Food Chem. 2014, 147, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Goupy, P.; Vian, M.A.; Chemat, F.; Caris-Veyrat, C. Identification and quantification of flavonols, anthocyanins and lutein diesters in tepals of Crocus sativus by ultra performance liquid chromatography coupled to diode array and ion trap mass spectrometry detections. Ind. Crop. Prod. 2013, 44, 496–510. [Google Scholar] [CrossRef]
- Termentzi, A.; Kokkalou, E. LC-DAD-MS (ESI+) analysis and antioxidant capacity of Crocus sativus petal extracts. Planta Med. 2008, 74, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Vignolini, P.; Heimler, D.; Pinelli, P.; Ieri, F.; Sciullo, A.; Romani, A. Characterization of By-product of Saffron (Crocus sativus L.) Production. Nat. Prod. Commun. 2008, 3. [Google Scholar] [CrossRef]
- Montoro, P.; Tuberoso, C.I.G.; Maldini, M.; Cabras, P.; Pizza, C. Qualitative Profile and Quantitative Determination of Flavonoids from Crocus sativus L. Petals by LC-MS/MS. Nat. Prod. Commun. 2008, 3. [Google Scholar] [CrossRef]
- Zorzetto, C.; Sanchez Mateo, C.C.; Rabanal, R.M.; Lupidi, G.; Petrelli, D.; Vitali, L.A.; Bramucci, M.; Quassinti, L.; Caprioli, G.; Papa, F.; et al. Phytochemical analysis and in vitro biological activity of three Hypericum species from the Canary Islands (Hypericum reflexum, Hypericum canariense and Hypericum grandifolium). Fitoterapia 2015, 100, 95–109. [Google Scholar] [CrossRef]
- Singleton, V.L.; Rossi, J.A. Colorimetry of Total Phenolics with Phosphomolybdic-Phosphotungstic Acid Reagents. Am. J. Enol. Vitic. 1965, 16, 144–158. [Google Scholar]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Venditti, A.; Bianco, A.; Quassinti, L.; Bramucci, M.; Lupidi, G.; Damiano, S.; Papa, F.; Vittori, S.; Maleci Bini, L.; Giuliani, C.; et al. Phytochemical Analysis, Biological Activity, and Secretory Structures of Stachys annua (L.) L. subsp. annua (Lamiaceae) from Central Italy. Chem. Biodivers. 2015, 12, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Ornano, L.; Venditti, A.; Ballero, M.; Sanna, C.; Quassinti, L.; Bramucci, M.; Lupidi, G.F.P.; Vittori, S.; Maggi, F.; Bianco, A. Chemopreventive and antioxidant activity of the chamazulene-rich essential oil obtained from Artemisia arborescens L. growing on the Isle of La Maddalena, Sardinia, Italy. Chem. Biodivers. 2013, 10, 1464–1474. [Google Scholar] [CrossRef]
- Manuela, C.; Maria Rosa, G.; Dolores Vargas, P.; Gianni, S.; Roberta, C.; Piera, D.M. Quantification of phenolic compounds in different types of crafts beers, worts, starting and spent ingredients by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2020, 1612, 460622. [Google Scholar] [CrossRef]
