Mitochondria and Aging—The Role of Exercise as a Countermeasure



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
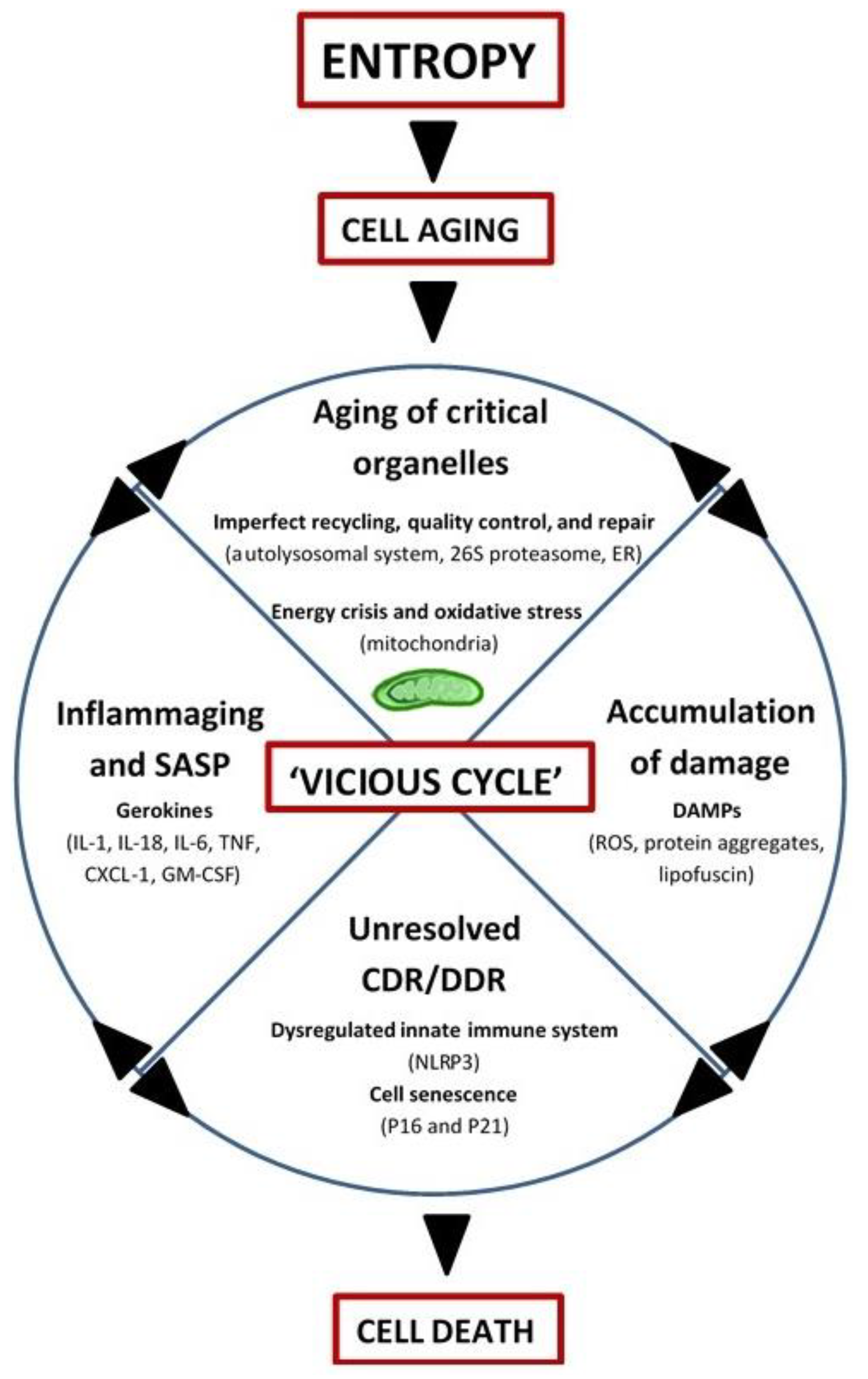
2. Integrated Systems Hypothesis of Aging
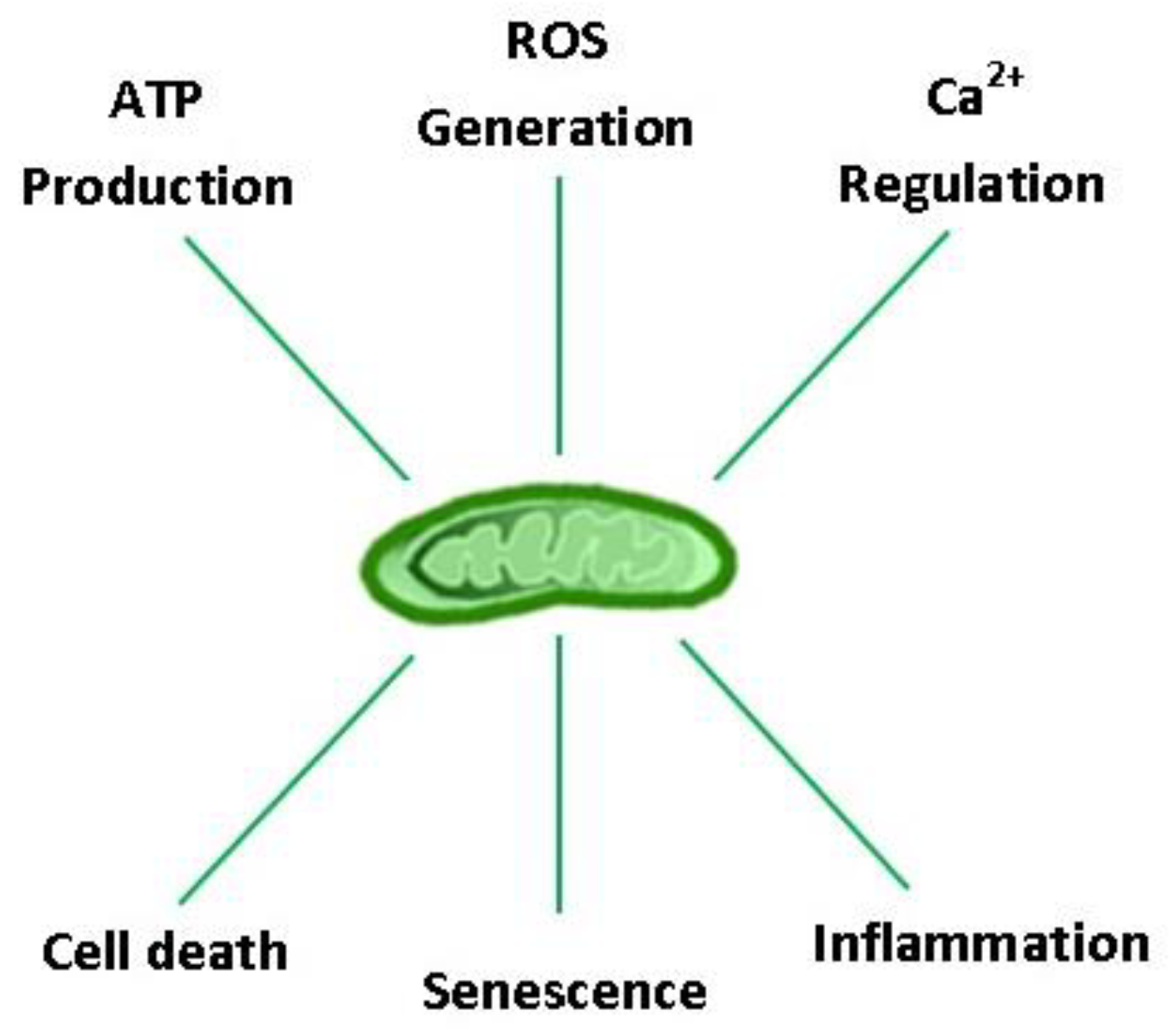
3. Mitochondrial Respiration: Yin and Yang of Aerobic Life
4. Mitochondrial Aging
4.1. Oxidative Stress, mtDNA Mutagenesis, Apoptosis, and Respiration
4.2. Garbage Catastrophe—The Role of Mitochondria
4.3. Inflammaging—The Role of Mitochondria
5. The Anti-Aging Benefits of Physical Activity
5.1. Acute Exercise is Hormesis
5.2. Mitochondrial Rejuvenation
5.3. Intracellular Garbage Clearance
5.4. Boosting the Immune System
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kurland, C.G.; Andersson, S.G.E. Origin and Evolution of the Mitochondrial Proteome. Microbiol. Mol. Biol. Rev. 2000, 64, 786–820. [Google Scholar] [CrossRef]
- Holland, H. The oxygenation of atmosphere and oceans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 903–915. [Google Scholar] [CrossRef]
- Lane, N. Bioenergetic Constraints on the Evolution of Complex Life. Cold Harb. Perspect. Boil. 2014, 6, a015982. [Google Scholar] [CrossRef] [PubMed]
- Stamati, K.; Mudera, V.; Cheema, U. Evolution of oxygen utilization in multicellular organisms and implications for cell signalling in tissue engineering. J. Tissue Eng. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Burini, R.C.; Leonard, W.R. The evolutionary roles of nutrition selection and dietary quality in the human brain size and encephalization. Nutrire 2018, 43, 19. [Google Scholar] [CrossRef]
- Wood, B.; Collard, M. The Human Genus. Science 1999, 284, 65–71. [Google Scholar] [CrossRef]
- Bramble, D.M.; Lieberman, D.E. Endurance running and the evolution of Homo. Nature 2004, 432, 345. [Google Scholar] [CrossRef] [PubMed]
- Pontzer, H. Economy and Endurance in Human Evolution. Curr. Biol. 2017, 27, R613–R621. [Google Scholar] [CrossRef]
- Kodama, S.; Saito, K.; Tanaka, S.; Maki, M.; Yachi, Y.; Asumi, M.; Sugawara, A.; Totsuka, K.; Shimano, H.; Ohashi, Y.; et al. Cardiorepiratory fitness as a quantitative predictor of all-cause mortality and cardiovascular events in healthy men and women: A meta-analysis. JAMA 2009, 301, 2024–2035. [Google Scholar] [CrossRef] [PubMed]
- Reimers, C.D.; Knapp, G.; Reimers, A.K. Does physical activity increase life expectancy? A review of the literature. J. Aging Res. 2012, 9, 2012. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Brellenthin, A.; Thompson, P.; Sui, X.; Lee, I.; Lavie, C. Running as a key lifestyle medicine for longevity. Prog. Cardiovasc. Dis. 2017, 60, 45–55. [Google Scholar] [CrossRef]
- Pimentel, A.E.; Gentile, C.L.; Tanaka, H.; Seals, D.R.; Gates, P.E. Greater rate of decline in maximal aerobic capacity with age in endurance-trained than in sedentary men. J. Appl. Physiol. 2003, 94, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Desouza, C.A.; Jones, P.P.; Stevenson, E.T.; Davy, K.P.; Seals, D.R. Greater rate of decline in maximal aerobic capacity with age in physically active vs. sedentary healthy women. J. Appl. Physiol. 1997, 83, 1947–1953. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: The Mitochondrial-Lysosomal Axis Theory of Aging. Antioxid. Redox Signal. 2010, 12, 503–535. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; MacNeil, L.; Kitaoka, Y.; Suri, R.; Young, S.; Kaczor, J.; Nates, N.; Ansari, M.; Wong, T.; Ahktar, M.; et al. Combined aerobic exercise and enzyme replacement therapy rejuvenates the mitochondrial-lysosomal axis and alleviates autophagic blockage in Pompe disease. Free Radic. Biol. Med. 2015, 87, 98–112. [Google Scholar] [CrossRef]
- Hyttinena, J.M.; Amadiob, M.; Viiri, J.; Pascaleb, A.; Salminenc, A.; Kaarnirantaa, K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. [Google Scholar] [CrossRef]
