From Powerhouse to Perpetrator—Mitochondria in Health and Disease

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
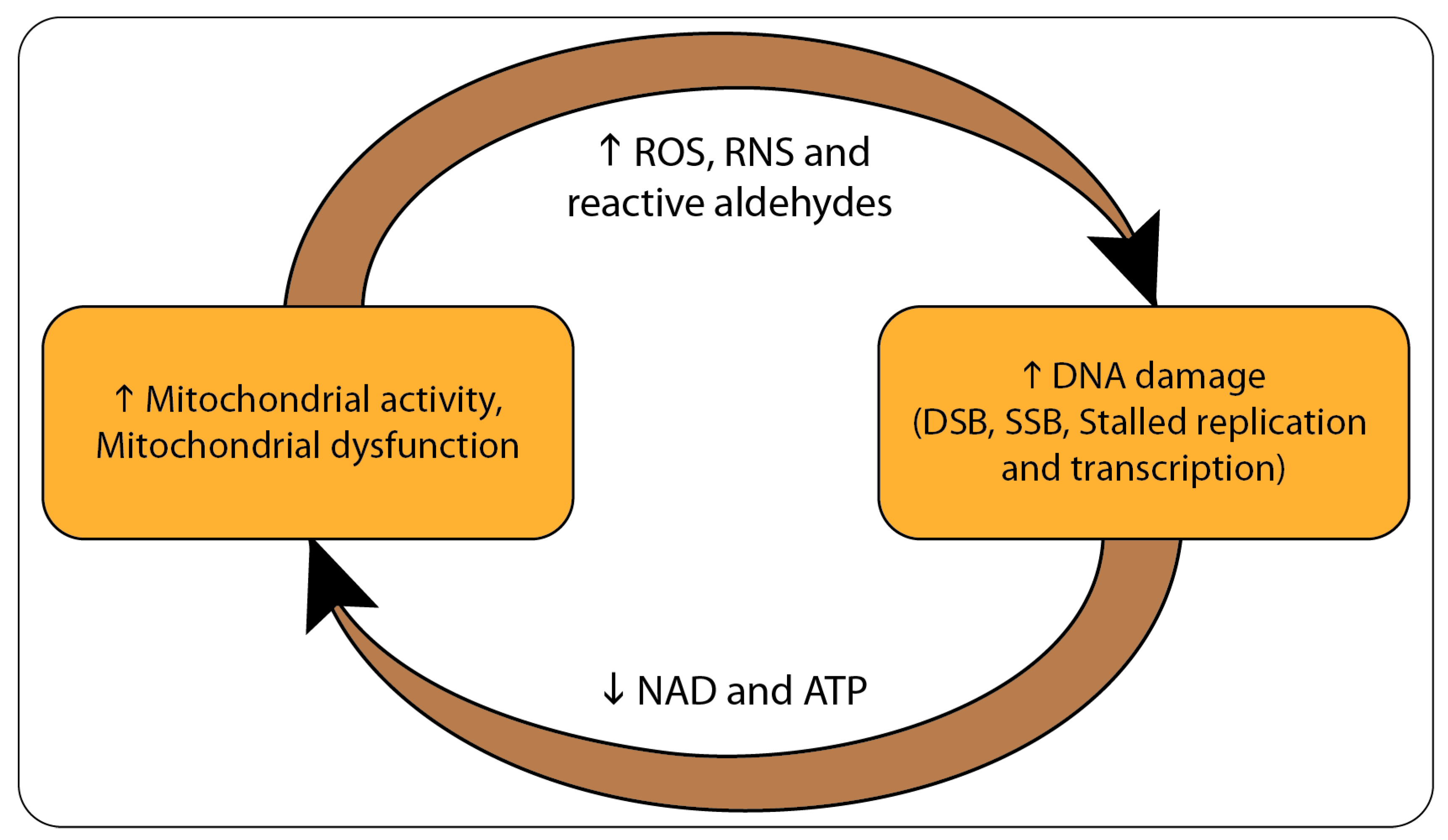
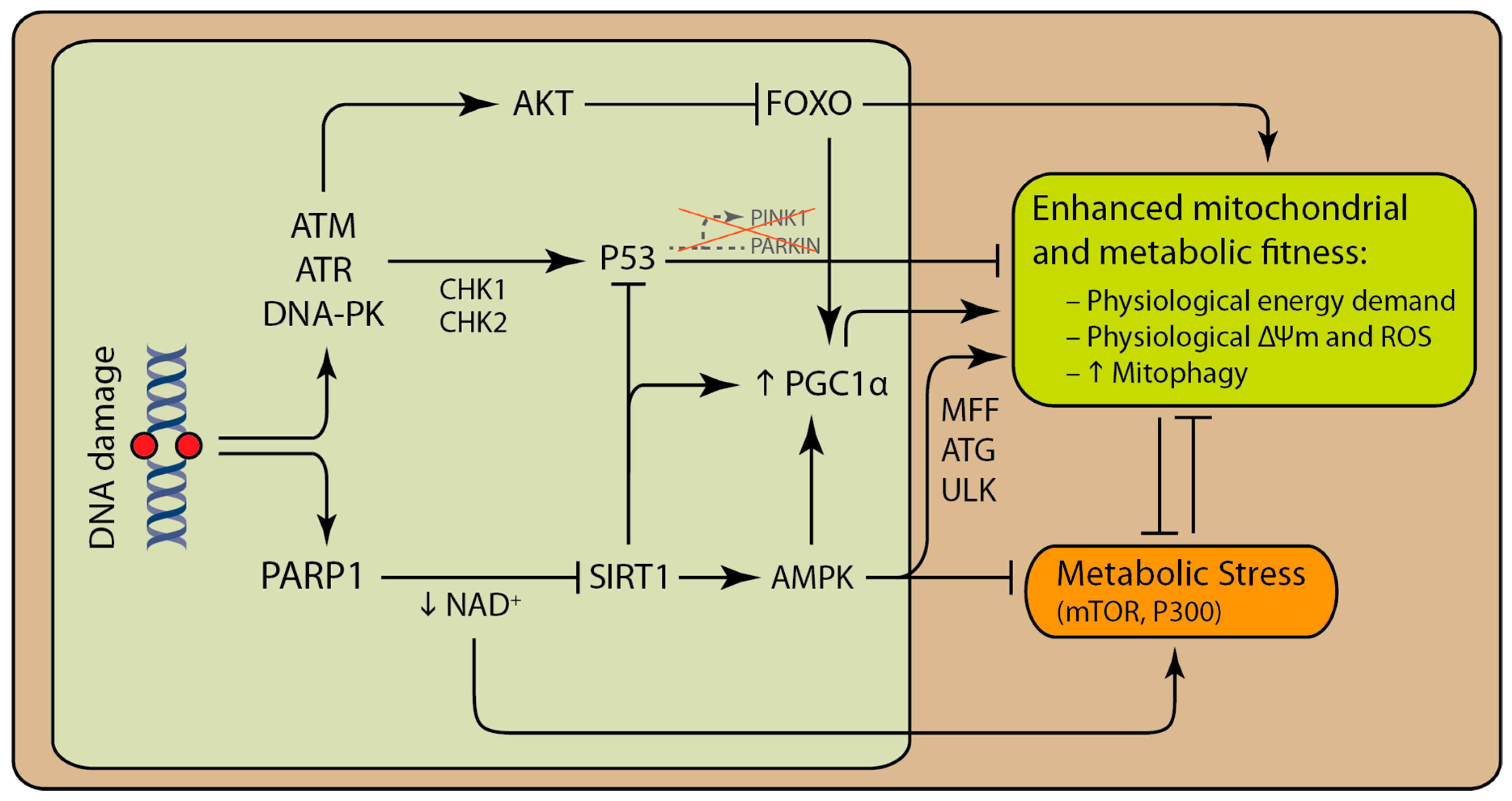
2. From DNA Damage to Mitochondrial Dysfunction
3. PARP Modulates Mitochondrial Function and Cellular Metabolism
4. DNA Damage can Activate Both Pro-Survival and Pro-Death Pathways That Involve the Mitochondria
5. Mito-Nuclear Signaling in Aging and Cancer
6. Mitochondrial ROS Are Involved in Signaling and Determine Cell Fate
7. Mitochondria Influence Post-Translational Modifications (PTMs) and Epigenetic Marks
8. Regulation of dNTP Pools
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Xie, X.-H.; Chen, C.-H.; Peng, X.; Zhang, P.; Yang, C.; Wang, Y.-T. Molecular Regulation Mechanisms and Interactions Between Reactive Oxygen Species and Mitophagy. DNA Cell Biol. 2019, 38, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Aunan, J.R.; Cho, W.C.; Søreide, K. The Biology of Aging and Cancer: A Brief Overview of Shared and Divergent Molecular Hallmarks. Aging Dis. 2017, 8, 628. [Google Scholar] [CrossRef] [PubMed]
- Burcham, P.C. Internal hazards: Baseline DNA damage by endogenous products of normal metabolism. Mutat. Res./Genet. Toxicol. Environ. Mutagenesis 1999, 443, 11–36. [Google Scholar] [CrossRef]
- De Bont, R.; van Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- White, R.R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Mol. Cell 2016, 63, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Fan, M.; Candas, D.; Jiang, G.; Papadopoulos, S.; Tian, L.; Woloschak, G.; Grdina, D.J.; Li, J.J. CDK1 Enhances Mitochondrial Bioenergetics for Radiation-Induced DNA Repair. Cell Rep. 2015, 13, 2056–2063. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD(+) Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef]
- Fakouri, N.B.; Durhuus, J.A.; Regnell, C.E.; Angleys, M.; Desler, C.; Olive, M.H.; Martín-Pardillos, A.; Tsaalbi-Shtylik, A.; Thomsen, K.; Lauritzen, M.; et al. Rev1 contributes to proper mitochondrial function via the PARP-NAD+-SIRT1-PGC1α axis. Sci. Rep. 2017, 7, 12480. [Google Scholar] [CrossRef]
- Rivera-Torres, J.; Acín-Perez, R.; Cabezas-Sánchez, P.; Osorio, F.G.; Gonzalez-Gómez, C.; Megias, D.; Cámara, C.; López-Otín, C.; Enríquez, J.A.; Luque-García, J.L.; et al. Identification of mitochondrial dysfunction in Hutchinson–Gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J. Proteom. 2013, 91, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef]
- Qian, W.; Choi, S.; Gibson, G.A.; Watkins, S.C.; Bakkenist, C.J.; Van Houten, B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J. Cell Sci. 2012, 125, 5745–5757. [Google Scholar] [CrossRef] [PubMed]
- Temelie, M.; Savu, D.I.; Moisoi, N. Intracellular and Intercellular Signalling Mechanisms following DNA Damage Are Modulated By PINK1. Oxid. Med. Cell. Longev. 2018, 2018, 1–15. [Google Scholar] [CrossRef]
- Desler, C.; Hansen, T.L.; Frederiksen, J.B.; Marcker, M.L.; Singh, K.K.; Juel Rasmussen, L. Is There a Link between Mitochondrial Reserve Respiratory Capacity and Aging? J. Aging Res. 2012, 2012, 192503. [Google Scholar] [CrossRef]