- Tarozzi, A.; Morroni, F.; Merlicco, A.; Hrelia, S.; Angeloni, C.; Cantelli-Forti, G.; Hrelia, P. Sulforaphane as an inducer of glutathione prevents oxidative stress-induced cell death in a dopaminergic-like neuroblastoma cell line. J. Neurochem. 2009, 111, 1161–1171. [Google Scholar] [CrossRef]
- Tarozzi, A.; Marchesl, A.; Hrelia, S.; Angeloni, C.; Andrisano, V.; Fiori, J.; Cantelli-Forti, G.; Hrella, P. Protective Effects of Cyanidin-3-O-β-glucopyranoside Against UVA-induced Oxidative Stress in Human Keratinocytes. Photochem. Photobiol. 2005, 81, 623–629. [Google Scholar] [CrossRef]
- Iliass, L.; Touria, L.; Cédric, D.; Sohely, S.; Pierre, V.; Ennouamane, S.; Véronique, M. The waste of saffron crop, a cheap source of bioactive compounds. J. Funct. Foods 2017, 35, 341–351. [Google Scholar] [CrossRef]
- Menghini, L.; Leporini, L.; Vecchiotti, G.; Locatelli, M.; Carradori, S.; Ferrante, C.; Zengin, G.; Recinella, L.; Chiavaroli, A.; Leone, S.; et al. Crocus sativus L. stigmas and byproducts: Qualitative fingerprint, antioxidant potentials and enzyme inhibitory activities. Food Res. Int. 2018, 109, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Biancamaria, S.; Gabriele, R.; Silvia, G.; Matteo, B.; Pascual, M.d.; Fernandez, J.A.; Luigi, L.; Marco, T. Identification of phenolic markers for saffron authenticity and origin: An untargeted metabolomics approach. Food Res. Int. 2019, 126, 108584. [Google Scholar] [CrossRef]
- Ferreres, F.; Llorach, R.; Gil-Izquierdo, A. Characterization of the interglycosidic linkage in di-, tri-, tetra- and pentaglycosylated flavonoids and differentiation of positional isomers by liquid chromatography/electrospray ionization tandem mass spectrometry. J. Mass Spectrom. 2004, 39, 312–321. [Google Scholar] [CrossRef]
- Nørbæk, R.; Brandt, K.; Nielsen, J.K.; Ørgaard, M.; Jacobsen, N. Flower pigment composition of Crocus species and cultivars used for a chemotaxonomic investigation. Biochem. Syst. Ecol. 2002, 30, 763–791. [Google Scholar] [CrossRef]
- Oyvind, M.A.; Kenneth, R.M. Flavonoids; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar] [CrossRef]
- Giupponi, L.; Ceciliani, G.; Leoni, V.; Panseri, S.; Pavlovic, R.; Lingua, G.; di Filippo, A.; Giorgi, A. Quality traits of saffron produced in Italy: Geographical area effect and good practices. J. Appl. Bot. Food Qual. 2019, 92, 336–342. [Google Scholar]
