
Chromatin Accessibility Dynamics Reveal Conserved Transcriptional Regulatory Networks During Insect Metamorphosis in Harmonia axyridis and Drosophila melanogaster


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
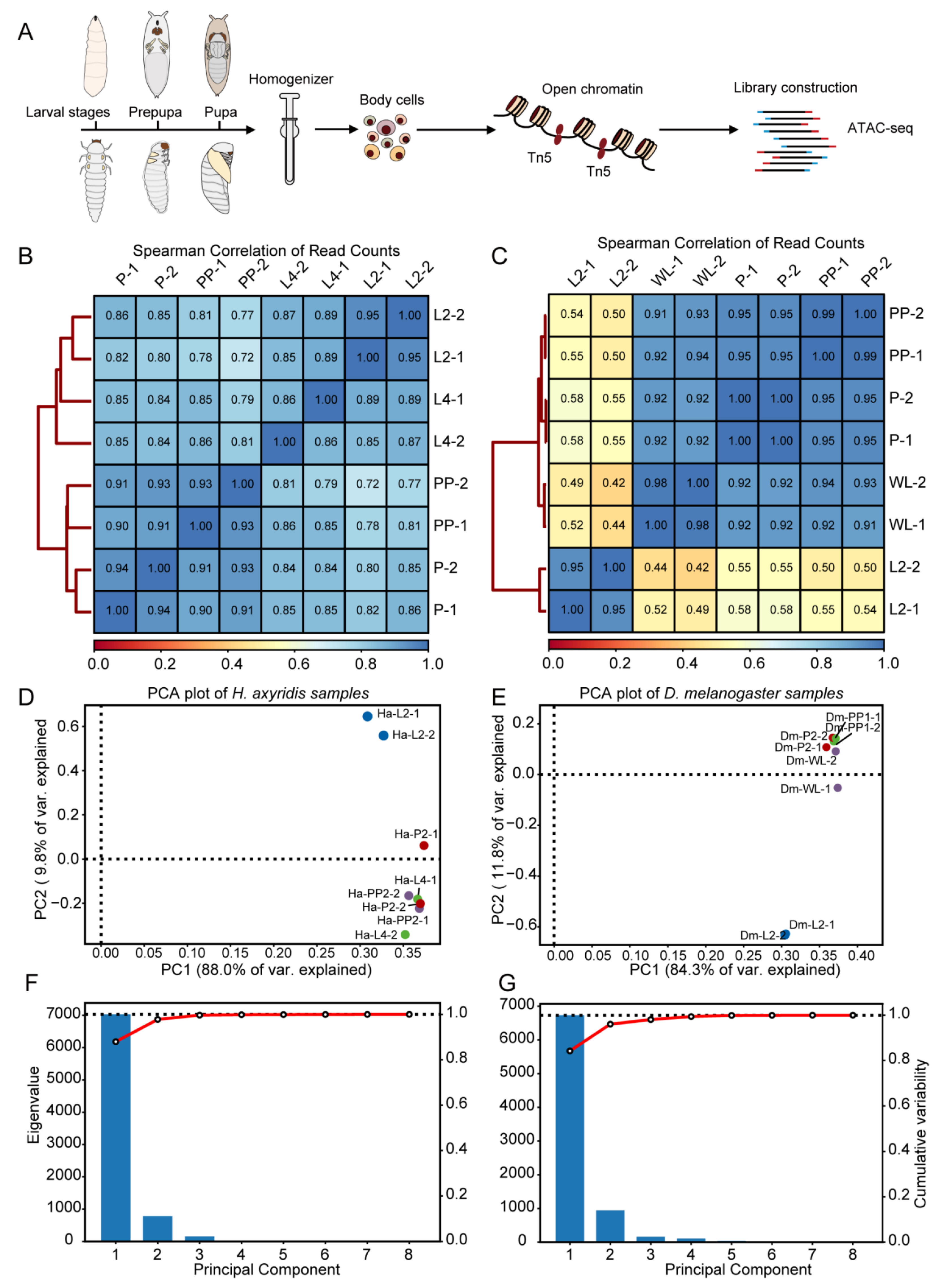
2.1. Sample Collection
2.2. ATAC-Seq Library Preparation
2.3. Nuclei Quality Assessment
2.4. ATAC-Seq Library Construction
2.5. ATAC-Seq Analysis
2.6. Gene Regulatory Network Construction
3. Results
3.1. ATAC-Seq Data Quality Assessment
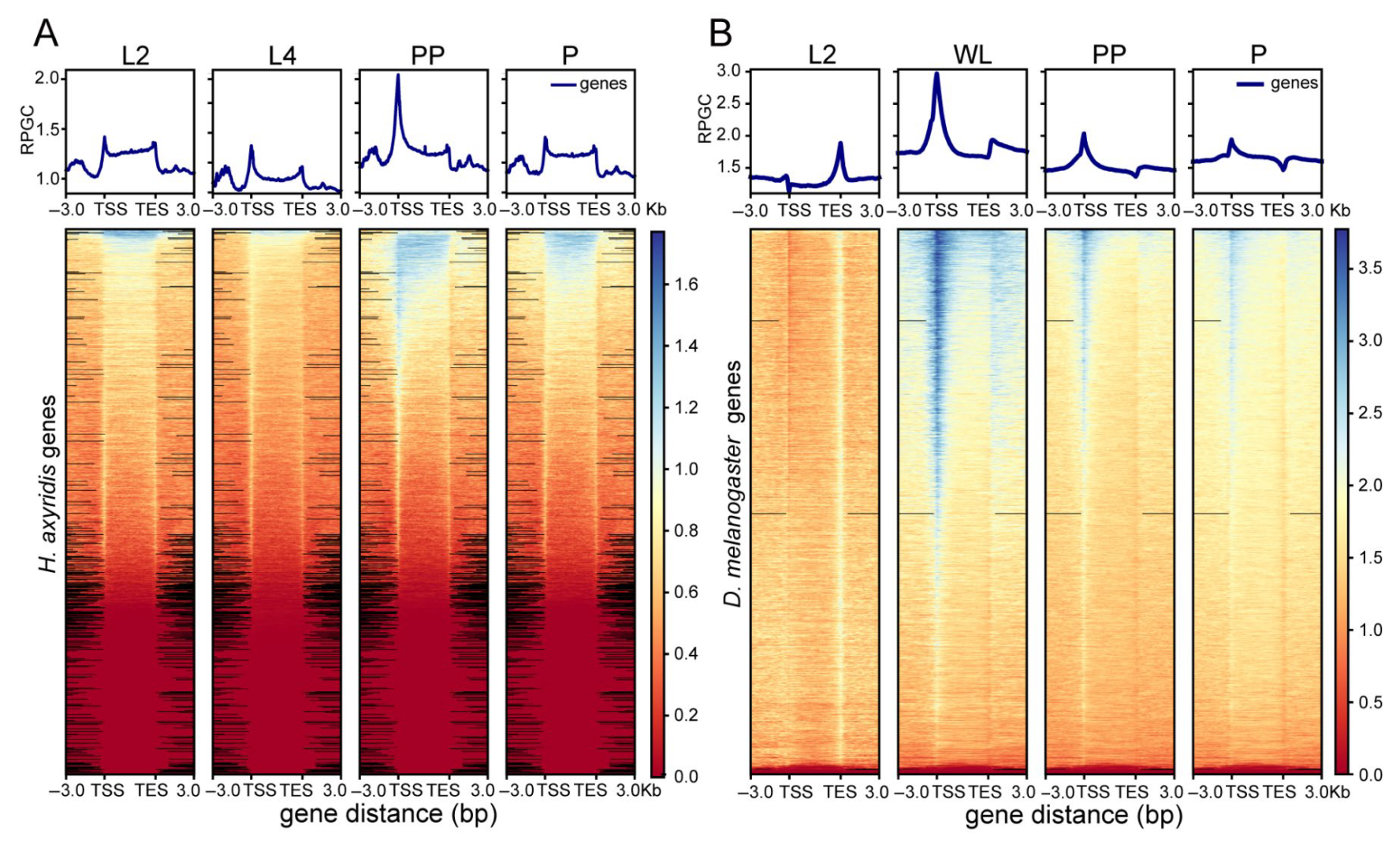
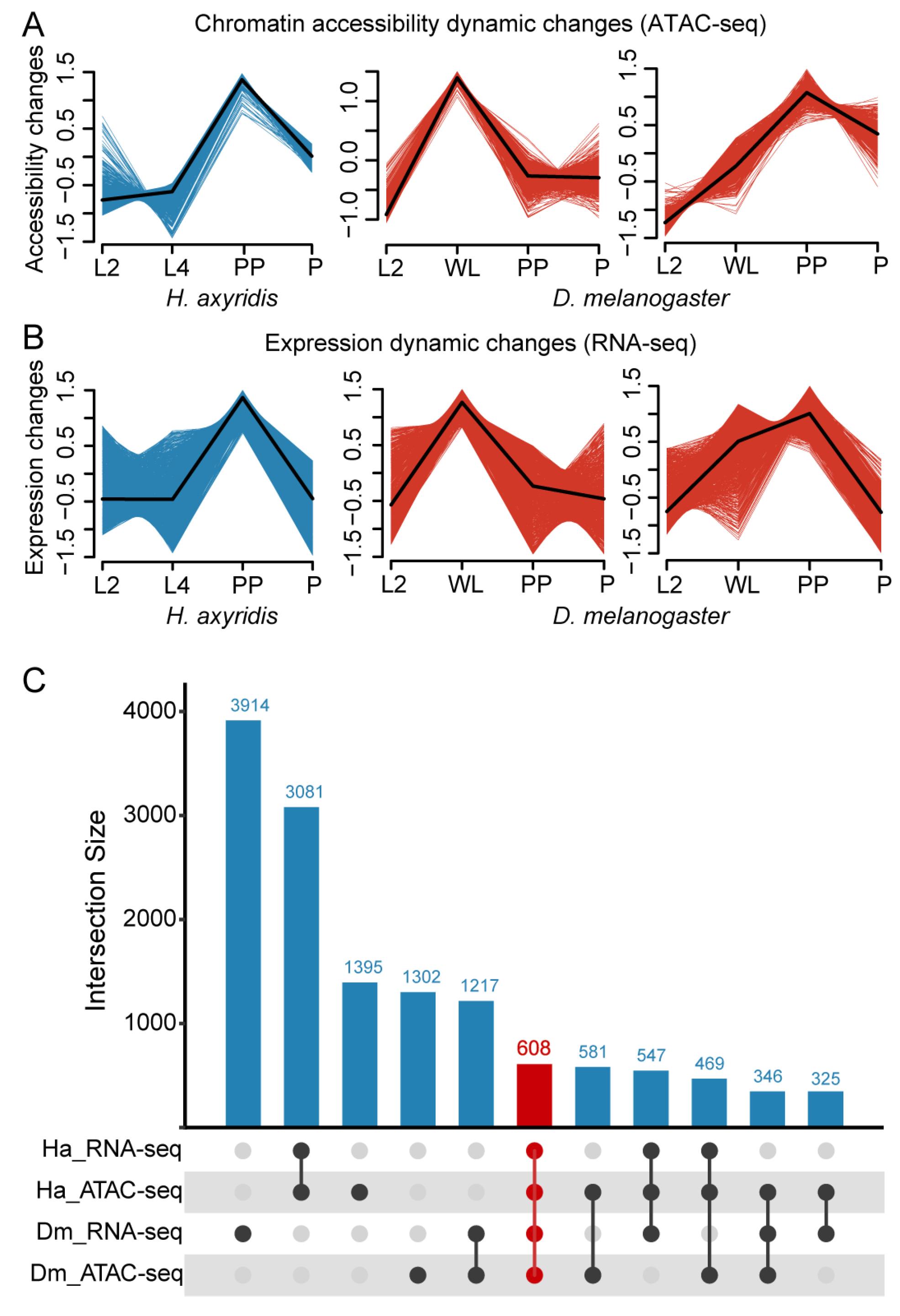
3.2. Chromatin Accessibility Pattern Analysis
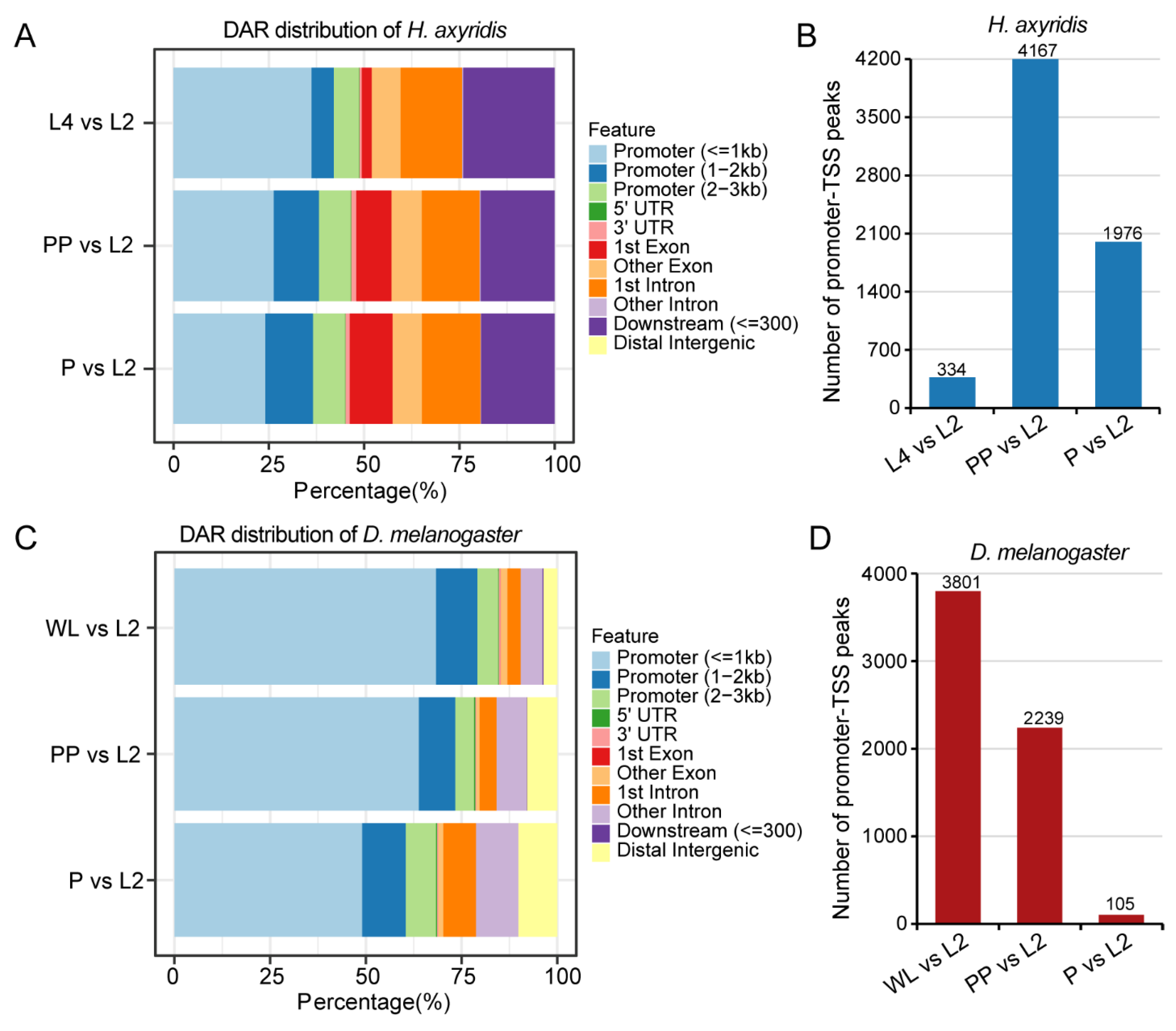
3.3. Differential Accessibility Regions Analysis
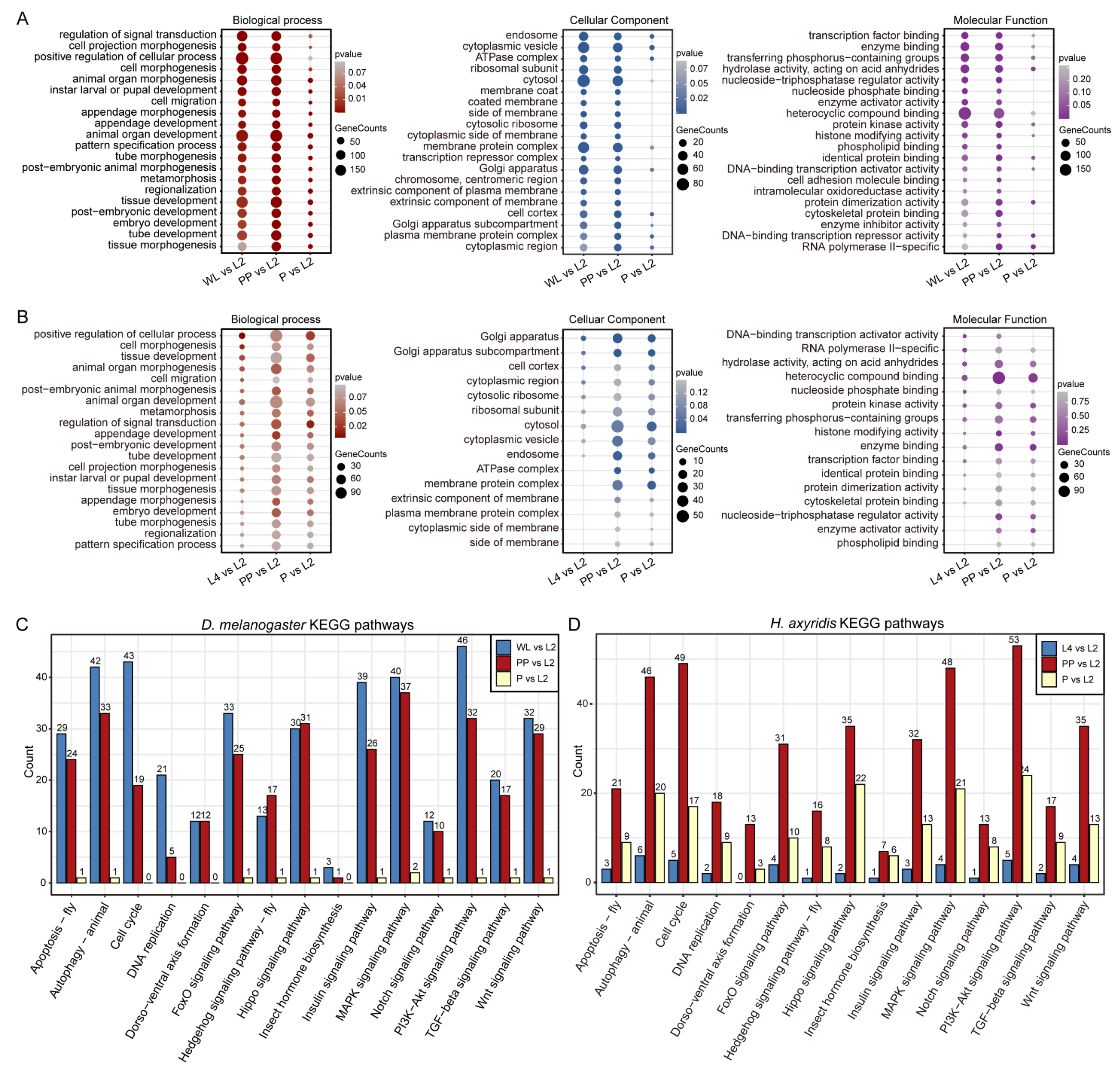
3.4. Identification of Conserved Metamorphosis-Associated Genes via Integration of Chromatin Accessibility and Expression Dynamics
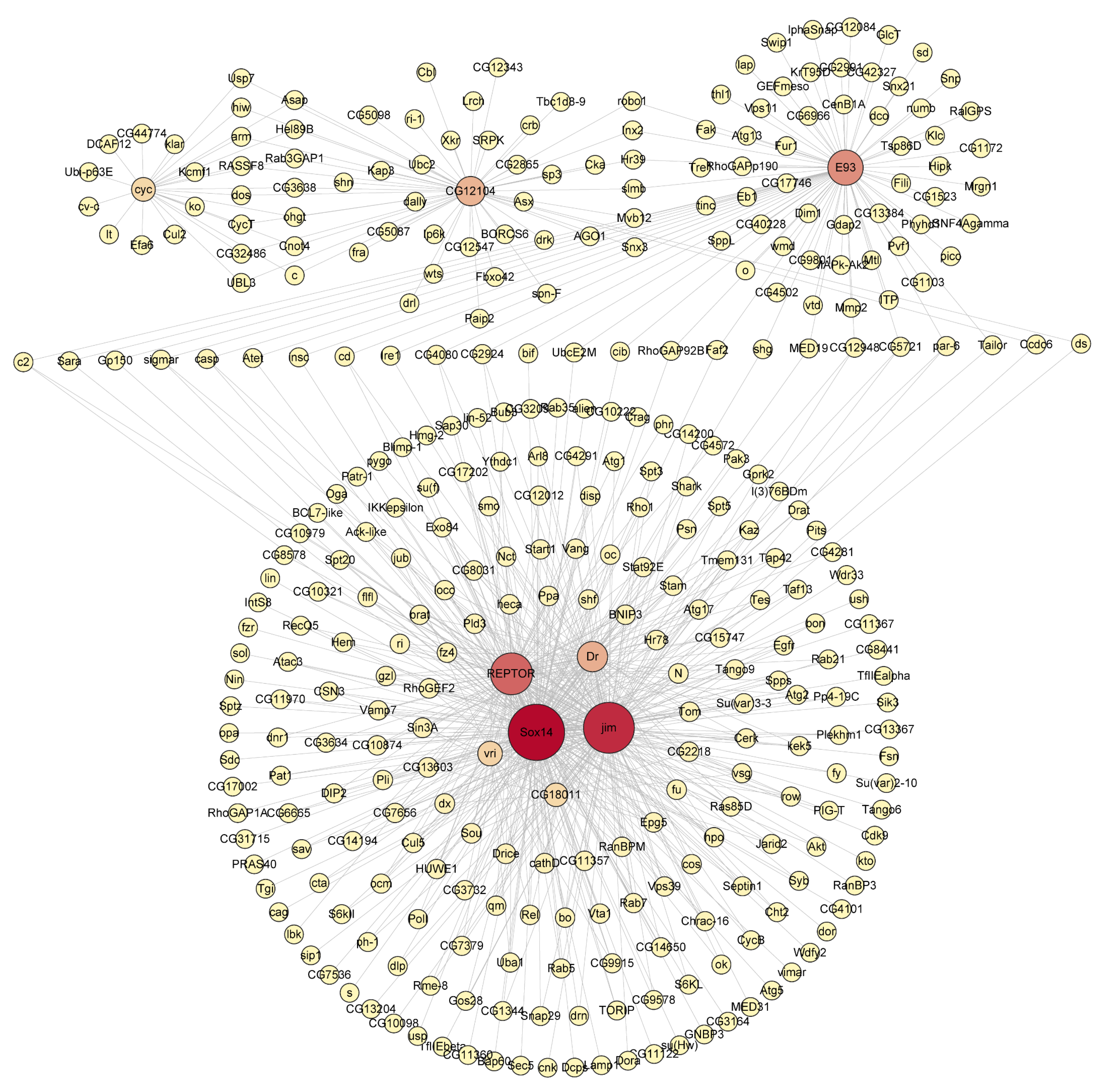
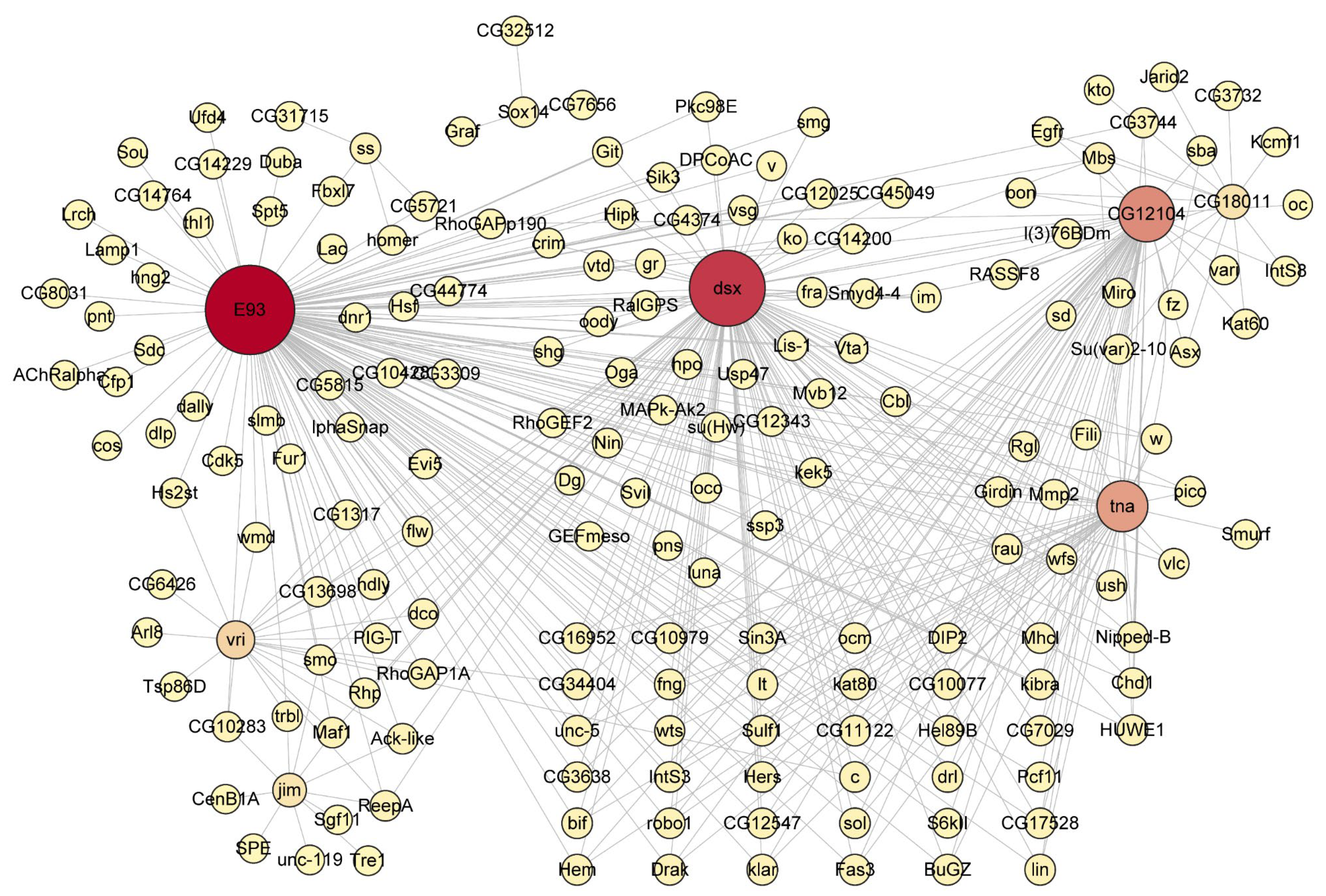
3.5. The Metamorphosis-Associated Transcriptional Network Architectures in D. melanogaster and H. axyridis
4. Discussion