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011, 30, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Bozulic, L.; Surucu, B.; Hynx, D.; Hemmings, B.A. PKBα/Akt1 Acts Downstream of DNA-PK in the DNA Double-Strand Break Response and Promotes Survival. Mol. Cell 2008, 30, 203–213. [Google Scholar] [CrossRef]
- Park, S.-J.; Gavrilova, O.; Brown, A.L.; Soto, J.E.; Bremner, S.; Kim, J.; Xu, X.; Yang, S.; Um, J.-H.; Koch, L.G.; et al. DNA-PK Promotes the Mitochondrial, Metabolic, and Physical Decline that Occurs During Aging. Cell Metab. 2017, 25, 1135–1146.e7. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Kang, J.-G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Bensaad, K.; Vousden, K.H. p53: New roles in metabolism. Trends Cell Biol. 2007, 17, 286–291. [Google Scholar] [CrossRef]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4, 2308. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.B.; Kinoshita, C.; Kinoshita, Y.; Morrison, R.S. p53 and mitochondrial function in neurons. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1186–1197. [Google Scholar] [CrossRef]
- Bai, P.; Nagy, L.; Fodor, T.; Liaudet, L.; Pacher, P. Poly(ADP-ribose) polymerases as modulators of mitochondrial activity. Trends Endocrinol. Metab. 2015, 26, 75–83. [Google Scholar] [CrossRef]
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012, 26, 417–432. [Google Scholar] [CrossRef]
- Krietsch, J.; Rouleau, M.; Pic, É.; Ethier, C.; Dawson, T.M.; Dawson, V.L.; Masson, J.-Y.; Poirier, G.G.; Gagné, J.-P. Reprogramming cellular events by poly(ADP-ribose)-binding proteins. Mol. Asp. Med. 2013, 34, 1066–1087. [Google Scholar] [CrossRef]
- Davidovic, L.; Vodenicharov, M.; Affar, E.B.; Poirier, G.G. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp. Cell Res. 2001, 268, 7–13. [Google Scholar] [CrossRef]
- Bürkle, A.; Virag, L. Poly(ADP-ribose): PARadigms and PARadoxes. Mol. Asp. Med. 2013, 34, 1046–1065. [Google Scholar] [CrossRef]
- Langelier, M.-F.; Planck, J.L.; Roy, S.; Pascal, J.M. Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: Structural and functional insights into DNA-dependent PARP-1 activity. J. Biol. Chem. 2011, 286, 10690–10701. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.A.E.; Timinszky, G.; Arribas-Bosacoma, R.; Kozlowski, M.; Hassa, P.O.; Hassler, M.; Ladurner, A.G.; Pearl, L.H.; Oliver, A.W. The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat. Struct. Mol. Biol. 2012, 19, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, A.J.; Timinszky, G.; Kong, S.E.; Jin, J.; Cai, Y.; Swanson, S.K.; Washburn, M.P.; Florens, L.; Ladurner, A.G.; Conaway, J.W.; et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. USA 2009, 106, 13770–13774. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016, 17, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef]
- Maryanovich, M.; Zaltsman, Y.; Ruggiero, A.; Goldman, A.; Shachnai, L.; Zaidman, S.L.; Porat, Z.; Golan, K.; Lapidot, T.; Gross, A. An MTCH2 pathway repressing mitochondria metabolism regulates haematopoietic stem cell fate. Nat. Commun. 2015, 6, 7901. [Google Scholar] [CrossRef]
