
Integrated Bioinformatics Analysis and Cellular Experimental Validation Identify Lipoprotein Lipase Gene as a Novel Biomarker for Tumorigenesis and Prognosis in Lung Adenocarcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. LUAD Data Acquisition
2.2. Acquisition of Druggable Target Genes
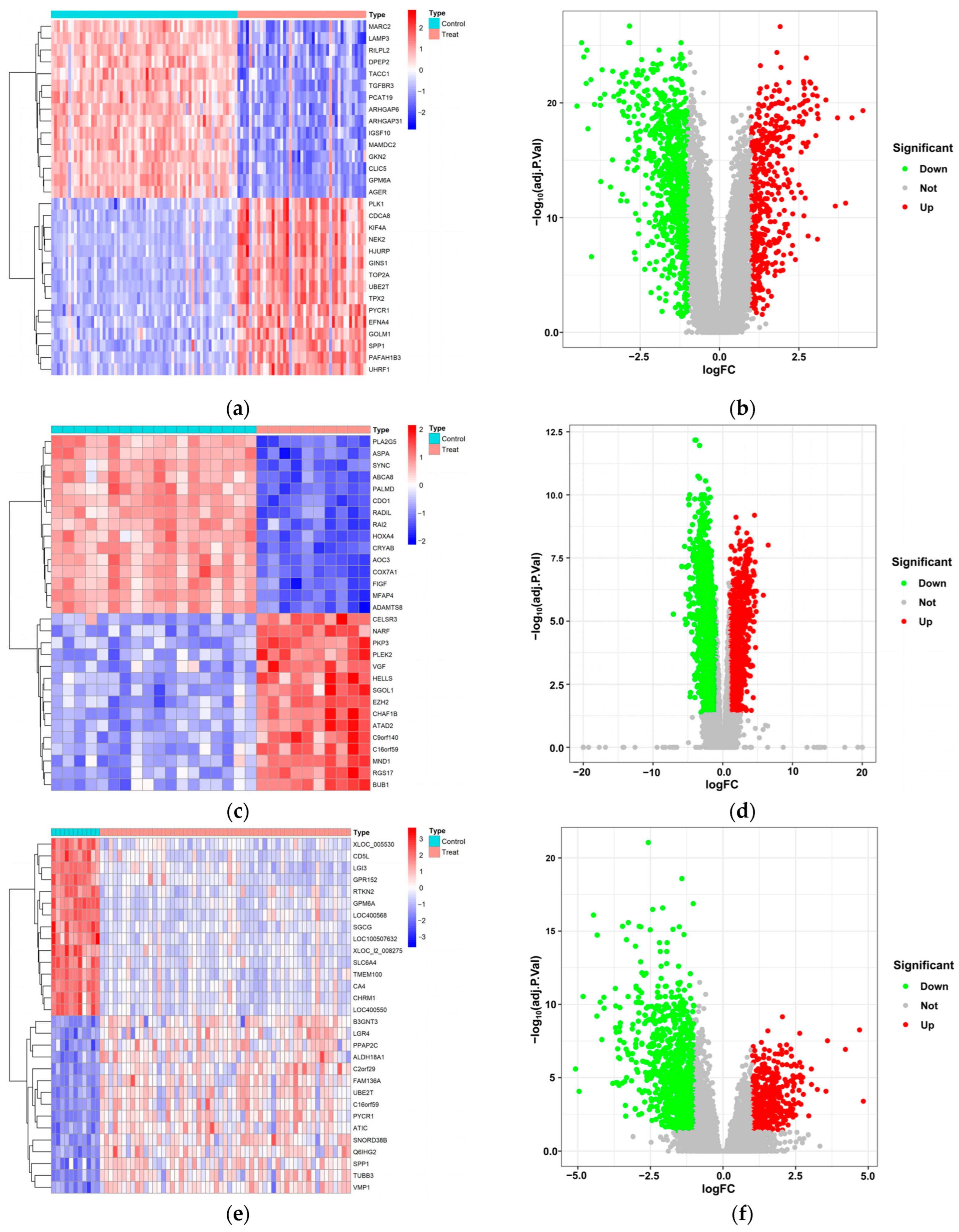
2.3. Extraction of DEGs in LUAD
2.4. The Identification of Druggable Target Gene eQTLs and LUAD GWAS