4.1. Chromatin Accessibility Dynamics as a Driver of Metamorphosis
4.2. Species-Specific and Conserved Patterns in Metamorphic Regulation
4.3. Key Transcription Factors in Metamorphic Regulation
4.4. Integrated Analysis of Chromatin Accessibility and Gene Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Truman, J. The Evolution of Insect Metamorphosis. Curr. Biol. 2019, 29, R1252–R1268. [Google Scholar] [CrossRef] [PubMed]
- Belles, X. Insect Metamorphosis.; Academic Press: London, UK, 2020; pp. 131–176. [Google Scholar] [CrossRef]
- Truman, J.; Riddiford, L. The Evolution of Insect Metamorphosis: A Developmental and Endocrine View. Philos. T. R. Soc. B 2019, 374, 1783. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, N.; Rewitz, K.F.; O’Connor, M.B. Ecdysone Control of Developmental Transitions: Lessons from Drosophila Research. Annu. Rev. Entomol. 2013, 58, 497–516. [Google Scholar] [CrossRef] [PubMed]
- Riddiford, L.M.; Hiruma, K.; Zhou, X.; Nelson, C.A. Insights into the Molecular Basis of the Hormonal Control of Molting and Metamorphosis from Manduca sexta and Drosophila melanogaster. Insect Biochem. Mol. Biol. 2003, 33, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Jindra, M.; Palli, S.R.; Riddiford, L.M. The Juvenile Hormone Signaling Pathway in Insect Development. Annu. Rev. Entomol. 2013, 58, 181–204. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhou, S. Post-Transcriptional Regulation of Insect Metamorphosis and Oogenesis. Cell. Mol. Life Sci. 2020, 77, 1893–1909. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Liu, Z.; Li, C.; Wei, X.; Lin, Y.; You, L.; Zhu, Z.; Deng, H.; Feng, Q.; Huang, Y.; et al. Role of Juvenile Hormone Receptor Methoprene-Tolerant 1 In Silkworm Larval Brain Development and Domestication. Zool. Res. 2021, 42, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Kayukawa, T.; Nagamine, K.; Ito, Y.; Nishita, Y.; Ishikawa, Y.; Shinoda, T. Krüppel Homolog 1 Inhibits Insect Metamorphosis via Direct Transcriptional Repression of Broad-Complex, a Pupal Specifier Gene. J. Biol. Chem. 2016, 291, 1751–1762. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Liu, C.; Liu, W.; Wang, D.; Wang, J.; Zhao, X. Methoprene-Tolerant 1 Regulates Gene Transcription to Maintain Insect Larval Status. J. Mol. Endocrinol. 