- Feng, J.; Park, J.; Cron, P.; Hess, D.; Hemmings, B.A. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J. Biol. Chem. 2004, 279, 41189–41196. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Park, J.; Feng, J.; Li, Y.; Hammarsten, O.; Brazil, D.P.; Hemmings, B.A. DNA-dependent protein kinase-mediated phosphorylation of protein kinase B requires a specific recognition sequence in the C-terminal hydrophobic motif. J. Biol. Chem. 2009, 284, 6169–6174. [Google Scholar] [CrossRef]
- Toulany, M.; Maier, J.; Iida, M.; Rebholz, S.; Holler, M.; Grottke, A.; Jüker, M.; Wheeler, D.L.; Rothbauer, U.; Rodemann, H.P. Akt1 and Akt3 but not Akt2 through interaction with DNA-PKcs stimulate proliferation and post-irradiation cell survival of K-RAS-mutated cancer cells. Cell Death Discov. 2017, 3, 17072. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Turner, K.M.; Yung, W.K.A.; Chen, K.; Zhang, W. Role of AKT signaling in DNA repair and clinical response to cancer therapy. Neuro-Oncology 2014, 16, 1313–1323. [Google Scholar] [CrossRef]
- Stronach, E.A.; Chen, M.; Maginn, E.N.; Agarwal, R.; Mills, G.B.; Wasan, H.; Gabra, H. DNA-PK mediates AKT activation and apoptosis inhibition in clinically acquired platinum resistance. Neoplasia 2011, 13, 1069–1080. [Google Scholar] [CrossRef]
- Robey, R.B.; Hay, N. Is Akt the “Warburg kinase”-Akt-energy metabolism interactions and oncogenesis. Semin. Cancer Biol. 2009, 19, 25–31. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Kops, G.J.P.L.; de Ruiter, N.D.; De Vries-Smits, A.M.M.; Powell, D.R.; Bos, J.L.; Burgering, B.M.T. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature 1999, 398, 630–634. [Google Scholar] [CrossRef]
- Webb, A.E.; Brunet, A. FOXO transcription factors: Key regulators of cellular quality control. Trends Biochem. Sci. 2014, 39, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.W. Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yen, K.; Cohen, P. Humanin: A harbinger of mitochondrial-derived peptides? Trends Endocrinol. Metab. 2013, 24, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Son, J.M.; Benayoun, B.A.; Lee, C. The Mitochondrial-Encoded Peptide MOTS-c Translocates to the Nucleus to Regulate Nuclear Gene Expression in Response to Metabolic Stress. Cell Metab. 2018, 28, 516–524. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Sundaram, R.K.; Oeck, S.; Corso, C.D.; Liu, Y.; Noorbakhsh, S.; Niger, M.; Boeke, M.; Ueno, D.; Kalathil, A.N.; et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat. Genet. 2018, 50, 1086–1092. [Google Scholar] [CrossRef]
- Li, X.; Egervari, G.; Wang, Y.; Berger, S.L.; Lu, Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol. 2018, 19, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Sivanand, S.; Rhoades, S.; Jiang, Q.; Lee, J.V.; Benci, J.; Zhang, J.; Yuan, S.; Viney, I.; Zhao, S.; Carrer, A.; et al. Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Mol. Cell 2017, 67, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Desler, C.; Munch-Petersen, B.; Stevnsner, T.; Matsui, S.-I.; Kulawiec, M.; Singh, K.K.; Rasmussen, L.J. Mitochondria as determinant of nucleotide pools and chromosomal stability. Mutat. Res. 2007, 625, 112–124. [Google Scholar] [CrossRef]