2.5. Mendelian Randomization Study Based on Summary Data
2.6. Co-Localization Analysis
2.7. Pan-Cancer Analysis
2.8. TCGA Data Analysis
2.9. Survival Analysis
2.10. Diagnosis Value Analysis
2.11. TMB and MSI Analysis
2.12. Cell Lines
2.13. Wound Healing Assay
2.14. Evaluation of the Effects of Activated LPL on A549 Cells Using the IncuCyte Live-Cell Imaging System
2.15. Cell Viability Assay
2.16. Quantitative Real-Time PCR (qRT-PCR)
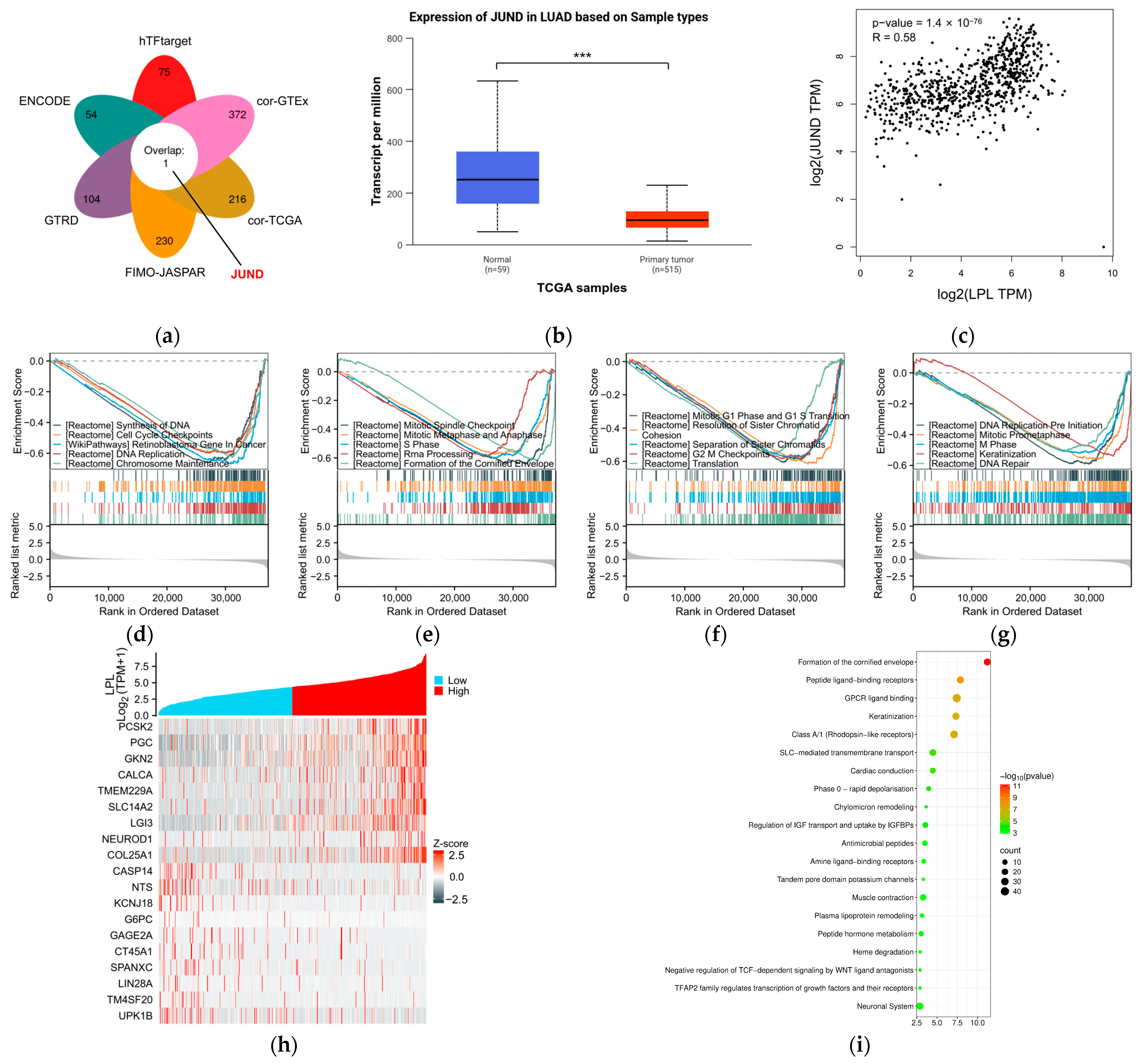
2.17. Transcription Factor Prediction of LPL
2.18. Differential Expression and Pathway Enrichment Analyses
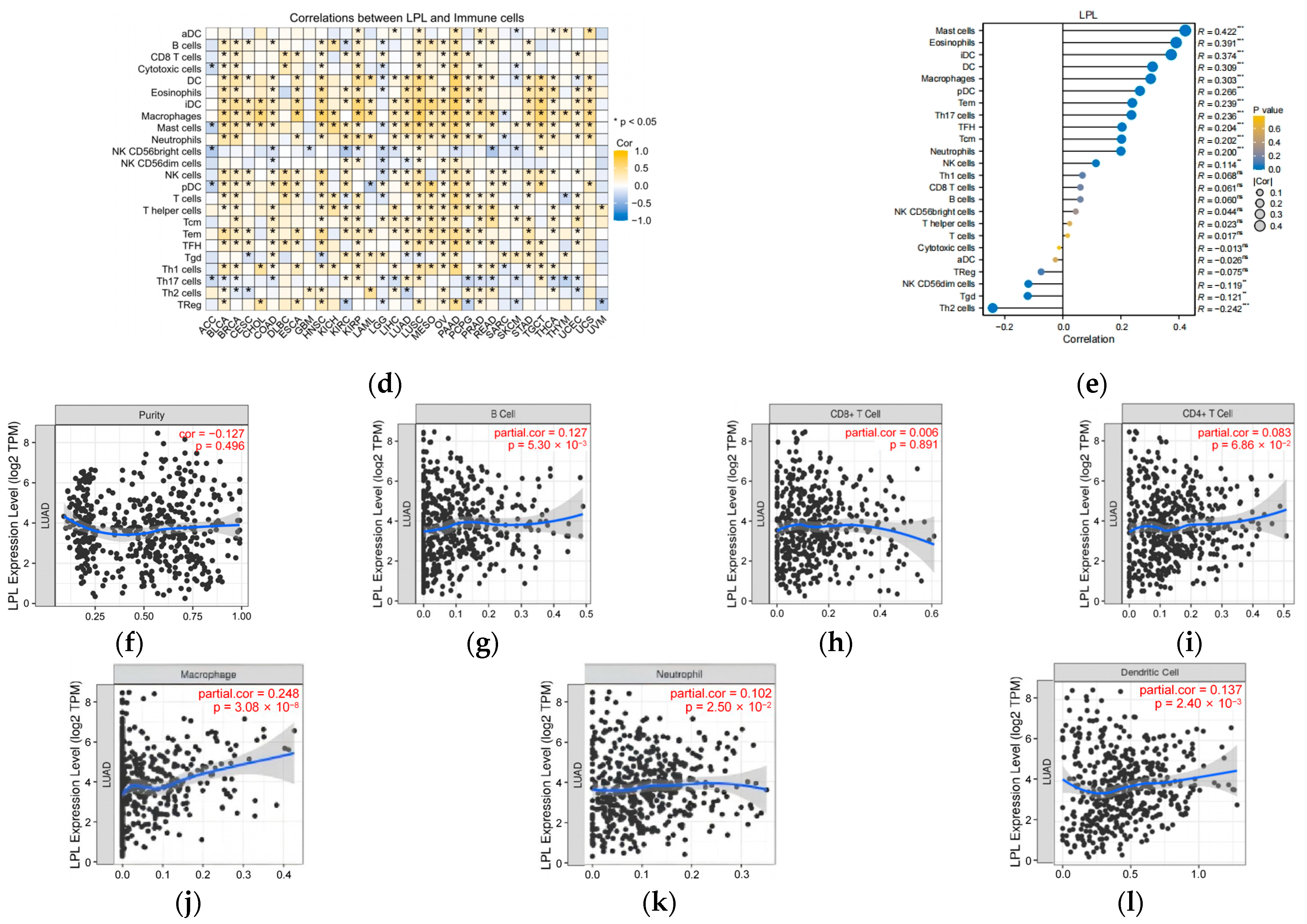
