

A High-Quality Chromosome-Level Genome Assembly and Comparative Analyses Provide Insights into the Adaptation of Chrysomya megacephala (Fabricius, 1794) (Diptera: Calliphoridae)
 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Taxon Sampling and Sequencing
2.2. Genome Assembly
2.3. Genome Annotation
2.4. Phylogenomics and Gene Family Evolution Analyses
2.5. Detoxification and Chemoreception Gene Family Annotation
3. Results and Discussion
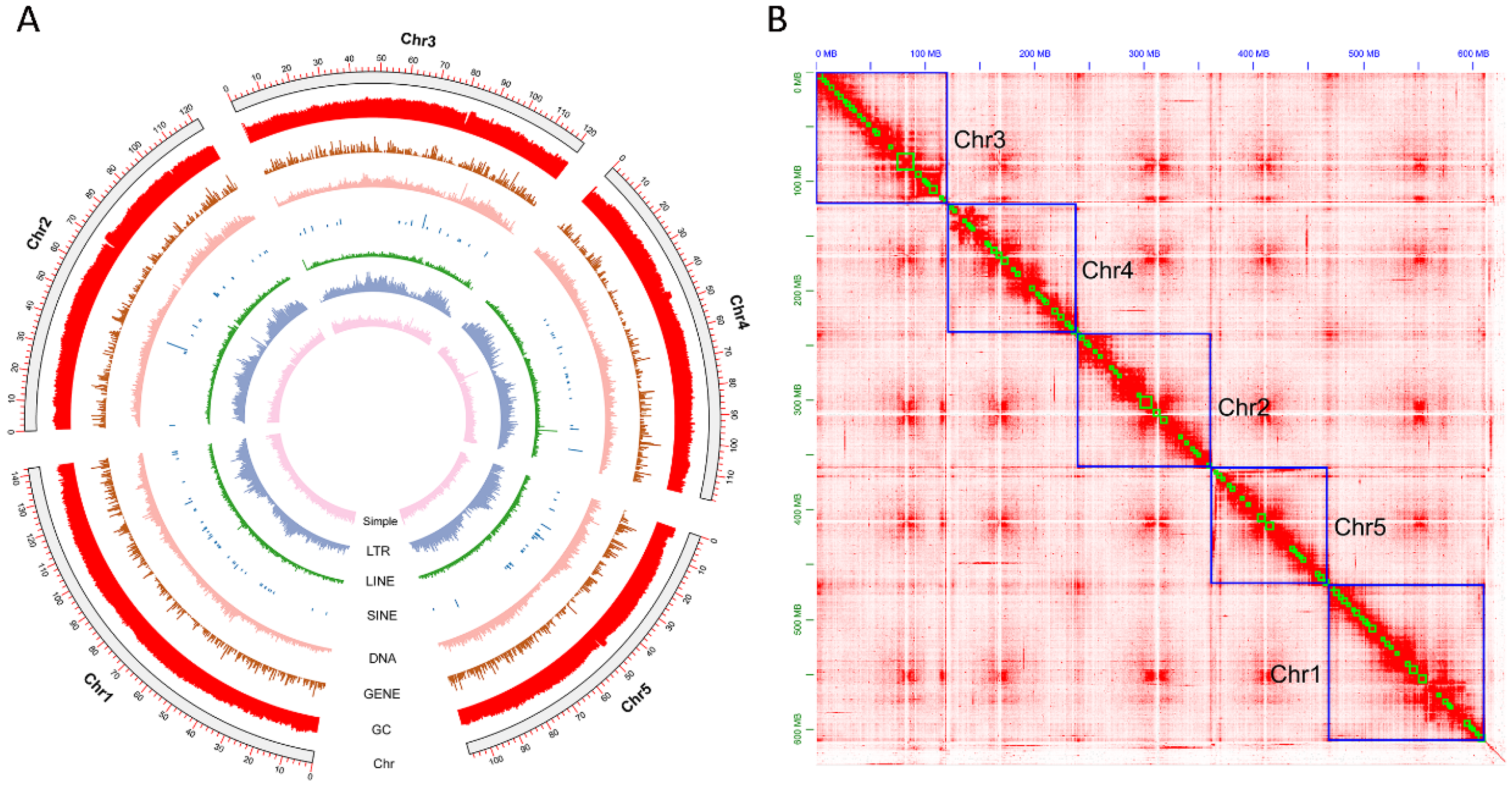
3.1. Genome Assembly
3.2. Genome Annotation
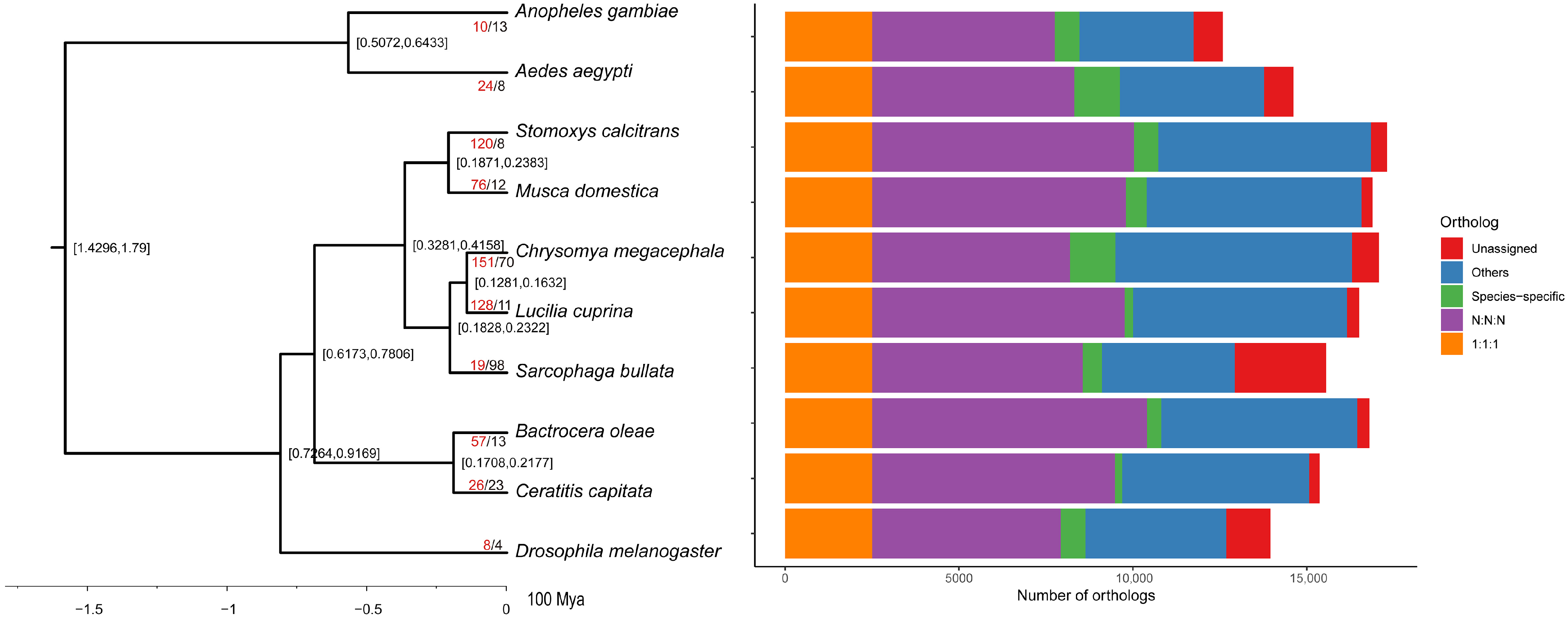
3.3. Phylogenetic and Gene Family Evolution Analyses
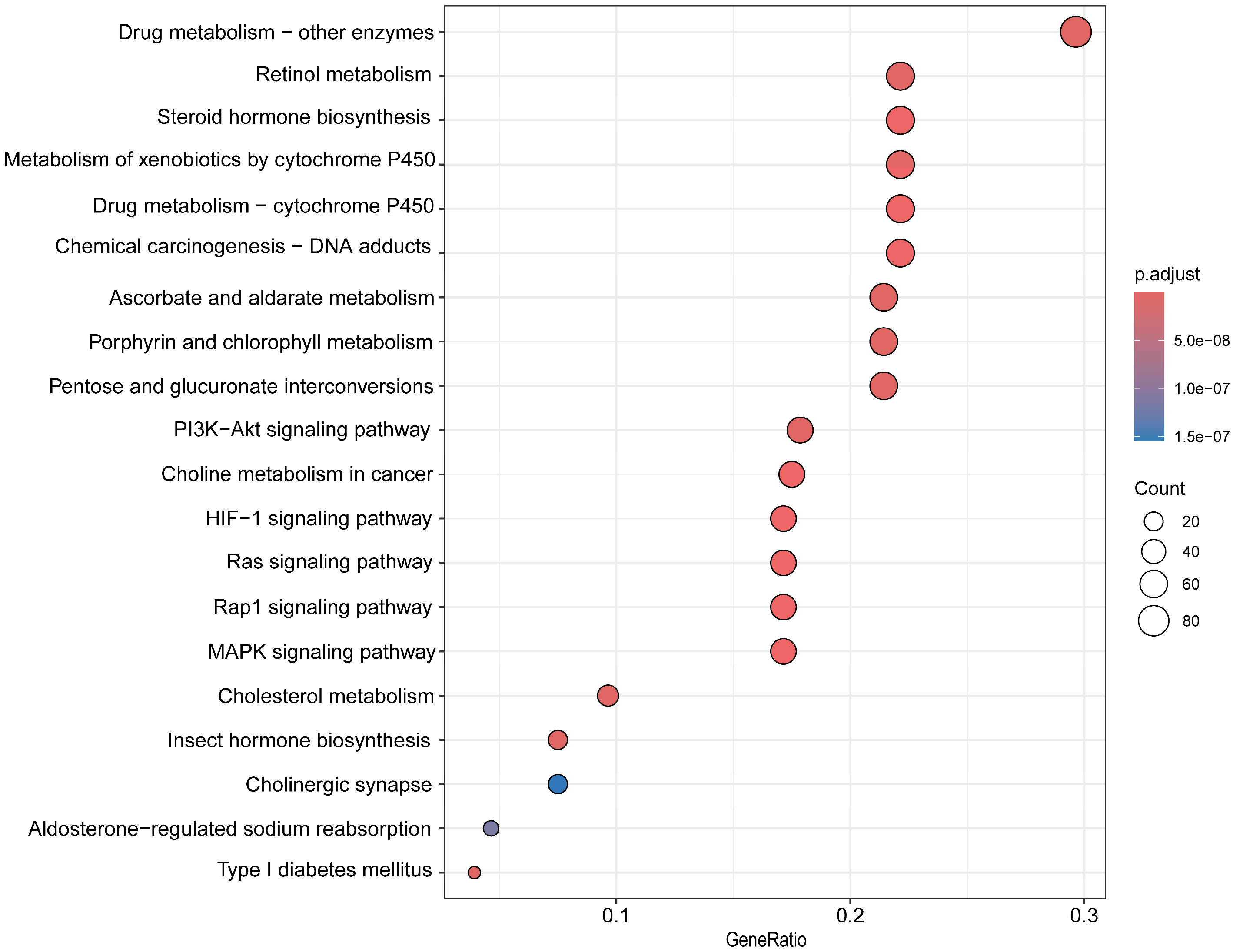
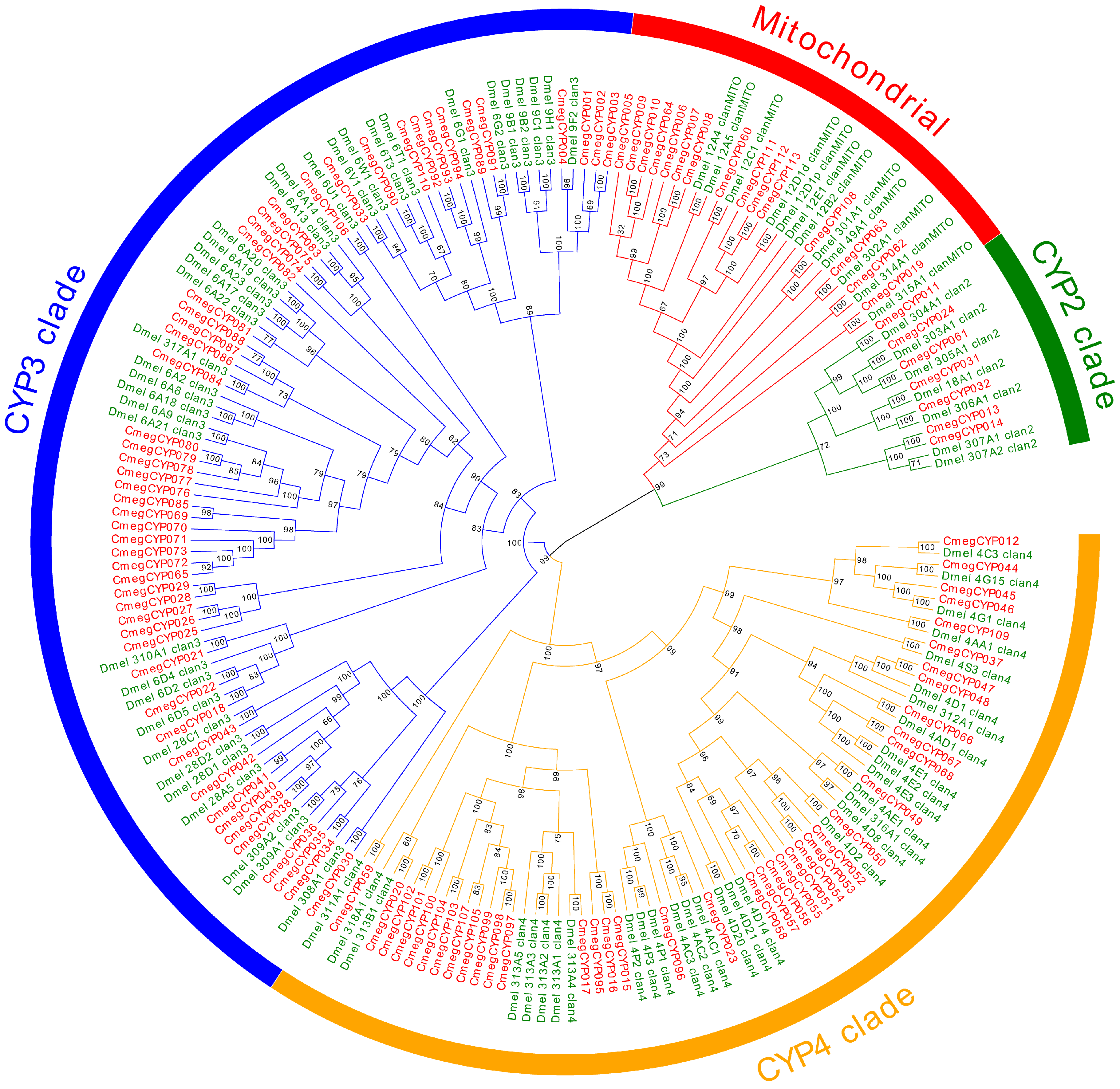
3.4. Detoxification Gene Family Annotation
3.5. Chemoreception Gene Family Annotation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Henssge, C.; Madea, B.J. Estimation of the time since death. Forensic Sci. Int. 2007, 165, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Amendt, J.; Campobasso, C.P.; Gaudry, E.; Reiter, C.; LeBlanc, H.N.; JR Hall, M.J. Best practice in forensic entomology-standards and guidelines. Int. J. Leg. Med. 2007, 121, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Gruner, S.V. Forensic Entomology: International Dimensions and Frontiers. Fla. Entomol. 