- Rasmussen, A.K.; Chatterjee, A.; Rasmussen, L.J.; Singh, K.K. Mitochondria-mediated nuclear mutator phenotype in Saccharomyces cerevisiae. Nucleic Acids Res. 2003, 31, 3909–3917. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell 2016, 61, 199–209. [Google Scholar] [CrossRef]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta Bioenerget. 2012, 1817, 1833–1838. [Google Scholar] [CrossRef]
- Brace, L.E.; Vose, S.C.; Stanya, K.; Gathungu, R.M.; Marur, V.R.; Longchamp, A.; Treviño-Villarreal, H.; Mejia, P.; Vargas, D.; Inouye, K.; et al. Increased oxidative phosphorylation in response to acute and chronic DNA damage. NPJ Aging Mech. Dis. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Trifunovic, A.; Hansson, A.; Wredenberg, A.; Rovio, A.T.; Dufour, E.; Khvorostov, I.; Spelbrink, J.N.; Wibom, R.; Jacobs, H.T.; Larsson, N.-G. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2005, 102, 17993–17998. [Google Scholar] [CrossRef]
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 2016, 100, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Maryanovich, M.; Gross, A. A ROS rheostat for cell fate regulation. Trends Cell Biol. 2013, 23, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Folmes, C.D.L.; Dzeja, P.P.; Nelson, T.J.; Terzic, A. Metabolic Plasticity in Stem Cell Homeostasis and Differentiation. Cell Stem Cell 2012, 11, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A.; Woodbine, L.; Marchetti, C.; Dawson, S.; Lambe, T.; Liu, C.; Rodrigues, N.P.; Crockford, T.L.; Cabuy, E.; Vindigni, A.; et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature 2007, 447, 686–690. [Google Scholar] [CrossRef]
- Niedernhofer, L.J. DNA repair is crucial for maintaining hematopoietic stem cell function. DNA Repair 2008, 7, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Forsberg, E.C.; et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 2014, 512, 198–202. [Google Scholar] [CrossRef]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The world of protein acetylation. Biochim. Biophys. Acta Proteins Proteom. 2016, 1864, 1372–1401. [Google Scholar] [CrossRef] [PubMed]
- Hauer, M.H.; Gasser, S.M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017, 31, 2204–2221. [Google Scholar] [CrossRef]
- Zane, L.; Sharma, V.; Misteli, T. Common features of chromatin in aging and cancer: Cause or coincidence? Trends Cell Biol. 2014, 24, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Wellen, K.E.; Rabinowitz, J.D. Metabolic control of methylation and acetylation. Curr. Opin. Chem. Biol. 2016, 30, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef]
- Ali, I.; Conrad, R.J.; Verdin, E.; Ott, M. Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev. 2018, 118, 1216–1252. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, D.C.; Sorum, A.W.; Guasch, L.; Nicklaus, M.C.; Meier, J.L. Metabolic Regulation of Histone Acetyltransferases by Endogenous Acyl-CoA Cofactors. Chem. Biol. 2015, 22, 1030–1039. [Google Scholar] [CrossRef]