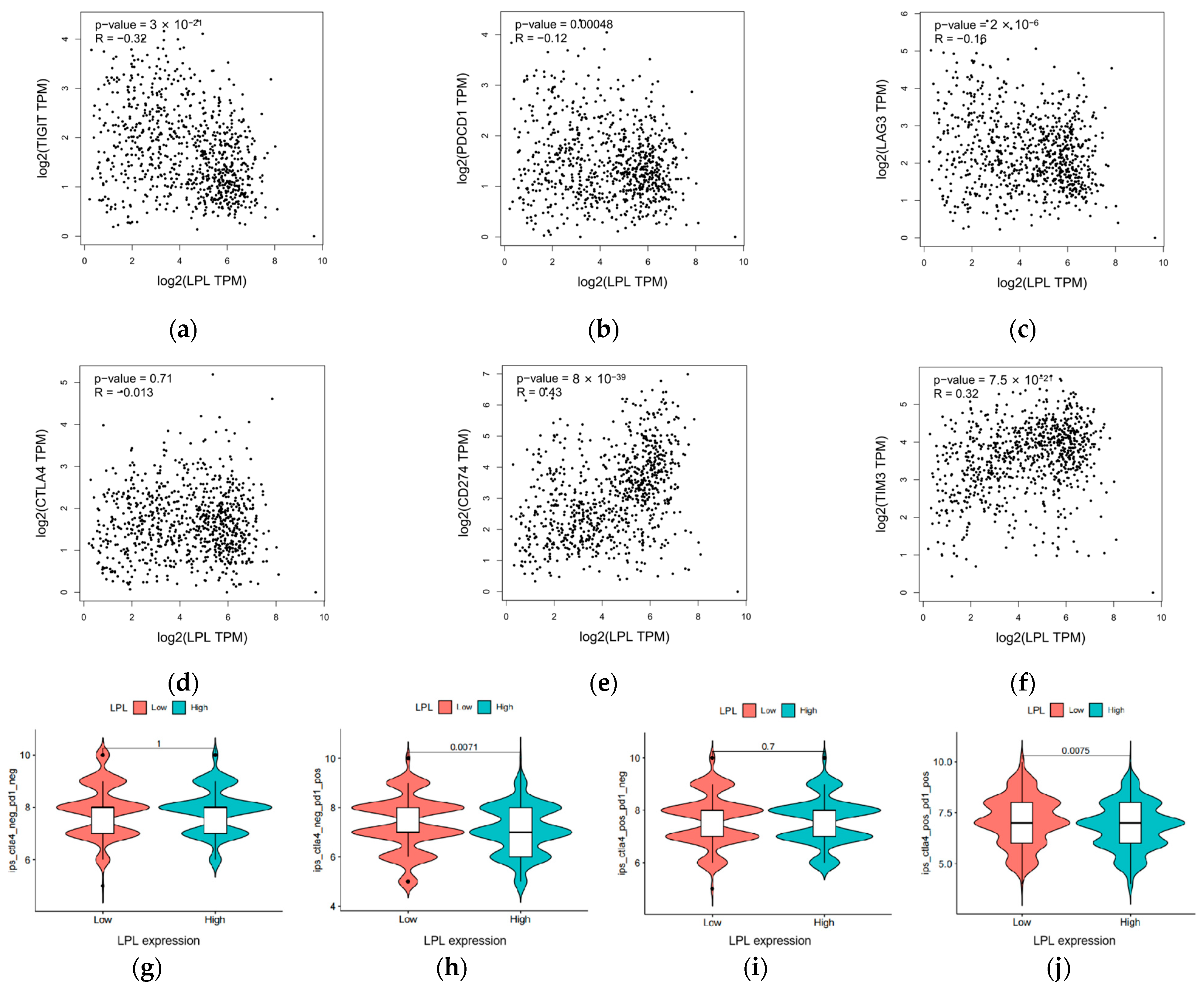
2.19. Assessment of Immunotherapy Response, Immune Checkpoint Expression, and Immune Cell Infiltration
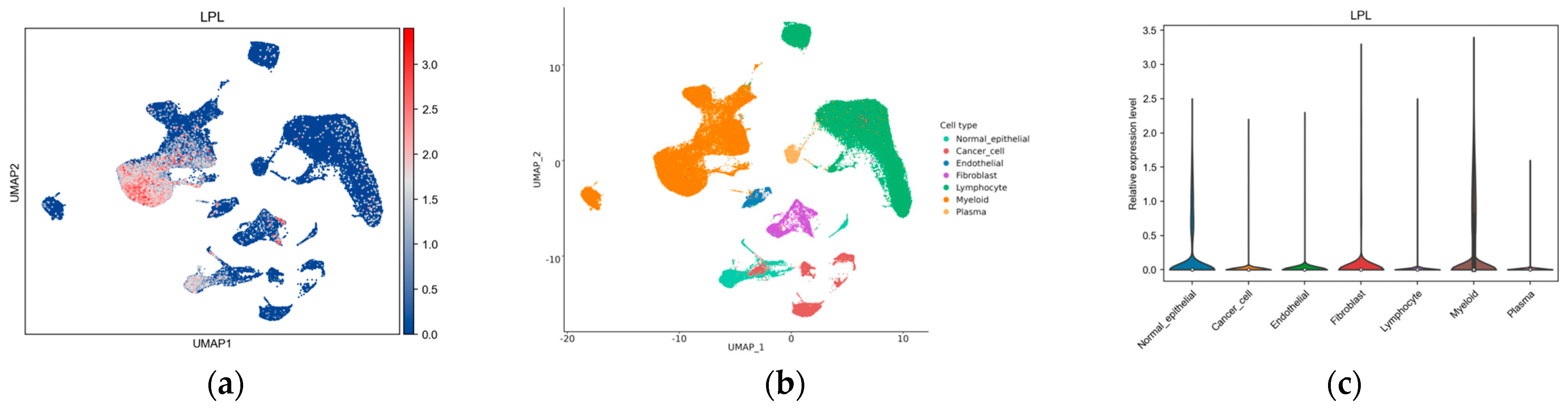