- Bernerd, F.; Marionnet, C.; Duval, C. Solar ultraviolet radiation induces biological alterations in human skin in vitro: Relevance of a well-balanced UVA/UVB protection. Indian J. Dermatol. Venereol. Leprol. 2012, S15–S23. [Google Scholar] [CrossRef]
- Gęgotek, A.; Bielawska, K.; Biernacki, M.; Zaręba, I.; Surażyński, A.; Skrzydlewska, E. Comparison of protective effect of ascorbic acid on redox and endocannabinoid systems interactions in in vitro cultured human skin fibroblasts exposed to UV radiation and hydrogen peroxide. Arch. Dermatol. Res. 2017, 309, 285–303. [Google Scholar] [CrossRef]
- Moan, J.; Grigalavicius, M.; Baturaite, Z.; Dahlback, A.; Juzeniene, A. The relationship between UV exposure and incidence of skin cancer. Photodermatol. Photoimmunol. Photomed. 2015, 31, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Barolet, D.; Christiaens, F.; Hamblin, M.R. Infrared and skin: Friend or foe. J. Photochem. Photobiol. B Biol. 2016, 155, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.-W.; Hung, Y.-C.; Lin, T.-Y.; Fang, J.-Y.; Yang, P.-M.; Chen, M.-H.; Pan, T.-L. Comparison of the Biological Impact of UVA and UVB upon the Skin with Functional Proteomics and Immunohistochemistry. Antioxidants 2019, 8, 569. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.; Chen, L. Polyphenols and bioavailability: An update. Crit. Rev. Food Sci. Nutr. 2019, 59, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Calò, R.; Visone, C.M.; Marabini, L. Thymol and Thymus vulgaris L. activity against UVA- and UVB-induced damage in NCTC 2544 cell line. Mutat. Res. Genet. Toxicol. Environ. Mutagenesis 2015, 791, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Serini, S.; Cassano, R.; Facchinetti, E.; Amendola, G.; Trombino, S.; Calviello, G. Anti-Irritant and Anti-Inflammatory Effects of DHA Encapsulated in Resveratrol-Based Solid Lipid Nanoparticles in Human Keratinocytes. Nutrients 2019, 11, 1400. [Google Scholar] [CrossRef] [PubMed]
- Abruzzo, A.; Armenise, N.; Bigucci, F.; Cerchiara, T.; Gösser, M.B.; Samorì, C.; Galletti, P.; Tagliavini, E.; Brown, D.M.; Johnston, H.J.; et al. Surfactants from itaconic acid: Toxicity to HaCaT keratinocytes in vitro, micellar solubilization, and skin permeation enhancement of hydrocortisone. Int. J. Pharm. 2017, 524, 9–15. [Google Scholar] [CrossRef]
- Burlando, B.; Parodi, A.; Volante, A.; Bassi, A.M. Comparison of the irritation potentials of Boswellia serrata gum resin and of acetyl-11-keto-β-boswellic acid by in vitro cytotoxicity tests on human skin-derived cell lines. Toxicol. Lett. 2008, 177, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Bonarska-Kujawa, D.; Pruchnik, H.; Kleszczyńska, H. Interaction of selected anthocyanins with erythrocytes and liposome membranes. Cell Mol. Biol. Lett. 2012, 17, 289–308. [Google Scholar] [CrossRef]