- Nilsson, M.I.; Bourgeois, J.M.; Nederveen, J.P.; Leite, M.R.; Hettinga, B.P.; Bujak, A.L.; May, L.; Lin, E.; Crozier, M.; Rusiecki, D.R.; et al. Lifelong aerobic exercise protects against inflammaging and cancer. PLoS ONE 2019, 14, e0210863. [Google Scholar] [CrossRef]
- Mehta, M.M.; Weinberg, S.E.; Chandel, N.S. Mitochondrial control of immunity: Beyond ATP. Nat. Rev. Immunol. 2017, 17, 608. [Google Scholar] [CrossRef]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef]
- Garatachea, N.; Pareja-Galeano, H.; Sanchis-Gomar, F.; Santos-Lozano, A.; Fiuza-Luces, C.; Moran, M.; Emanuele, E.; Joyner, M.J.; Lucia, A. Exercise attenuates the major hallmarks of aging. Rejuvenation Res. 2014, 18, 57–89. [Google Scholar] [CrossRef] [PubMed]
- Fiuza-Luces, C.; Garatachea, N.; Berger, N.; Lucia, A. Exercise is the real polypill. Physiology 2013, 28, 330–358. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, Z.A. An attempt at a rational classification of theories of ageing. Biol. Rev. 1990, 65, 375–398. [Google Scholar]
- Schrodinger, E. What is life. In The Physical Aspect of the Living Cell; McMillan Co.: New York, NY, USA, 1947. [Google Scholar]
- Bortz, W.M. Aging as entropy. Exp. Gerontol. 1986, 21, 321–328. [Google Scholar] [CrossRef]
- Hayflick, L. Aging: The reality: Anti-aging is an oxymoron. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2004, 59, B573–B578. [Google Scholar] [CrossRef]
- Hayflick, L. Entropy explains aging, genetic determinism explains longevity, and undefined terminology explains misunderstanding both. PLoS Genet. 2007, 3, e220. [Google Scholar] [CrossRef]
- Harman, D. The biologic clock: The mitochondria. J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Sanz, A.; Pamplona, R.; Barja, G. Is the mitochondrial free radical theory of aging intact? Antioxid. Redox Signal. 2006, 8, 582–599. [Google Scholar] [CrossRef]
- Miquel, J.; Economos, A.C.; Fleming, J.; Johnson, J.E.J. Mitochondrial role in cell aging. Exp. Gerontol. 1980, 15, 575–591. [Google Scholar] [CrossRef]
- Bokov, A.; Chaudhuri, A.; Richardson, A. The role of oxidative damage and stress in aging. Mech. Ageing Dev. 2004, 125, 811–826. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘Garb-Aging’. Trends Endocrinol. Metab. 2017, 28, 199–209. [Google Scholar] [CrossRef]
- Gray, M. Origin and evolution of organelle genomes. Curr. Opin. Genet. Dev. 1993, 3, 884–890. [Google Scholar] [CrossRef]
- Calvo, S.; Clauser, K.; Mootha, V. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, D.F.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef]
- Harper, M.; Bevilacqua, L.; Hagopian, K.; Weindruch, R.; Ramsey, J. Ageing, oxidative stress, and mitochondrial uncoupling. Acta Physiol. Scand. 2004, 182, 321–331. [Google Scholar]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.-S.; Benoit, B.; Brand, M.D. Mitochondrial and cytosolic sources of hydrogen peroxide in resting C2C12 myoblasts. Free Radic. Biol. Med. 2019, 130, 140–150. [Google Scholar] [CrossRef]
- Maggiorani, D.; Manzella, N.; Edmondson, D.E.; Mattevi, A.; Parini, A.; Binda, C.; Mialet-Perez, J. Monoamine Oxidases, Oxidative Stress, and Altered Mitochondrial Dynamics in Cardiac Ageing. Oxidative Med. Cell. Longev. 2017, 2017, 3017947. [Google Scholar] [CrossRef] [PubMed]
- De Grey, A.D. HO2•: The Forgotten Radical. DNA Cell Biol. 2002, 21, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Hormesis defined. Ageing Res. Rev. 2008, 7, 1–7. [Google Scholar] [CrossRef]
- Kohen, R.; Nyska, A. Invited review: Oxidation of biological systems: Oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef]
- Sohal, R.; Ku, H.; Agarwal, S.; Forster, M.; Lal, H. Oxidative damage, mitochondrial oxidant generation, and antioxidant defenses during aging and in response to food restriction in the mouse. Mech. Ageing Dev. 1994, 74, 121–133. [Google Scholar] [CrossRef]