2014, 53, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Kayukawa, T.; Jouraku, A.; Ito, Y.; Shinoda, T. Molecular Mechanism Underlying Juvenile Hormone-Mediated Repression of Precocious Larval–Adult Metamorphosis. Proc. Natl. Acad. Sci. USA 2017, 114, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of Native Chromatin for Fast and Sensitive Epigenomic Profiling of Open Chromatin, DNA-Binding Proteins and Nucleosome Position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Davie, K.; Jacobs, J.; Atkins, M.; Potier, D.; Christiaens, V.; Halder, G.; Aerts, S. Discovery of Transcription Factors and Regulatory Regions Driving In Vivo Tumor Development by ATAC-Seq and FAIRE-Seq Open Chromatin Profiling. PLoS Genet. 2015, 11, e1004994. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.J.; Weilert, M.; Krueger, S.; Pampari, A.; Liu, H.; Yang, A.W.H.; Morrison, J.A.; Hughes, T.R.; Rushlow, C.A.; Kundaje, A.; et al. Chromatin Accessibility in the Drosophila Embryo Is Determined by Transcription Factor Pioneering and Enhancer Activation. Dev. Cell 2023, 58, 1898–1916. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Song, L.; Wang, X.; Cheng, H.; Wang, C.; Meyer, C.A.; Liu, T.; Tang, M.; Aluru, S.; Yue, F.; et al. Fast Alignment and Preprocessing of Chromatin Profiles with Chromap. Nat. Commun. 2021, 12, 6566. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.; Whitwham, A.; Keane, T.; McCarthy, S.; Davies, R.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2020, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, F.; Dündar, F.; Diehl, S.; Grüning, B.A.; Manke, T. deepTools: A Flexible Platform for Exploring Deep-Sequencing Data. Nucleic. Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, J.M. Improved Peak-Calling with MACS2. BioRxiv 2018. [Google Scholar] [CrossRef]
- Kumar, L.; Futschik, M.E. Mfuzz: A Software Package for Soft Clustering of Microarray Data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Shu, R.; Zhang, C.; Xiao, Y.; Jing, D.; Tang, J.; Cao, Z.; Chen, X.; Mei, Y.; Li, F. Developmental Correspondence of Juvenile Stages across the Locust, Harlequin Ladybird, and Diamondback Moth. iScience 2024, 27, 110898. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin Accessibility and the Regulatory Epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Lluch, S.; Blanco, E.; Tilgner, H.; Curado, J.; Ruiz-Romero, M.; Corominas, M.; Guigó, R. Absence of Canonical Marks of Active Chromatin in Developmentally Regulated Genes. Nat. Genet. 2015, 47, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Ureña, E.; Manjón, C.; Franch-Marro, X.; Martín, D. Transcription Factor E93 Specifies Adult Metamorphosis in Hemimetabolous and Holometabolous Insects. Proc. Natl. Acad. Sci. USA 2014, 111, 7024–7029. [Google Scholar] [CrossRef] [PubMed]
- Ureña, E.; Chafino, S.; Manjón, C.; Franch-Marro, X.; Martín, D. The Occurrence of the Holometabolous Pupal Stage Requires the Interaction between E93, Krüppel-Homolog 1 and Broad-Complex. PLoS Genet. 2016, 12, e1006020. [Google Scholar] [CrossRef] [PubMed]
- Kirilly, D.; Gu, Y.; Huang, Y.; Wu, Z.; Bashirullah, A.; Low, B.C.; Kolodkin, A.L.; Wang, H.; Yu, F. A Genetic Pathway Composed of Sox14 and Mical Governs Severing of Dendrites during Pruning. Nat. Neurosci. 2009, 12, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Tiebe, M.; Lutz, M.; De La Garza, A.; Buechling, T.; Boutros, M.; Teleman, A.A. REPTOR and REPTOR-BP Regulate Organismal Metabolism and Transcription Downstream of TORC1. Dev. Cell 2015, 33, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.S.; Wolfner, M.F. A Molecular Analysis of Doublesex, a Bifunctional Gene That Controls Both Male and Female Sexual Differentiation in Drosophila melanogaster. Gene Dev. 1988, 2, 477–489. [Google Scholar] [CrossRef] [PubMed]
- King-Jones, K.; Thummel, C.S. Nuclear Receptors—A Perspective from Drosophila. Nat. Rev. Genet. 2005, 6, 311–323. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Name | Domain |
|---|---|---|
| dsx | doublesex | DM |
| E93 | Ecdysone-induced protein 93F | Ets |
| REPTOR | Repressed by TOR | TF_bZIP |
| Sox14 | Sox box protein 14 | HMG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, J.; Zhou, H.; Cheng, Z.; Mei, Y.; Lu, Y.; Chen, X. Chromatin Accessibility Dynamics