- Verjee, S.; Garo, E.; Pelaez, S.; Fertig, O.; Hamburger, M.; Butterweck, V. Saffron Flower Extract Promotes Scratch Wound Closure of Keratinocytes and Enhances VEGF Production. Planta Med. 2017, 83, 1176–1183. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Batches | Total Phenol Content (mg GAE/l) | ABTS μmol TEA/mL) | FRAP (μmol TEA/mL) | DPPH (μmol TEA/mL) |
|---|---|---|---|---|
| A1 (EtOH/H2O Alba) | 151.138 ± 15.205 | 411.222 ± 26.352 | 467.118 ± 10.726 | 518.443 ± 16.288 |
| A2 (H2O Alba) | 1020.951 ± 70.802 | 154.175 ± 4.164 | 419.985 ± 10.811 | 167.229 ± 2.1004 |
| A3 (Gly Alba) | 55.995 ± 0.058 | 888.907 ± 109.053 | 808.927 ± 189.782 | 878.107 ± 10.127 |
| C1 (EtOH/H2O Sibillini) | 204.571 ± 4.885 | 795.218 ± 20.614 | 488.909 ± 10.556 | 684.315 ± 10.822 |
| C2 (H2O Sibillini) | 1400.950 ± 13.369 | 114.884 ± 2.312 | 409.029 ± 7.236 | 110.312 ± 7.588 |
| C3 (Gly Sibillini) | 62.485 ± 7.225 | 1040.150 ± 12.604 | 846.637 ± 16.258 | 998.123 ± 10.556 |
| Peak Number | R.T. | Ionization Mode | Ions | Ions | Error ppm | Formula | Principal Putative ion | MSC % * | MSC % * | Identification |
|---|---|---|---|---|---|---|---|---|---|---|
| Experimental | Theoretical | ChemSpider | PubChem | |||||||
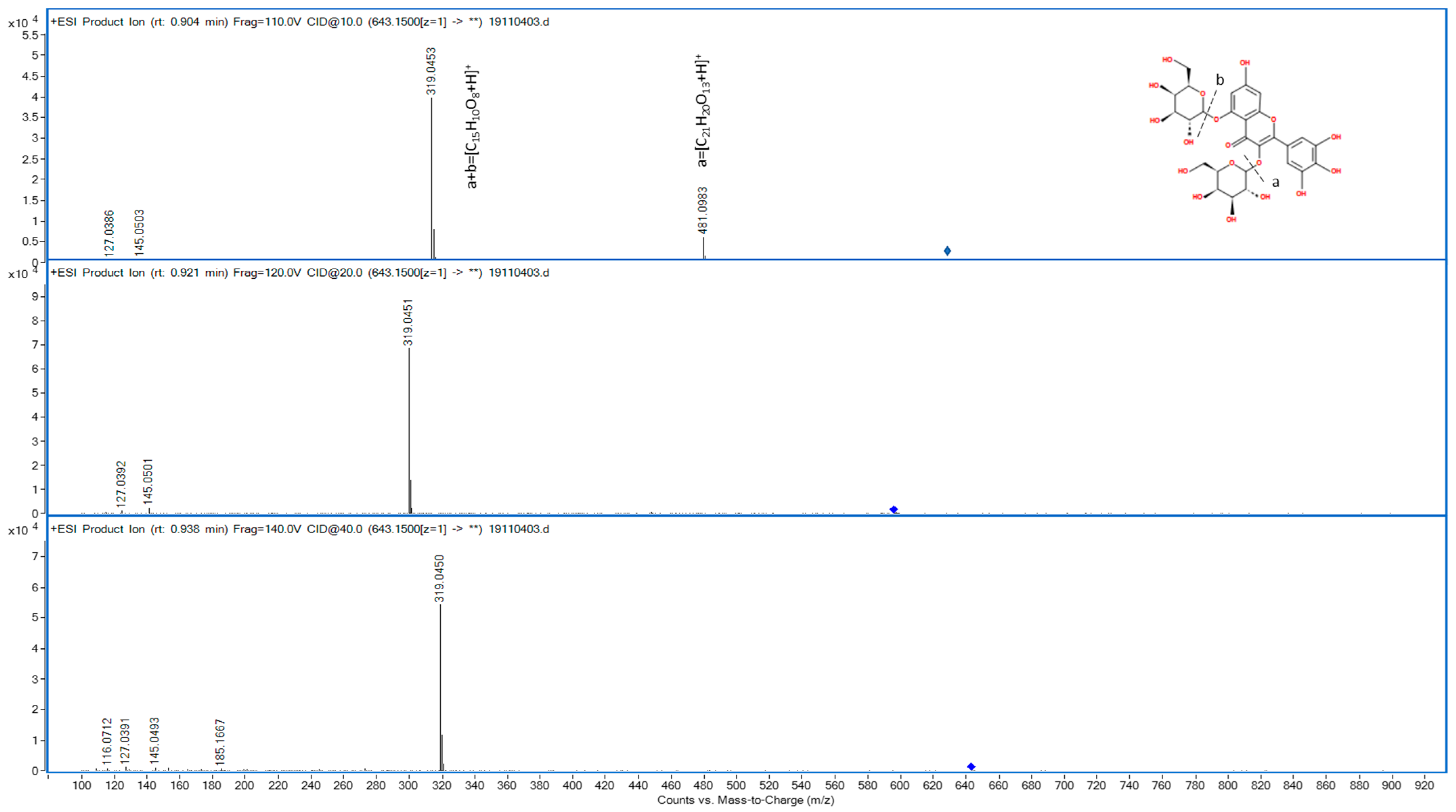
| F3 | 0.895 | Positive | 643.1507 (P) | 643.151 | −0.4665 | [C27H30O18+H]+ | Aglycone-diglucoside | 76.9 | 77.6 | Myricetin-diglucoside |
| 481.0983 (F) | 481.0982 | 0.2079 | Aglycone-glucoside | 99.4 | 99.4 | |||||
| 319.0453 (F) | 319.0454 | −0.3134 | Aglycone | 98.5 | 99.6 | |||||
| F7 | 1.162 | Positive | 713.1930(P) | 713.1929 | 0.1402 | [C31H36O19+H]+ [C31H36O19+Na]+ | Glycoside | 75.7 | 75.7 | Primflasine |
| 735.1745(A) | 735.1748 | −0.4081 | Sodium adduct | 97.7 | 97.7 | |||||
| 551.1399(F) | 551.1395 | 0.7258 | Aglycone-glucoside | 99.6 | 99.6 | |||||
| 287.0553(F) | 287.055 | 1.0451 | Aglycone | |||||||
| Negative | 711.1776(P) | 711.1778 | −0.2812 | [C31H36O19]− | Glycoside | 77.9 | 77.9 | |||
| 667.1500(F) | 667.1516 | −2.3983 | C2H4O | 93.9 | 93.9 | |||||
| 651.1565(F) | 651.1567 | −0.3071 | C2H4O2 | 96.7 | 96.7 | |||||
| 609.1456(F) | 609.1461 | −0.8208 | C4H6O3 | 94.1 | 94.1 | |||||
| 284.0325(F) | 284.0326 | −0.3521 | Aglycone | 99.3 | 99.3 |