- Sohal, R.; Sohal, B.; Orr, W. Mitochondrial superoxide and hydrogen peroxide generation, protein oxidative damage, and longevity in different species of flies. Free Radic. Biol. Med. 1995, 19, 499–504. [Google Scholar] [CrossRef]
- Ku, H.; Brunk, U.; Sohal, R. Relationship between mitochondrial superoxide and hydrogen peroxide production and longevity of mammalian species. Free Radic. Biol. Med. 1993, 15, 621–627. [Google Scholar] [CrossRef]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Saleem, A.; Hood, D.A. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008, 7, 2–12. [Google Scholar] [CrossRef]
- Dai, D.F.; Chen, T.; Wanagat, J.; Laflamme, M.; Marcinek, D.J.; Emond, M.J.; Ngo, C.P.; Prolla, T.A.; Rabinovitch, P.S. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 2010, 9, 536–544. [Google Scholar] [CrossRef]
- Hiona, A.; Sanz, A.; Kujoth, G.C.; Pamplona, R.; Seo, A.Y.; Hofer, T.; Someya, S.; Miyakawa, T.; Nakayama, C.; Samhan-Arias, A.K.; et al. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS ONE 2010, 5, e11468. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Trifunovic, A.; Hansson, A.; Wredenberg, A.; Rovio, A.T.; Dufour, E.; Khvorostov, I.; Spelbrink, J.N.; Wibom, R.; Jacobs, H.T.; Larsson, N.G. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2005, 102, 17993–17998. [Google Scholar] [CrossRef]
- Marzetti, E.; Privitera, G.; Simili, V.; Wohlgemuth, S.E.; Aulisa, L.; Pahor, M.; Leeuwenburgh, C. Multiple Pathways to the Same End: Mechanisms of Myonuclear Apoptosis in Sarcopenia of Aging. Sci. J. 2010, 10, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Alway, S.E.; Siu, P.M. Nuclear apoptosis contributes to sarcopenia. Exerc. Sport Sci. Rev. 2008, 36, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Sahin, E.; DePinho, R.A. Axis of ageing: Telomeres, p53 and mitochondria. Nat. Rev. Mol. Cell Biol. 2013, 13, 397–404. [Google Scholar] [CrossRef]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, M.; Hattori, K.; Sugiyama, S.; Ozawa, T. Age-associated oxygen damage and mutations in mitochondrial DNA in human hearts. Biochem. Biophys. Res. Commun. 1992, 189, 979–985. [Google Scholar] [CrossRef]
- Hayakawa, M.; Torii, K.; Sugiyama, S.; Tanaka, M.; Ozawa, T. Age-associated accumulation of 8-hydroxydeoxyguanosine in mitochondrial DNA of human diaphragm. Biochem. Biophys. Res. Commun. 1991, 179, 1023–1029. [Google Scholar] [CrossRef]
- Mecocci, P.; Fan, G.; Fulle, S.; MacGarvey, U.; Shinobu, L.; Polidori, M.C.; Cherubini, A.; Vecchiet, J.; Senin, U.; Beal, M.F. Age-dependent increases in oxidative damage to DNA, lipids, and proteins in human skeletal muscle. Free Radic. Biol. Med. 1999, 26, 303–308. [Google Scholar] [CrossRef]
- Parise, G.; Kaczor, J.J.; Mahoney, D.J.; Phillips, S.M.; Tarnopolsky, M.A. Oxidative stress and the mitochondrial theory of aging in human skeletal muscle. Exp. Gerontol. 2004, 39, 1391–1400. [Google Scholar]
- Beregi, E.; Regius, O. Comparative morphological study of age related mitochondrial changes of the lymphocytes and skeletal muscle cells. Acta Morphol. Hung. 1987, 35, 219–224. [Google Scholar]
- Greco, M.; Villani, G.; Mazzucchelli, F.; Bresolin, N.; Papa, S.; Attardi, G. Marked aging-related decline in efficiency of oxidative phosphorylation in human skin fibroblasts. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 1706–1708. [Google Scholar] [CrossRef]
- Papa, S. Mitochondrial oxidative phosphorylation changes in the life span. Molecular aspects and physiopathological implications. Biochim. Biophys. Acta (BBA) Bioenerg. 1996, 1276, 87–105. [Google Scholar] [CrossRef]
- Short, K.; Bigelow, M.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.L.; Robinson, M.M.; Nair, K.S. Skeletal muscle aging and the mitochondrion. Trends Endocrinol. Metab. 2013, 24, 247–256. [Google Scholar] [CrossRef]