Reveal Conserved Transcriptional Regulatory Networks During Insect Metamorphosis in Harmonia axyridis and Drosophila melanogaster. Biology 2025, 14, 912. https://doi.org/10.3390/biology14080912
Tang J, Zhou H, Cheng Z, Mei Y, Lu Y, Chen X. Chromatin Accessibility Dynamics Reveal Conserved Transcriptional Regulatory Networks During Insect Metamorphosis in Harmonia axyridis and Drosophila melanogaster. Biology. 2025; 14(8):912. https://doi.org/10.3390/biology14080912
Chicago/Turabian StyleTang, Jiejing, Hang Zhou, Ziqi Cheng, Yang Mei, Yueqi Lu, and Xi Chen. 2025. "Chromatin Accessibility Dynamics Reveal Conserved Transcriptional Regulatory Networks During Insect Metamorphosis in Harmonia axyridis and Drosophila melanogaster" Biology 14, no. 8: 912. https://doi.org/10.3390/biology14080912
APA StyleTang, J., Zhou, H., Cheng, Z., Mei, Y., Lu, Y., & Chen, X. (2025). Chromatin Accessibility Dynamics Reveal Conserved Transcriptional Regulatory Networks During Insect Metamorphosis in Harmonia axyridis and Drosophila melanogaster. Biology, 14(8), 912. https://doi.org/10.3390/biology14080912