2015, 98, 1015–1016. [Google Scholar] [CrossRef]
- Tomberlin, J.K.; Mohr, R.; Benbow, M.E.; Tarone, A.M.; Vanlaerhoven, S.J. A roadmap for bridging basic and applied research in forensic entomology. Annu. Rev. Entomol. 2011, 56, 401–421. [Google Scholar] [CrossRef] [PubMed]
- Benecke, M.; Lessig, R.J. Child neglect and forensic entomology. Forensic Sci. Int. 2001, 120, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.; Wall, R. Genetic relationships between blowflies (Calliphoridae) of forensic importance. Forensic Sci. Int. 2001, 120, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.D.; Sperling, F.A. DNA-based identification of forensically important Chrysomyinae (Diptera: Calliphoridae). Forensic Sci. Int. 2001, 120, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.S. Minimum and maximum development rates of some forensically important Calliphoridae (Diptera). J. Forensic Sci. 2000, 45, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Harvey, M.; Gaudieri, S.; Villet, M.H.; Dadour, I.J. A global study of forensically significant calliphorids: Implications for identification. Forensic Sci. Int. 2008, 177, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.Q.; Zhao, J.M. Flies of China; Liaoning Sciene and Technology Press: Shenyang, China, 1996; 384p. [Google Scholar]
- Ren, L.; Shang, Y.; Yang, L.; Wang, S.; Wang, X.; Chen, S.; Guo, Y. Chromosome-level de novo genome assembly of Sarcophaga peregrina provides insights into the evolutionary adaptation of flesh flies. Mol. Ecol. Resour. 2021, 21, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.D. Chrysomya megacephala (Diptera: Calliphoridae) has reached the continental United States: Review of its biology, pest status, and spread around the world. J. Med. Entomol. 1991, 28, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Sukontason, K.; Piangjai, S.; Siriwattanarungsee, S.; Sukontason, K.L. Morphology and developmental rate of blowflies Chrysomya megacephala and Chrysomya rufifacies in Thailand: Application in forensic entomology. Parasitol. Res. 2008, 102, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Čičková, H.; Newton, G.L.; Lacy, R.C.; Kozánek, M.J. The use of fly larvae for organic waste treatment. Waste Manag. 