- Petersen, K.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.; Devries, M.; Safdar, A.; Hamadeh, M.; Tarnopolsky, M. The effect of aging onhuman skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 119–128. [Google Scholar] [CrossRef]
- Lanza, I.; Nair, K.S. Mitochondrial function as a determinant of life span. Pflug. Arch. Eur. J. Physiol. 2010, 459, 277–289. [Google Scholar] [CrossRef]
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Johnson, F.; Sinclair, D.; Guarente, L. Molecular biology of aging. Cell 1999, 96, 291–302. [Google Scholar] [CrossRef]
- Aiken, J.; Bua, E.; Cao, Z.; Lopez, M.; Wanagat, J.; McKenzie, D.; McKiernan, S. Mitochondrial DNA deletion mutations and sarcopenia. Ann. N. Y. Acad. Sci. 2002, 959, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Bua, E.; Johnson, J.; Herbst, A.; Delong, B.; McKenzie, D.; Salamat, S.; Aiken, J.M. Mitochondrial DNA deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 2006, 79, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Flint Beal, M.; Wallace, D.C. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Katayama, M.; Tanaka, M.; Yamamoto, H.; Ohbayashi, T.; Nimura, Y.; Ozawa, T. Deleted mitochondrial DNA in the skeletal muscle of aged individuals. Biochem. Int. 1991, 25, 47–56. [Google Scholar] [PubMed]
- Lee, H.-C.; Pang, C.-Y.; Hsu, H.-S.; Wei, Y.-H. Differential accumulations of 4977 bp deletion in mitochondrial DNA of various tissues in human ageing. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1994, 1226, 37–43. [Google Scholar] [CrossRef]
- Melov, S.; Shoffner, J.M.; Kaufman, A.; Wallace, D.C. Marked increase in the number and variety of mitochondrial DNA rearrangements in aging human skeletal muscle. Nucleic Acids Res. 1995, 23, 4122–4126. [Google Scholar] [CrossRef]
- Lee, H.-C.; Pang, C.-Y.; Hsu, H.-S.; Weia, Y.-H. Ageing-associated tandem duplications in the D-loop of mitochondrial DNA of human muscle. FEBS Lett. 1994, 354, 79–83. [Google Scholar] [CrossRef]
- Zhang, C.F.; Linnane, A.W.; Nagley, P. Occurrence of a particular base substitution (3243 A to G) in mitochondrial DNA of tissues of ageing humans. Biochem. Biophys. Res. Commun. 1993, 195, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Wanagat, J.; McKiernan, S.H.; Aiken, J.M. Mitochondrial DNA deletion mutations are concomitant with ragged red regions of individual, aged muscle fibers: Analysis by laser-capture microdissection. Nucleic Acids Res. 2001, 29, 4502–4508. [Google Scholar] [CrossRef]
- Pak, J.W.; Herbst, A.; Bua, E.; Gokey, N.; McKenzie, D.; Aiken, J.M. Mitochondrial DNA mutations as a fundamental mechanism in physiological declines associated with aging. Aging Cell 2003, 2, 1–7. [Google Scholar] [CrossRef]
- Wanagat, J.; Cao, Z.; Pathare, P.; Aiken, J.M. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2001, 15, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Brierley, E.J.; Johnson, M.A.; Lightowlers, R.N.; James, O.F.; Turnbull, D.M. Role of mitochondrial DNA mutations in human aging: Implications for the central nervous system and muscle. Ann. Neurol. 1998, 43, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Reeve, A.K.; Samuels, D.C.; Chinnery, P.F.; Blackwood, J.K.; Taylor, R.W.; Wanrooij, S.; Spelbrink, J.N.; Lightowlers, R.N.; Turnbull, D.M. What causes mitochondrial DNA deletions in human cells? Nat. Genet. 2008, 40, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Chabi, B.; de Camaret, B.M.; Chevrollier, A.; Boisgard, S.; Stepien, G. Random mtDNA deletions and functional consequence in aged human skeletal muscle. Biochem. Biophys. Res. Commun. 2005, 332, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R.; Bottoni, P.; Giardina, B. Advances in Mitochondrial Medicine; Springer Publishing: Dordrecht, The Netherlands, 2012. [Google Scholar]