| A1 | A2 | A3 | C1 | C2 | C3 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Identification | Conc. 1 | SD% | Conc. 1 | SD% | Conc. 1 | SD% | Conc. 1 | SD% | Conc. 1 | SD% | Conc. 1 | SD% |
| Kaempferol 3,7, 4′- tri-O-glucoside | 0.24 | 0.1 | 2.18 | 0.3 | 1.48 | 0.1 | 1.99 | 0.2 | 1.37 | 0.9 | 1.35 | 0.1 |
| Kaempferol 3,7-O-diglucoside | 0.03 | 0.3 | 0.29 | 0.1 | 0.17 | 0.1 | 0.15 | 7.1 | 0.20 | 8.9 | 0.12 | 0.1 |
| Myricetin diglucoside | 0.09 | 0.1 | 0.19 | 0.2 | 0.16 | 0.3 | 0.19 | 0.1 | 0.30 | 0.3 | 0.14 | 0.2 |
| Quercetin 3,7-O-diglucoside | 0.02 | 0.4 | 0.22 | 0.4 | 0.12 | 1.7 | 0.12 | 6.3 | 0.17 | 0.2 | 0.10 | 0.1 |
| Isorhamnetin 3,7-O-diglucoside | 0.07 | 0.5 | 0.53 | 0.3 | 0.53 | 1.9 | 0.70 | 1.3 | 0.77 | 4.3 | 0.63 | 0.1 |
| Quercetin 3-O-sophoroside | 0.69 | 0.1 | 1.36 | 0.1 | 1.32 | 0.2 | 1.22 | 0.5 | 1.46 | 1.1 | 0.96 | 0.1 |
| Primfrasine | 0.12 | 0.3 | 0.72 | 0.1 | 0.56 | 0.4 | 0.57 | 1.0 | 0.75 | 0.9 | 0.51 | 0.1 |
| Kaempferol 3-O-sophoroside | 12.84 | 0.1 | 17.53 | 0.1 | 17.16 | 0.2 | 18.10 | 0.1 | 21.91 | 0.2 | 12.11 | 0.1 |
| Kaempferol 3-O-rutinoside | 0.04 | 0.1 | 0.08 | 0.2 | 0.06 | 10.5 | 0.07 | 1.7 | 0.11 | 3.7 | 0.05 | 0.1 |
| Isorhamnetin 3-O-rutinoside | 0.25 | 0.2 | 0.46 | 0.2 | 0.65 | 1.0 | 0.42 | 0.1 | 0.57 | 0.4 | 0.38 | 0.1 |
| Quercetin 3-O-glucoside | 0.08 | 0.2 | 0.23 | 0.1 | 0.17 | 0.1 | 0.26 | 0.2 | 0.43 | 0.1 | 0.22 | 0.3 |
| Kaempferol 3-O-(6″- acetyl-glucoside) 7-O-glycoside | 0.01 | 2.9 | 0.01 | 0.1 | 0.01 | 0.1 | 0.01 | 1.9 | 0.02 | 8.3 | 0.01 | 0.1 |
| Kaempferol 3-glucoside | 0.12 | 0.7 | 0.41 | 2.6 | 0.29 | 0.6 | 0.14 | 0.5 | 0.03 | 0.5 | 0.07 | 1.0 |
| Isorhamnetin 3-O-glucoside | 0.33 | 0.1 | 0.74 | 0.1 | 0.54 | 0.1 | 1.24 | 0.1 | 1.45 | 0.1 | 0.98 | 0.1 |
| Kaempferol 3-sophoroside-7-rhamnoside | 0.09 | 0.1 | 0.18 | 0.3 | 0.06 | 1.3 | 0.14 | 1.0 | 0.25 | 1.3 | 0.13 | 0.8 |
| Kaempferol 3-O-(6″-acetyl-galactoside) or Kaempferol 3-O-(6″-acetyl-glucoside) | 0.03 | 0.2 | 0.08 | 1.9 | 0.04 | 2.7 | 0.05 | 0.2 | 0.10 | 6.4 | 0.04 | 0.7 |
| Kaempferol 3-O-(6″-acetyl-galactoside) or Kaempferol 3-O-(6″-acetyl-glucoside) | 0.03 | 7.1 | 0.06 | 0.7 | 0.04 | 0.9 | 0.02 | 0.1 | 0.01 | 2.2 | 0.01 | 1.8 |
| Quercetin 3-O-glucoside-7-O-rhamnoside | 0.02 | 0.8 | 0.04 | 0.2 | 0.05 | 2.1 | 0.10 | 0.1 | 0.05 | 0.1 | 0.08 | 0.2 |
| Kaempferol 3-O-glucoside-7-O-rhamnoside | <LOQ | - | 0.01 | 10.7 | 0.01 | 0.1 | 0.03 | 1.4 | 0.01 | 2.4 | 0.02 | 5.3 |
| Isorhamnetin 3-O-glucoside. 7-O-rhamnoside | 0.04 | 1.2 | 0.01 | 1.4 | 0.01 | 0.5 | 0.02 | 1.3 | 0.01 | 0.1 | 0.02 | 6.0 |
| Kaempferol | 0.11 | 2.8 | 0.03 | 7.0 | 0.13 | 6.1 | 0.10 | 5.8 | 0.28 | 1.7 | 0.07 | 9.1 |