2015, 35, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, D.; Huang, M.; Hu, X.; Shen, J.; Zhao, Z.; Chen, J.J. Chrysomya megacephala (Fabricius) larvae: A new biodiesel resource. Appl. Energ. 2012, 94, 349–354. [Google Scholar] [CrossRef]
- Hu, X. Bio-translating food waste by Chrysomya megacephala Larvae. Ph.D. Thesis, Sun Yet-sen University, Guangzhou, China, 2012. [Google Scholar]
- Zito, P.; Sajeva, M.; Raspi, A.; Dötterl, S.J. Dimethyl disulfide and dimethyl trisulfide: So similar yet so different in evoking biological responses in saprophilous flies. Chemoecology 2014, 24, 261–267. [Google Scholar] [CrossRef]
- Xu, L.; Jiang, H.B.; Yu, J.L.; Pan, D.; Tao, Y.; Lei, Q.; Chen, Y.; Liu, Z.; Wang, J.J. Two odorant receptors regulate 1-octen-3-ol induced oviposition behavior in the oriental fruit fly. Commun. Biol. 2023, 6, 176. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.F. The organization of the chemosensory system in Drosophila melanogaster: A rewiew. Cell Tissue Res. 1994, 275, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Field, L.M.; Pickett, J.A.; Wadhams, L.J. Molecular studies in insect olfaction. Insect Mol. Biol. 2000, 9, 545–551. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Li, Z.; Yin, Y.; Na, N.; Gao, X.J. Time of day-specific changes in metabolic detoxification and insecticide tolerance in the house Fly, Musca domestica L. Front. Physiol. 2022, 12, 803682. [Google Scholar] [CrossRef]
- Rane, R.V.; Walsh, T.K.; Pearce, S.L.; Jermiin, L.S.; Gordon, K.H.; Richards, S.; Oakeshott, J.G. Are feeding preferences and insecticide resistance associated with the size of detoxifying enzyme families in insect herbivores? Curr. Opin. Insect Sci. 2016, 13, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.R.; Backes, W.L. Formation of P450·P450 complexes and their effect on P450 function. Pharmacol. Ther. 2012, 133, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Tsubota, T.; Shiotsuki, T.J. Genomic and phylogenetic analysis of insect carboxyl/cholinesterase genes. J. Pestic. Sci. 2010, 35, 310–314. [Google Scholar] [CrossRef]
- Bushnell, B. BBtools. 2014. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 10 March 2025).