- Alway, S.; Myers, M.; Mohamed, J. Regulation of satellite cell function in sarcopenia. Front. Aging Neurosci. 2014, 6, 246. [Google Scholar] [CrossRef]
- Dickinson, J.M.; Volpi, E.; Rasmussen, B.B. Exercise and nutrition to target protein synthesis impairments in aging skeletal muscle. Exerc. Sport Sci. Rev. 2013, 41, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Breen, L.; Phillips, S.M. Skeletal muscle protein anabolism in elderly: Intervention to counteract the “anabolic resistance” of ageing. Nutr. Metab. 2011, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Selby, A.; Rankin, D.; Patel, R.; Atherton, P.; Hildebrandt, W.; Williams, J.; Smith, K.; Seynnes, O.; Hiscock, N.; et al. Age-related differences in the dose–response relationship of muscle protein synthesis to resistance exercise in young and old men. J. Physiol. 2009, 587, 211–217. [Google Scholar] [CrossRef]
- Churchward-Venne, T.A.; Breen, L.; Phillips, S.M. Alterations in human muscle protein metabolism with aging: Protein and exercise as countermeasures to offset sarcopenia. BioFactors 2013, 40, 199–205. [Google Scholar] [CrossRef]
- Linnane, A.; Ozawa, T.; Marzuki, S.; Tanaka, M. Mitochondrial DNA mutations as an important contributor to aging and degenerative diseases. Lancet 1989, 333, 642–645. [Google Scholar] [CrossRef]
- Fischer, F.; Hamann, A.; Osiewacz, H.D. Mitochondrial quality control: An integrated network of pathways. Trends Biochem. Sci. 2012, 37, 284–292. [Google Scholar] [CrossRef]
- Jensen, M.B.; Jasper, H. Mitochondrial Proteostasis in the Control of Aging and Longevity. Cell Metab. 2014, 20, 214–225. [Google Scholar] [CrossRef]
- Lim, J.-A.; Li, L.; Raben, N. Pompe disease: From pathophysiology to therapy and back again. Front. Aging Neurosci. 2014, 6, 177. [Google Scholar] [CrossRef]
- Rubinsztein, D.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef]
- Höhn, A.; König, J.; Grune, T. Protein oxidation in aging and the removal of oxidized proteins. J. Proteom. 2013, 92, 132–159. [Google Scholar] [CrossRef] [PubMed]
- Rajawat, Y.S.; Hilioti, Z.; Bossis, I. Aging: Central role for autophagy and the lysosomal degradative system. Ageing Res. Rev. 2009, 8, 199–213. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Droge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining “clean” cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef]
- Zhang, C.; Cuervo, A.M. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med. 2008, 14, 959–965. [Google Scholar] [CrossRef]
- Gray, D.A.; Woulfe, J. Lipofuscin and aging: A matter of toxic waste. Sci. Aging Knowl. Knowl. Environ. 2005, 2005, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Melis, J.P.; Jonker, M.J.; Vijg, J.; Hoeijmakers, J.H.; Breit, T.M.; Van Steeg, H. Aging on a different scale chronological versus pathology-related aging. Aging 2013, 5, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Höhn, A.; Grune, T. Lipofuscin: Formation, effects and role of macroautophagy. Redox Biol. 2013, 1, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Hütter, E.; Skovbro, M.; Lener, B.; Prats, C.; Rabøl, R.; Dela, F.; Jansen-Dürr, P. Oxidative stress and mitochondrial impairment can be separated from lipofuscin accumulation in aged human skeletal muscle. Aging Cell 2007, 6, 245–256. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T. Lysosomal iron, iron chelation, and cell death. Antioxid. Redox Signal. 2013, 18, 888–898. [Google Scholar] [CrossRef]
- Mrschtik, M.; Ryan, K.M. Lysosomal proteins in cell death and autophagy. FEBS J. 2015, 282, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Latz, E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur. J. Immunol. 2010, 40, 620–623. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Álvarez-Rodríguez, L.; López-Hoyos, M.; Muñoz-Cacho, P.; Martínez-Taboada, V.M. Aging is associated with circulating cytokine dysregulation. Cell. Immunol. 2012, 273, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Krabbe, K.S.; Pedersen, M.; Bruunsgaard, H. Inflammatory mediators in the elderly. Exp. Gerontol. 2004, 39, 687–699. [Google Scholar] [CrossRef]