- Katada, S.; Imhof, A.; Sassone-Corsi, P. Connecting Threads: Epigenetics and Metabolism. Cell 2012, 148, 24–28. [Google Scholar] [CrossRef]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.W.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.-F.; Lim, H.-W.; Liu, S.; et al. Akt-Dependent Metabolic Reprogramming Regulates Tumor Cell Histone Acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef]
- Metzger, E.; Schüle, R. The expanding world of histone lysine demethylases. Nat. Struct. Mol. Biol. 2007, 14, 252–254. [Google Scholar] [CrossRef] [PubMed]
- Forneris, F.; Binda, C.; Vanoni, M.A.; Mattevi, A.; Battaglioli, E. Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 2005, 579, 2203–2207. [Google Scholar] [CrossRef]
- Metzger, E.; Imhof, A.; Patel, D.; Kahl, P.; Hoffmeyer, K.; Friedrichs, N.; Müller, J.M.; Greschik, H.; Kirfel, J.; Ji, S.; et al. Phosphorylation of histone H3T6 by PKCβI controls demethylation at histone H3K4. Nature 2010, 464, 792–796. [Google Scholar] [CrossRef]
- He, Y.-F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hu, D.; Chen, H.; Shi, G.; Fetahu, I.S.; Wu, F.; Rabidou, K.; Fang, R.; Tan, L.; Xu, S.; et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 2018, 559, 637–641. [Google Scholar] [CrossRef]
- Pai, C.-C.; Kearsey, S.E. A Critical Balance: dNTPs and the Maintenance of Genome Stability. Genes 2017, 8, 57. [Google Scholar] [CrossRef]
- Kunz, B.A. Mutagenesis and deoxyribonucleotide pool imbalance. Mutat. Res. 1988, 200, 133–147. [Google Scholar] [CrossRef]
- Kunz, B.A.; Kohalmi, S.E.; Kunkel, T.A.; Mathews, C.K.; Mclntosh, E.M.; Reidy, J.A. Deoxyribonucleoside triphosphate levels: A critical factor in the maintenance of genetic stability. Mutat. Res. Toxicol. 1994, 318, 1–64. [Google Scholar] [CrossRef]
- Mathews, C.K. DNA precursor metabolism and genomic stability. FASEB J. 2006, 20, 1300–1314. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Abdulovic, A.L.; Viberg, J.; Nilsson, A.K.; Kunkel, T.A.; Chabes, A. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 2011, 39, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Reichard, P. Interactions between deoxyribonucleotide and DNA synthesis. Annu. Rev. Biochem. 1988, 57, 349–374. [Google Scholar] [CrossRef]
- Meuth, M. The molecular basis of mutations induced by deoxyribonucleoside triphosphate pool imbalances in mammalian cells. Exp. Cell Res. 1989, 181, 305–316. [Google Scholar] [CrossRef]
- Meuth, M. The genetic consequences of nucleotide precursor pool imbalance in mammalian cells. Mutat. Res. Mol. Mech. Mutagen. 1984, 126, 107–112. [Google Scholar] [CrossRef]
- Haracska, L.; Prakash, S.; Prakash, L. Replication pastO6-Methylguanine by Yeast and Human DNA Polymerase η. Mol. Cell. Biol. 2000, 20, 8001–8007. [Google Scholar] [CrossRef]