2.20. Single-Cell Analysis
2.21. Statistical Analysis
3. Results
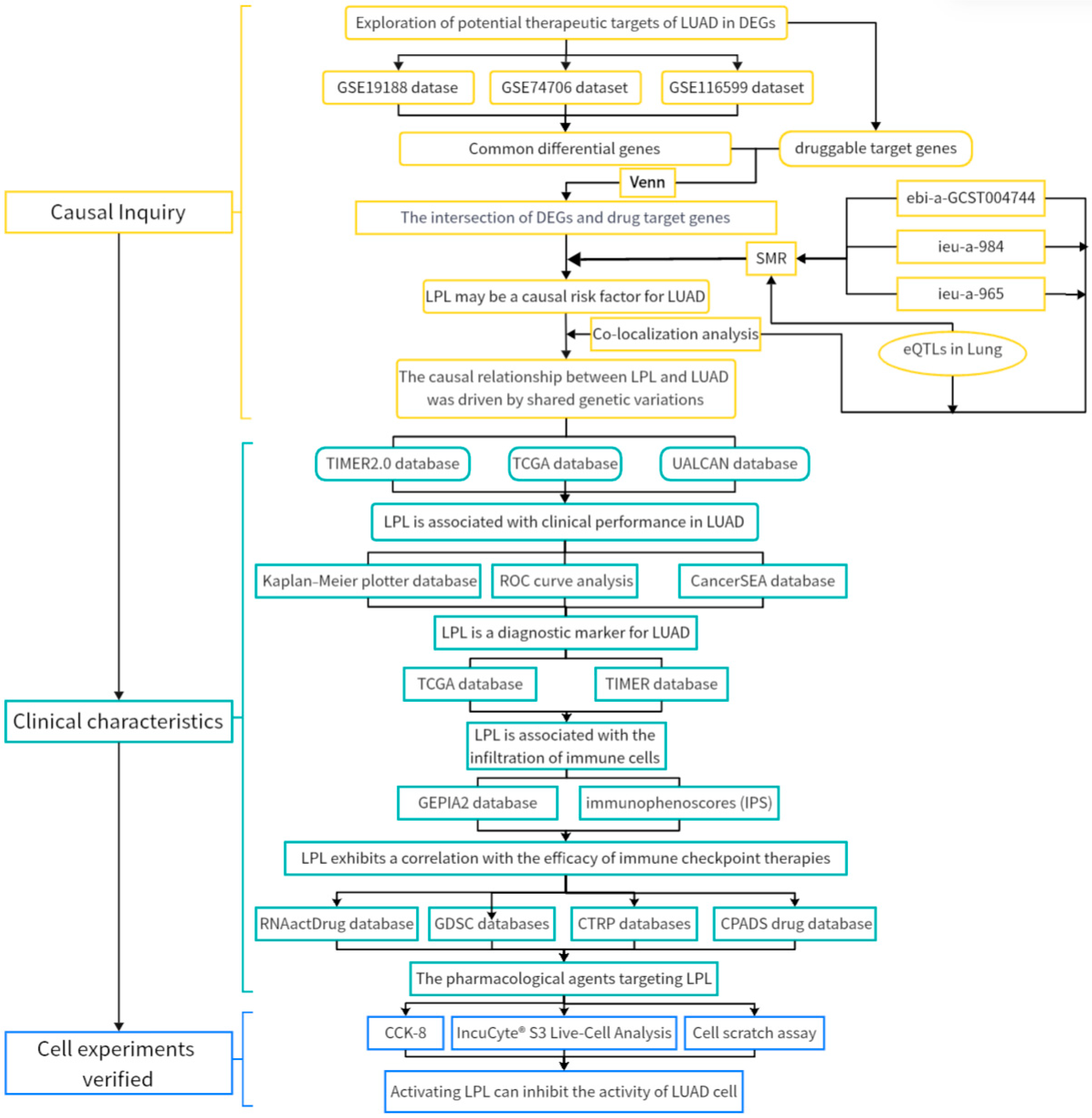
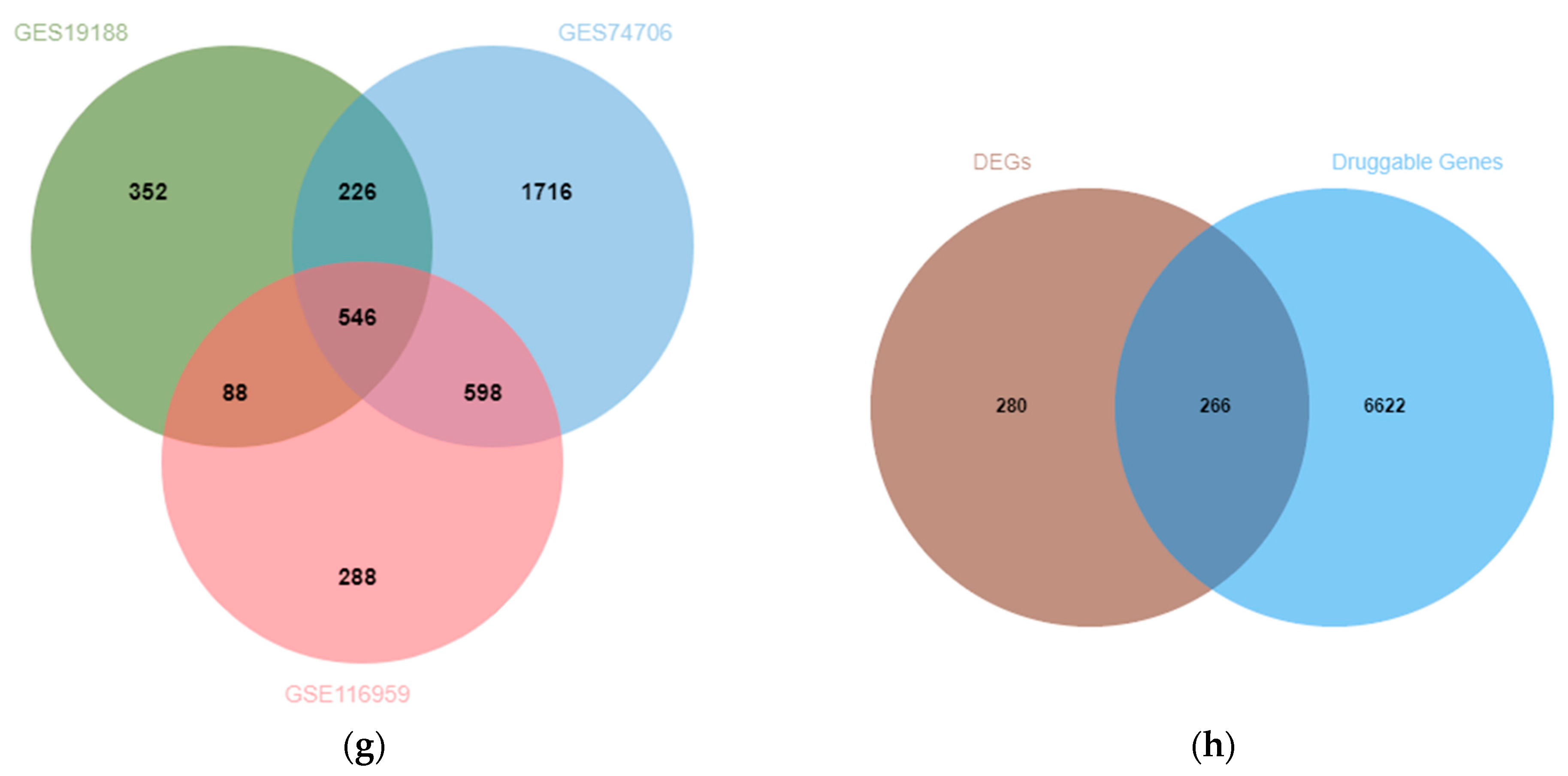
3.1. Exploration of Potential Therapeutic Targets of LUAD in DEGs