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods. 2021, 18, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; McCarthy, S.A.; Wood, J.; Howe, K.; Wang, Y.; Durbin, R. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 2020, 36, 2896–2898. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, M.; Söding, J.J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Kojima, K.K.; Kohany, O.J. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. Dna 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Hubley, R.; Finn, R.D.; Clements, J.; Eddy, S.R.; Jones, T.A.; Bao, W.; Smit, A.F.; Wheeler, T.J. The Dfam database of repetitive DNA families. Nucleic Acids Res. 2016, 44, D81–D89. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013–2015. Available online: http://www.repeatmasker.org (accessed on 10 March 2025).
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic. Acids. Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef] [PubMed]
- Holt, C.; Yandell, M.J. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. Bmc Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [PubMed]
- Brůna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M.J. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. Nar. Genom. Bioinform. 2021, 3, lqaa108. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B.J. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, W309–W312. [Google Scholar] [CrossRef] [PubMed]
- Brůna, T.; Lomsadze, A.; Borodovsky, M.J. GeneMark-EP+: Eukaryotic gene prediction with self-training in the space of genes and proteins. Nar. Genom. Bioinform. 2020, 2, lqaa026. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 38, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Kriventseva, E.V.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Dias, R.; Simão, F.A.; Zdobnov, E.M. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids. Res. 2019, 47, D807–D811. [Google Scholar] [CrossRef] [PubMed]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M.J. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed]
- Keilwagen, J.; Hartung, F.; Grau, J. GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data. In Gene Prediction; Springer: New York, NY, USA, 2019; pp. 161–177. [Google Scholar]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.J. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.J. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P.J. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.; Pethica, R.; Zhou, Y.; Talbot, C.; Vogel, C.; Madera, M.; Chothia, C.; Gough, J.J. SUPERFAMILY–sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 2009, 37, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.E.; Sillitoe, I.; Dawson, N.; Lam, S.D.; Clarke, T.; Lee, D.; Orengo, C.; Lees, J. Gene3D: Extensive prediction of globular domains in proteins. Nucleic Acids Res. 2018, 46, D435–D439. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; Von Mering, C.; Bork, P.J. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R.J. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S.J. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T.J. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Kück, P.; Longo, G.C. FASconCAT-G: Extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 2014, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate Maximum-Likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Reade, A.; Katz, K.; Naug, D. A capillary feeder (CAFE) assay to measure food consumption and diet choice of individual honey bees. J. Apic. Res. 2016, 55, 353–355. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Vizueta, J.; Sánchez-Gracia, A.; Rozas, J. Bitacora: A comprehensive tool for the identification and annotation of gene families in genome assemblies. Mol. Ecol. Resour. 