- Niida, H.; Shimada, M.; Murakami, H.; Nakanishi, M. Mechanisms of dNTP supply that play an essential role in maintaining genome integrity in eukaryotic cells. Cancer Sci. 2010, 101, 2505–2509. [Google Scholar] [CrossRef] [PubMed]
- Niida, H.; Katsuno, Y.; Sengoku, M.; Shimada, M.; Yukawa, M.; Ikura, M.; Ikura, T.; Kohno, K.; Shima, H.; Suzuki, H.; et al. Essential role of Tip60-dependent recruitment of ribonucleotide reductase at DNA damage sites in DNA repair during G1 phase. Genes Dev. 2010, 24, 333–338. [Google Scholar] [CrossRef]
- Tseng, L.-M.; Yin, P.-H.; Chi, C.-W.; Hsu, C.-Y.; Wu, C.-W.; Lee, L.-M.; Wei, Y.-H.; Lee, H.-C. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer 2006, 45, 629–638. [Google Scholar] [CrossRef]
- Yu, M.; Zhou, Y.; Shi, Y.; Ning, L.; Yang, Y.; Wei, X.; Zhang, N.; Hao, X.; Niu, R. Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in Chinese breast cancer patients. IUBMB Life 2007, 59, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Nomoto, S.; Fujii, T.; Kaneko, T.; Takeda, S.; Inoue, S.; Kanazumi, N.; Nakao, A. Correlation between copy number of mitochondrial DNA and clinico-pathologic parameters of hepatocellular carcinoma. Eur. J. Surg. Oncol. 2006, 32, 303–307. [Google Scholar] [CrossRef]
- Matsuyama, W.; Nakagawa, M.; Wakimoto, J.; Hirotsu, Y.; Kawabata, M.; Osame, M. Mitochondrial DNA mutation correlates with stage progression and prognosis in non-small cell lung cancer. Hum. Mutat. 2003, 21, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Lièvre, A.; Chapusot, C.; Bouvier, A.-M.; Zinzindohoué, F.; Piard, F.; Roignot, P.; Arnould, L.; Beaune, P.; Faivre, J.; Laurent-Puig, P. Clinical value of mitochondrial mutations in colorectal cancer. J. Clin. Oncol. 2005, 23, 3517–3525. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Mambo, E.; Sidransky, D. Mitochondrial DNA mutations in human cancer. Oncogene 2006, 25, 4663–4674. [Google Scholar] [CrossRef] [PubMed]
- Simonnet, H. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis 2002, 23, 759–768. [Google Scholar] [CrossRef]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Shidara, Y.; Yamagata, K.; Kanamori, T.; Nakano, K.; Kwong, J.Q.; Manfredi, G.; Oda, H.; Ohta, S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005, 65, 1655–1663. [Google Scholar] [CrossRef]
- Ralevic, V.; Burnstock, G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998, 50, 413–492. [Google Scholar] [PubMed]
- Smith, J.L. Enzymes of nucleotide synthesis. Curr. Opin. Struct. Biol. 1995, 5, 752–757. [Google Scholar] [CrossRef]
- Fang, J.; Uchiumi, T.; Yagi, M.; Matsumoto, S.; Amamoto, R.; Takazaki, S.; Yamaza, H.; Nonaka, K.; Kang, D. Dihydro-orotate dehydrogenase is physically associated with the respiratory complex and its loss leads to mitochondrial dysfunction. Biosci. Rep. 2013, 33, e00021. [Google Scholar] [CrossRef]
- Bader, B.; Knecht, W.; Fries, M.; Löffler, M. Expression, purification, and characterization of histidine-tagged rat and human flavoenzyme dihydroorotate dehydrogenase. Protein Expr. Purif. 1998, 13, 414–422. [Google Scholar] [CrossRef]