| Total amount | 15.23 | 25.35 | 23.56 | 25.61 | 30.26 | 17.98 | ||||||
| A1 | A2 | A3 | C1 | C2 | C3 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Identification | Conc. 1 | SD% | Conc. 1 | SD% | Conc. 1 | SD% | Conc. 1 | SD% | Conc. 1 | SD% | Conc. 1 | SD% |
| Delphinidin 3,5-O-diglucoside | 0.25 | 0.4 | 0.98 | 0.4 | 0.84 | 0.2 | 1.26 | 0.7 | 1.11 | 1.6 | 0.73 | 0.6 |
| Petunidin 3,5-O-diglucoside | 0.03 | 5.0 | 0.16 | 0.3 | 0.11 | 0.1 | 0.21 | 0.7 | 0.17 | 2.1 | 0.11 | 0.8 |
| Delphinidin 3-O-β-D-glucoside | 0.03 | 0.9 | 0.04 | 0.3 | 0.05 | 0.4 | 0.13 | 1.3 | 0.04 | 3.3 | 0.02 | 2.1 |
| Malvidin diglucoside | <LOQ | 4.0 | 0.01 | 0.8 | 0.01 | 0.1 | 0.03 | 1.2 | 0.01 | 9.4 | 0.01 | 5.7 |
| Delphinidin diglucoside | <LOQ | - | <LOQ | - | <LOQ | - | <LOQ | - | <LOQ | - | <LOQ | - |
| Delphinidin diglucoside | <LOQ | - | <LOQ | - | <LOQ | - | <LOQ | - | <LOQ | - | <LOQ | - |
| Total amount | 0.35 | 1.18 | 1.00 | 1.86 | 1.33 | 0.86 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gigliobianco, M.R.; Cortese, M.; Peregrina, D.V.; Villa, C.; Lupidi, G.; Pruccoli, L.; Angeloni, C.; Tarozzi, A.; Censi, R.; Di Martino, P. Development of New Extracts of Crocus sativus L. By-Product from Two Different Italian Regions as New Potential Active Ingredient in Cosmetic Formulations. Cosmetics 2021, 8, 51. https://doi.org/10.3390/cosmetics8020051
Gigliobianco MR, Cortese M, Peregrina DV, Villa C, Lupidi G, Pruccoli L, Angeloni C, Tarozzi A, Censi R, Di Martino P. Development of New Extracts of Crocus sativus L. By-Product from Two Different Italian Regions as New Potential Active Ingredient in Cosmetic Formulations. Cosmetics. 2021; 8(2):51. https://doi.org/10.3390/cosmetics8020051
Chicago/Turabian StyleGigliobianco, Maria Rosa, Manuela Cortese, Dolores Vargas Peregrina, Carla Villa, Giulio Lupidi, Letizia Pruccoli, Cristina Angeloni, Andrea Tarozzi, Roberta Censi, and Piera Di Martino. 2021. "Development of New Extracts of Crocus sativus L. By-Product from Two Different Italian Regions as New Potential Active Ingredient in Cosmetic Formulations" Cosmetics 8, no. 2: 51. https://doi.org/10.3390/cosmetics8020051
APA StyleGigliobianco, M. R., Cortese, M., Peregrina, D. V., Villa, C., Lupidi, G., Pruccoli, L., Angeloni, C., Tarozzi, A., Censi, R., & Di Martino, P. (2021). Development of New Extracts of Crocus sativus L. By-Product from Two Different Italian Regions as New Potential Active Ingredient in Cosmetic Formulations. Cosmetics, 8(2), 51. https://doi.org/10.3390/cosmetics8020051