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552–1562. [Google Scholar] [CrossRef]
- Singh, T.; Newman, A.B. Inflammatory markers in population studies of aging. Ageing Res. Rev. 2010, 10, 319–329. [Google Scholar] [CrossRef]
- Jo, E.; Lee, S.-R.; Park, B.-S.; Kim, J.-S. Potential mechanisms underlying the role of chronic inflammation in age-related muscle wasting. Aging Clin. Exp. Res. 2012, 24, 412–422. [Google Scholar]
- Simpson, R.J.; Lowder, T.W.; Spielmann, G.; Bigley, A.B.; LaVoy, E.C.; Kunz, H. Exercise and the aging immune system. Ageing Res. Rev. 2012, 11, 404–420. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of pro IL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Yazdi, A.; Drexler, S.; Tschopp, J. The role of the inflammasome in nonmyeloid cells. J. Clin. Immunol. 2010, 30, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef]
- Tschopp, J. Mitochondria: Sovereign of inflammation? Eur. J. Immunol. 2011, 41, 1196–1202. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Lukens, J.R.; Kanneganti, T.-D. Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends Mol. Med. 2014, 21, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Goto, S.; Naito, H.; Kaneko, T.; Chung, H.Y.; Radak, Z. Hormetic effects of regular exercise in aging: Correlation with oxidative stress. Appl. Physiol. Nutr. Metab. 2007, 32, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Zealley, B.; De Grey, A.D. Strategies for engineered negligible senescence. Gerontology 2013, 59, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Suliman, H.B. Redox regulation of mitochondrial biogenesis. Free Radic. Biol. Med. 2012, 53, 2043–2053. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef]
- He, C.; Sumpter, J.R.; Levine, B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 2012, 8, 1548–1551. [Google Scholar] [CrossRef]
- Pagano, A.F.; Py, G.; Bernardi, H.; Candau, R.B.; Sanchez, A.M.J. Autophagy and protein turnover signaling in slow-twitch muscle during exercise. Med. Sci. Sports Exerc. 2014, 46, 1314–1325. [Google Scholar] [CrossRef]
- Sanchez, A.M.; Bernardi, H.; Py, G.; Candau, R. Autophagy is essential to support skeletal muscle plasticity in response to endurance exercise. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R956–R969. [Google Scholar] [CrossRef]
- Powers, S.K.; Nelson, W.B.; Hudson, M.B. Exercise-induced oxidative stress in humans: Cause and consequences. Free Radic. Biol. Med. 2011, 51, 942–950. [Google Scholar] [CrossRef]
- Ji, L.L.; Gomez-Cabrera, M.-C.; Vina, J. Role of nuclear factor kB and mitogen-activated protein kinase signaling in exercise-induced antioxidant enzyme adaptation. Appl. Physiol. Nutr. Metab. 2007, 32, 930–935. [Google Scholar] [CrossRef]
- Gounder, S.S.; Kannan, S.; Devadoss, D.; Miller, C.J.; Whitehead, K.S.; Odelberg, S.J.; Firpo, M.A.; Paine, R., III; Hoidal, J.R.; Abel, E.D.; et al. Impaired transcriptional activity of Nrf2 in age-related myocardial oxidative stress is reversible by moderate exercise training. PLoS ONE 2012, 7, e45697. [Google Scholar] [CrossRef]
- MacNeil, L.G.; Safdar, A.; Baker, S.K.; Melov, S.; Tarnopolsky, M.A. Eccentric exercise affects Nrf2-mediated oxidative stress response in skeletal muscle by increasing nuclear Nrf2 content. Med. Sci. Sports Exerc. 2011, 43, 383. [Google Scholar] [CrossRef]
- Gleeson, M.; Bishop, N.C.; Stensel, D.J.; Lindley, M.R.; Mastana, S.S.; Nimmo, M.A. The anti-inflammatory effects of exercise: Mechanisms and implications for the prevention and treatment of disease. Nat. Rev. Immunol. 2011, 11, 607–615. [Google Scholar] [CrossRef]