- Löffler, M.; Jöckel, J.; Schuster, G.; Becker, C. Dihydroorotat-ubiquinone oxidoreductase links mitochondria in the biosynthesis of pyrimidine nucleotides. Mol. Cell. Biochem. 1997, 174, 125–129. [Google Scholar] [CrossRef]
- Beuneu, C.; Auger, R.; Löffler, M.; Guissani, A.; Lemaire, G.; Lepoivre, M. Indirect inhibition of mitochondrial dihydroorotate dehydrogenase activity by nitric oxide. Free Radic. Biol. Med. 2000, 28, 1206–1213. [Google Scholar] [CrossRef]
- Rawls, J.; Knecht, W.; Diekert, K.; Lill, R.; Löffler, M. Requirements for the mitochondrial import and localization of dihydroorotate dehydrogenase. Eur. J. Biochem. 2000, 267, 2079–2087. [Google Scholar] [CrossRef]
- Uhlin, U.; Eklund, H. Structure of ribonucleotide reductase protein R1. Nature 1994, 370, 533–539. [Google Scholar] [CrossRef]
- Jordan, A.; Reichard, P. Ribonucleotide reductases. Annu. Rev. Biochem. 1998, 67, 71–98. [Google Scholar] [CrossRef] [PubMed]
- Kashlan, O.B.; Scott, C.P.; Lear, J.D.; Cooperman, B.S. A comprehensive model for the allosteric regulation of mammalian ribonucleotide reductase. Functional consequences of ATP- and dATP-induced oligomerization of the large subunit. Biochemistry 2002, 41, 462–474. [Google Scholar] [CrossRef]
- Chabes, A.L.; Pfleger, C.M.; Kirschner, M.W.; Thelander, L. Mouse ribonucleotide reductase R2 protein: A new target for anaphase-promoting complex-Cdh1-mediated proteolysis. Proc. Natl. Acad. Sci. USA 2003, 100, 3925–3929. [Google Scholar] [CrossRef] [PubMed]
- Munch-Petersen, B.; Cloos, L.; Jensen, H.K.; Tyrsted, G. Human thymidine kinase 1. Regulation in normal and malignant cells. Adv. Enzyme Regul. 1995, 35, 69–89. [Google Scholar] [CrossRef]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.T.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar] [CrossRef]
- Clifford, R.; Louis, T.; Robbe, P.; Ackroyd, S.; Burns, A.; Timbs, A.T.; Wright Colopy, G.; Dreau, H.; Sigaux, F.; Judde, J.G.; et al. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood 2014, 123, 1021–1031. [Google Scholar] [CrossRef]
- Song, S.; Wheeler, L.J.; Mathews, C.K. Deoxyribonucleotide pool imbalance stimulates deletions in HeLa cell mitochondrial DNA. J. Biol. Chem. 2003, 278, 43893–43896. [Google Scholar] [CrossRef]
- López, L.C.; Akman, H.O.; García-Cazorla, A.; Dorado, B.; Martí, R.; Nishino, I.; Tadesse, S.; Pizzorno, G.; Shungu, D.; Bonilla, E.; et al. Unbalanced deoxynucleotide pools cause mitochondrial DNA instability in thymidine phosphorylase-deficient mice. Hum. Mol. Genet. 2009, 18, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Pontarin, G.; Gallinaro, L.; Ferraro, P.; Reichard, P.; Bianchi, V. Origins of mitochondrial thymidine triphosphate: Dynamic relations to cytosolic pools. Proc. Natl. Acad. Sci. USA 2003, 100, 12159–12164. [Google Scholar] [CrossRef] [PubMed]
- Desler, C.; Munch-Petersen, B.; Rasmussen, L.J. The Role of Mitochondrial dNTP Levels in Cells with Reduced TK2 Activity. Nucleosides Nucleotides Nucleic Acids 2006, 25, 1171–1175. [Google Scholar] [CrossRef] [PubMed]
- Pontarin, G.; Ferraro, P.; Bee, L.; Reichard, P.; Bianchi, V. Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc. Natl. Acad. Sci. USA 2012, 109, 13302–13307. [Google Scholar] [CrossRef]