2020, 20, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2012, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Xiong, M.; Lei, C.L.; Zhu, F. The developmental transcriptome of the synanthropic fly Chrysomya megacephala and insights into olfactory proteins. Bmc Genom. 2015, 16, 20. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.M.; Han, H.; Wang, M.; Jiang, Y.S.; Pi, Z.Y.; Qu, Y.H.; Liu, Z.H.; Cai, J.F. Characterized gene repertoires and functional gene reference for forensic entomology: Genomic and developmental transcriptomic analysis of Aldrichina grahami (Diptera: Calliphoridae). J. Med. Entomol. 2022, 59, 810–819. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, H.N.; Logan, J.G. Exploiting insect olfaction in forensic entomology. In Current Concepts in Forensic Entomology, 1st ed.; Amendt, J., Goff, M.L., Campobasso, C.P., Grassberger, M., Eds.; Springer: Dordrecht, The Netherlands, 2009; pp. 205–221. [Google Scholar]
- Feyereisen, R. Evolution of insect P450. Biochem. Soc. Trans. 2006, 34, 1252–1255. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, Z.; Chen, X.J. Regulation of insect P450s in response to phytochemicals. Curr. Opin. Insect Sci. 2021, 43, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, S.; Xie, X.; Dong, S.; Li, X. Identification of wild-type CYP321A2 and comparison of Allelochemical–induced expression profiles of CYP321A2 with its paralog CYP321A1 in Helicoverpa zea. Insects 2021, 12, 75. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | C. megacephala |
|---|---|
| Genome assembly | |
| Size (Mb) | 629.44 |
| Number of scaffolds | 97 |
| Number of chromosomes | 5 |
| Scaffold N50 length (Mb) | 121.37 |
| GC (%) | 33.74 |
| BUSCO completeness (%) | 98.90 |
| Protein-coding genes | |
| Gene number | 17,071 |
| Mean gene length (bp) | 9950.5 |
| BUSCO completeness (%) | 99.00 |
| Repetitive elements | |
| Size (Mb) | 244.26 (38.32%) |
| DNA transposons (Mb) | 20.96 (3.30%) |
| SINEs (kb) | 56.23 (0.00%) |
| LINEs (Mb) | 12.77 (2.03%) |
| LTRs (Mb) | 45.85 (7.27%) |
| Unclassified (Mb) | 138.36 (21.98%) |
| Number of ncRNA | 3740 |
| rRNA | 636 |
| miRNA | 78 |
| snRNA | 145 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, D.; Li, L.; Ma, J.; Jin, J.; Ding, C.; Fang, Q.; Jin, J.; Aishan, Z.; Li, X. A High-Quality Chromosome-Level Genome Assembly and Comparative Analyses Provide Insights into the Adaptation of Chrysomya megacephala (Fabricius, 1794) (Diptera: Calliphoridae). Biology 2025, 14, 913. https://doi.org/10.3390/biology14080913
Zhang D, Li L, Ma J, Jin J, Ding C, Fang Q, Jin J, Aishan Z, Li X. A High-Quality Chromosome-Level Genome Assembly and Comparative Analyses Provide Insights into the Adaptation of Chrysomya megacephala (Fabricius, 1794) (Diptera: Calliphoridae). Biology. 2025; 14(8):913. https://doi.org/10.3390/biology14080913
Chicago/Turabian StyleZhang, Dan, Liangliang Li, Junchao Ma, Jianfeng Jin, Chunli Ding, Qiang Fang, Jianjun Jin, Zhulidezi Aishan, and Xuebo Li. 2025. "A High-Quality Chromosome-Level Genome Assembly and Comparative Analyses Provide Insights into the Adaptation of Chrysomya megacephala (Fabricius, 1794) (Diptera: Calliphoridae)" Biology 14, no. 8: 913. https://doi.org/10.3390/biology14080913
APA StyleZhang, D., Li, L., Ma, J., Jin, J., Ding, C., Fang, Q., Jin, J., Aishan, Z., & Li, X. (2025). A High-Quality Chromosome-Level Genome Assembly and Comparative Analyses Provide Insights into the Adaptation of Chrysomya megacephala (Fabricius, 1794) (Diptera: Calliphoridae). Biology, 14(8), 913. https://doi.org/10.3390/biology14080913