- Petersen, A.; Pedersen, B. The anti-inflammatory effect of exercise. J. Appl. Physiol. 2005, 98, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.; Febbraio, M. Muscle as an endocrine organ: Focus on muscle-derived interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef]
- Bell, K.E.; Seguin, C.; Parise, G.; Baker, S.K.; Phillips, S.M. Day-to-day changes in muscle protein synthesis in recovery from resistance, aerobic, and high-intensity interval exercise in older men. J. Gerontol. Ser. A 2015, 70, 1024–1029. [Google Scholar] [CrossRef]
- Tapia, P.C. Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: Mitohormesis for health and vitality. Med. Hypotheses 2006, 66, 832–843. [Google Scholar]
- Menshikova, E.V.; Ritov, V.B.; Fairfull, L.; Ferrell, R.E.; Kelley, D.E.; Goodpaster, B.H. Effects of exercise on mitochondrial content and function in aging human skeletal muscle. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2006, 61, 534–540. [Google Scholar] [CrossRef]
- Short, K.; Vittone, J.; Bigelow, M. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes Care 2003, 52, 1888–1896. [Google Scholar] [CrossRef]
- Broskey, N.T.; Greggio, C.; Boss, A.; Boutant, M.; Dwyer, A.; Schlueter, L.; Hans, D.; Gremion, G.; Kreis, R.; Boesch, C.; et al. Skeletal muscle mitochondria in the elderly: Effects of physical fitness and exercise training. J. Clin. Endocrinol. Metab. 2015, 99, 1852–1861. [Google Scholar] [CrossRef]
- Conley, K.; Jubrias, S.; Cress, M.; Esselman, P. Elevated energy coupling and aerobic capacity improves exercise performance in endurance-trained elderly subjects. Exp. Physiol. 2013, 98, 899–907. [Google Scholar]
- Jubrias, S.A.; Esselman, P.C.; Price, L.B.; Cress, M.E.; Conley, K.E. Large energetic adaptations of elderly muscle to resistance and endurance training. J. Appl. Physiol. 2001, 90, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.A.; Lundby, C. Mitochondria express enhanced quality as well as quantity in association with aerobic fitness across recreationally active individuals up to elite athletes. J. Appl. Physiol. 2013, 114, 344–350. [Google Scholar] [CrossRef]
- Parise, G.; Brose, A.N.; Tarnopolsky, M.A. Resistance exercise training decreases oxidative damage to DNA and increases cytochrome oxidase activity in older adults. Exp. Gerontol. 2005, 40, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Parise, G.; Phillips, S.M.; Kaczor, J.J.; Tarnopolsky, M.A. Antioxidant enzyme activity is up-regulated after unilateral resistance exercise training in older adults. Free Radic. Biol. Med. 2005, 39, 289–295. [Google Scholar] [CrossRef]
- Ji, L.L.; Zhang, Y. Antioxidant and anti-inflammatory effects of exercise: Role of redox signaling. Free Radic. Res. 2014, 48, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Melov, S.; Tarnopolsky, M.A.; Beckman, K.; Felkey, K.; Hubbard, A. Resistance exercise reverses aging in human skeletal muscle. PLoS ONE 2007, 2, e465. [Google Scholar] [CrossRef]
- Tarnopolsky, M.; Zimmer, A.; Paikin, J.; Safdar, A.; Aboud, A.; Pearce, E.; Roy, B.; Doherty, T. Creatine monohydrate and conjugated linoleic acid improve strength and body composition following resistance exercise in older adults. PLoS ONE 2007, 2, e991. [Google Scholar] [CrossRef] [PubMed]
- Tarnopolsky, M.A. Mitochondrial DNA shifting in older adults following resistance exercise training. Appl. Physiol. Nutr. Metab. 2009, 34, 348–354. [Google Scholar] [CrossRef]
- Pesta, D.; Hoppel, F.; Macek, C.; Messner, H.; Faulhaber, M.; Kobel, C.; Parson, W.; Burtscher, M.; Schocke, M.; Gnaiger, E. Similar qualitative and quantitative changes of mitochondrial respiration following strength and endurance training in normoxia and hypoxia in sedentary humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1078–R1087. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.; Reidy, P.; Bhattarai, N.; Sidossis, L.; Rasmussen, B. Resistance exercise training alters mitochondrial function in human skeletal muscle. Med. Sci. Sports Exerc. 2014, 47, 1922. [Google Scholar] [CrossRef] [PubMed]