- Bourdon, A.; Minai, L.; Serre, V.; Jais, J.-P.; Sarzi, E.; Aubert, S.; Chrétien, D.; de Lonlay, P.; Paquis-Flucklinger, V.; Arakawa, H.; et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 2007, 39, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Pontarin, G.; Ferraro, P.; Rampazzo, C.; Kollberg, G.; Holme, E.; Reichard, P.; Bianchi, V. Deoxyribonucleotide metabolism in cycling and resting human fibroblasts with a missense mutation in p53R2, a subunit of ribonucleotide reductase. J. Biol. Chem. 2011, 286, 11132–11140. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Arakawa, H.; Yamaguchi, T.; Shiraishi, K.; Fukuda, S.; Matsui, K.; Takei, Y.; Nakamura, Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 2000, 404, 42–49. [Google Scholar] [CrossRef]
- James, S.J.; Miller, B.J.; McGarrity, L.J.; Morris, S.M. The effect of folic acid and/or methionine deficiency on deoxyribonucleotide pools and cell cycle distribution in mitogen-stimulated rat lymphocytes. Cell Prolif. 1994, 27, 395–406. [Google Scholar] [CrossRef]
- Micheli, V.; Camici, M.; G Tozzi, M.; L Ipata, P.; Sestini, S.; Bertelli, M.; Pompucci, G. Neurological Disorders of Purine and Pyrimidine Metabolism. Curr. Top. Med. Chem. 2011, 11, 923–947. [Google Scholar] [CrossRef]
- Desler, C.; Marcker, M.L.; Singh, K.K.; Rasmussen, L.J. The importance of mitochondrial DNA in aging and cancer. J. Aging Res. 2011, 2011, 407536. [Google Scholar] [CrossRef] [PubMed]
- Desler, C.; Frederiksen, J.H.; Angleys, M.; Maynard, S.; Keijzers, G.; Fagerlund, B.; Mortensen, E.L.; Osler, M.; Lauritzen, M.; Bohr, V.A.; et al. Increased deoxythymidine triphosphate levels is a feature of relative cognitive decline. Mitochondrion 2015, 25, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.; Hejl, A.-M.; Dinh, T.-S.T.; Keijzers, G.; Hansen, Å.M.; Desler, C.; Moreno-Villanueva, M.; Bürkle, A.; Rasmussen, L.J.; Waldemar, G.; et al. Defective mitochondrial respiration, altered dNTP pools and reduced AP endonuclease 1 activity in peripheral blood mononuclear cells of Alzheimer’s disease patients. Aging 2015, 7, 793–815. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fakouri, N.B.; Hansen, T.L.; Desler, C.; Anugula, S.; Rasmussen, L.J. From Powerhouse to Perpetrator—Mitochondria in Health and Disease. Biology 2019, 8, 35. https://doi.org/10.3390/biology8020035
Fakouri NB, Hansen TL, Desler C, Anugula S, Rasmussen LJ. From Powerhouse to Perpetrator—Mitochondria in Health and Disease. Biology. 2019; 8(2):35. https://doi.org/10.3390/biology8020035
Chicago/Turabian StyleFakouri, Nima B., Thomas Lau Hansen, Claus Desler, Sharath Anugula, and Lene Juel Rasmussen. 2019. "From Powerhouse to Perpetrator—Mitochondria in Health and Disease" Biology 8, no. 2: 35. https://doi.org/10.3390/biology8020035
APA StyleFakouri, N. B., Hansen, T. L., Desler, C., Anugula, S., & Rasmussen, L. J. (2019). From Powerhouse to Perpetrator—Mitochondria in Health and Disease. Biology, 8(2), 35. https://doi.org/10.3390/biology8020035