- Taivassalo, T.; Fu, K.; Johns, T.; Arnold, D.; Karpati, G.; Shoubridge, E.A. Gene shifting: A novel therapy for mitochondrial myopathy. Hum. Mol. Genet. 1999, 8, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.L.; Blakely, E.L.; Schaefer, A.M.; He, L.; Wyrick, P.; Haller, R.G.; Taylor, R.W.; Turnbull, D.M.; Taivassalo, T. Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain 2008, 131, 2832–2840. [Google Scholar] [CrossRef]
- Ji, L.L. Exercise at old age: Does it increase or alleviate oxidative stress? Ann. N. Y. Acad. Sci. 2001, 928, 236–247. [Google Scholar] [CrossRef]
- Izzotti, A. Genomic biomarkers and clinical outcomes of physical activity. Ann. N. Y. Acad. Sci. 2011, 1229, 103–114. [Google Scholar] [CrossRef]
- Couppe, C.; Svensson, R.B.; Grosset, J.-F.; Kovanen, V.; Nielsen, R.; Olsen, M.; Larsen, J.; Praet, S.E.; Skovgaard, D.; Hansen, M.; et al. Life-long endurance running is associated with reduced glycation and mechanical stress in connective tissue. Age 2013, 36, 9665. [Google Scholar] [CrossRef]
- Head, D.; Bugg, J.M.; Goate, A.M.; Fagan, A.M.; Mintun, M.A.; Benzinger, T.; Holtzman, D.M.; Morris, J.C. Exercise engagement as a moderator of the effects of apoe genotype on amyloid deposition. Arch. Neurol. 2012, 69, 636–643. [Google Scholar]
- Tam, B.T.; Siu, P.M. Autophagic cellular responses to physical exercise in skeletal muscle. Sports Med. 2014, 44, 625–640. [Google Scholar] [CrossRef]
- Erlich, A.T.; Brownlee, D.M.; Beyfuss, K.; Hood, D.A. Exercise induces TFEB expression and activity in skeletal muscle in a PGC-1α-dependent manner. Am. J. Physiol. Cell Physiol. 2018, 314, C62–C72. [Google Scholar] [CrossRef]
- Kim, Y.; Hood, D.A. Regulation of the autophagy system during chronic contractile activity-induced muscle adaptations. Physiol. Rep. 2017, 5, e13307. [Google Scholar] [CrossRef]
- Lira, V.A.; Okutsu, M.; Zhang, M.; Greene, N.P.; Laker, R.C.; Breen, D.S.; Hoehn, K.L.; Yan, Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 4184–4193. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Ostrowski, T.; Rohde, K.; Bruunsgaard, H. The cytokine response to strenuous exercise. Can. J. Physiol. Pharm. 1998, 76, 505–511. [Google Scholar] [CrossRef]
- Proske, U.; Morgan, D.L. Muscle damage from eccentric exercise: Mechanism, mechanical signs, adaptation and clinical applications. J. Physiol. 2001, 537, 333–345. [Google Scholar] [CrossRef]
- Greiwe, J.S.; Cheng, B.O.; Rubin, D.C.; Yarasheski, K.E.; Semenkovich, C.F. Resistance exercise decreases skeletal muscle tumor necrosis factor alpha in frail elderly humans. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2001, 15, 475–482. [Google Scholar]
- McFarlin, B.K.; Flynn, M.G.; Phillips, M.D.; Stewart, L.K.; Timmerman, K.L. Chronic resistance exercise training improves natural killer cell activity in older women. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 1315–1318. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nilsson, M.I.; Tarnopolsky, M.A. Mitochondria and Aging—The Role of Exercise as a Countermeasure. Biology 2019, 8, 40. https://doi.org/10.3390/biology8020040
Nilsson MI, Tarnopolsky MA. Mitochondria and Aging—The Role of Exercise as a Countermeasure. Biology. 2019; 8(2):40. https://doi.org/10.3390/biology8020040
Chicago/Turabian StyleNilsson, Mats I, and Mark A Tarnopolsky. 2019. "Mitochondria and Aging—The Role of Exercise as a Countermeasure" Biology 8, no. 2: 40. https://doi.org/10.3390/biology8020040
APA StyleNilsson, M. I., & Tarnopolsky, M. A. (2019). Mitochondria and Aging—The Role of Exercise as a Countermeasure. Biology, 8(2), 40. https://doi.org/10.3390/biology8020040