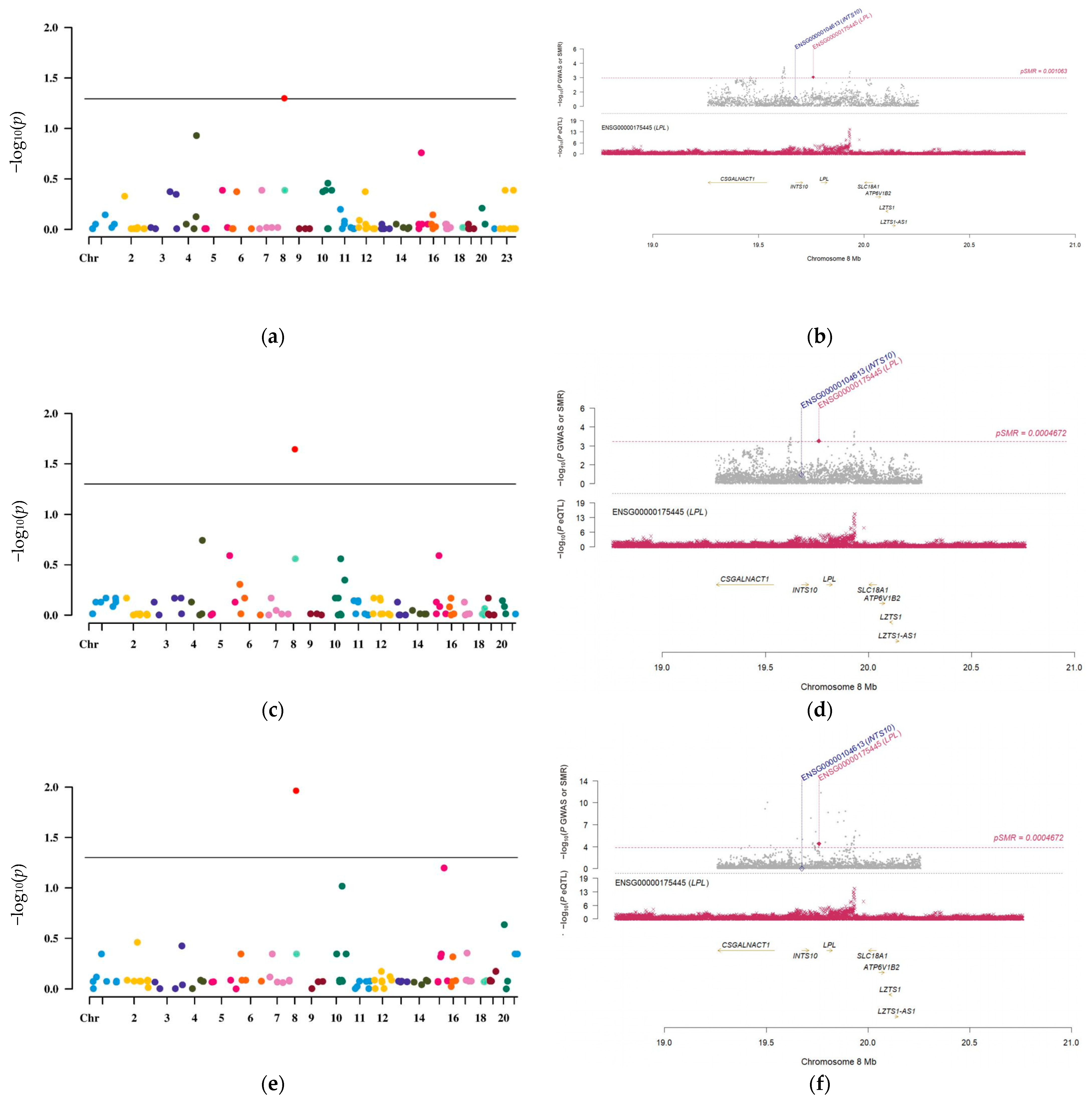
3.2. LPL Is a Potential Causal Risk Factor for LUAD
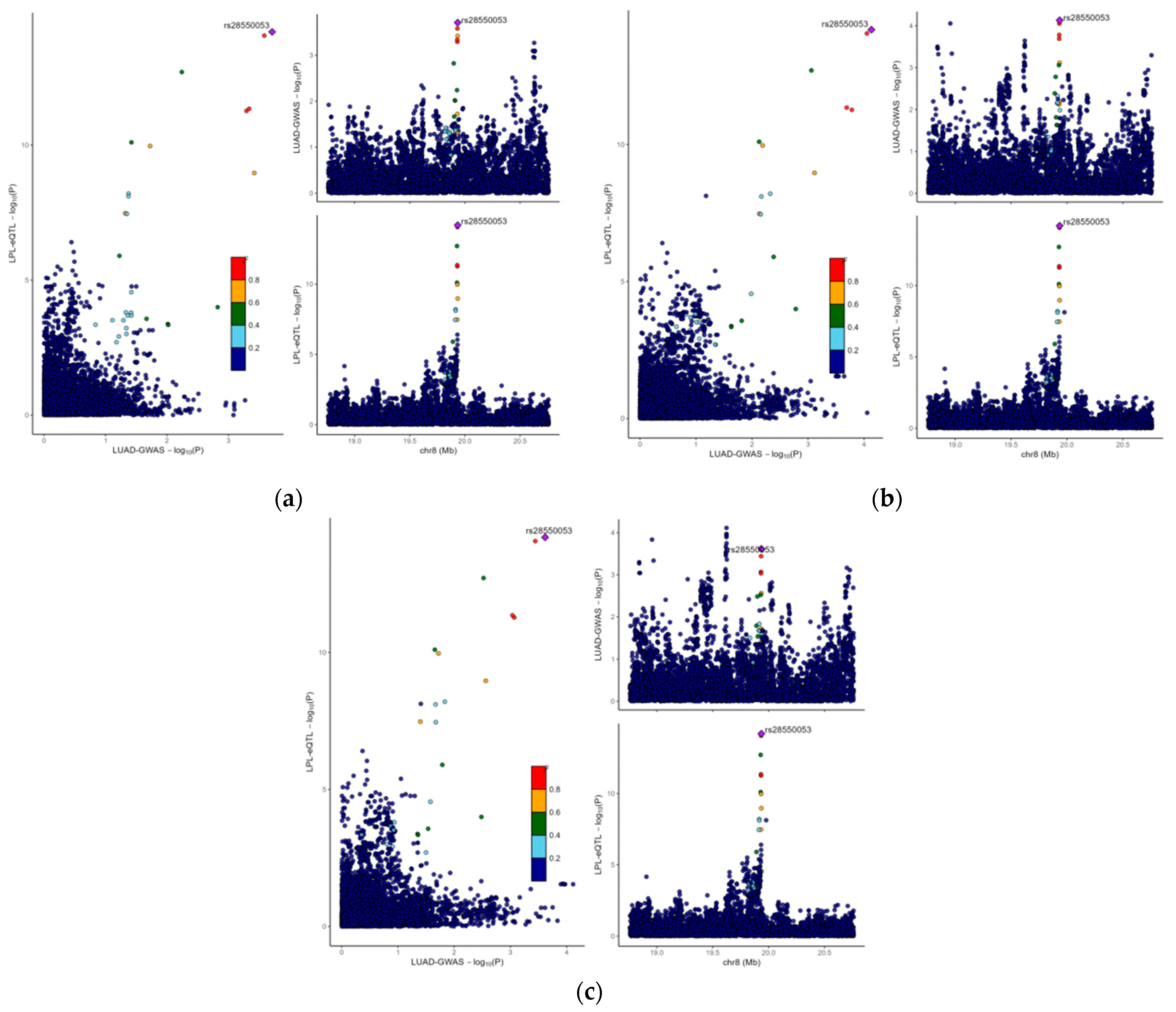
3.3. The Causal Link Between LPL and LUAD Is Shaped by Common Genetic Variations
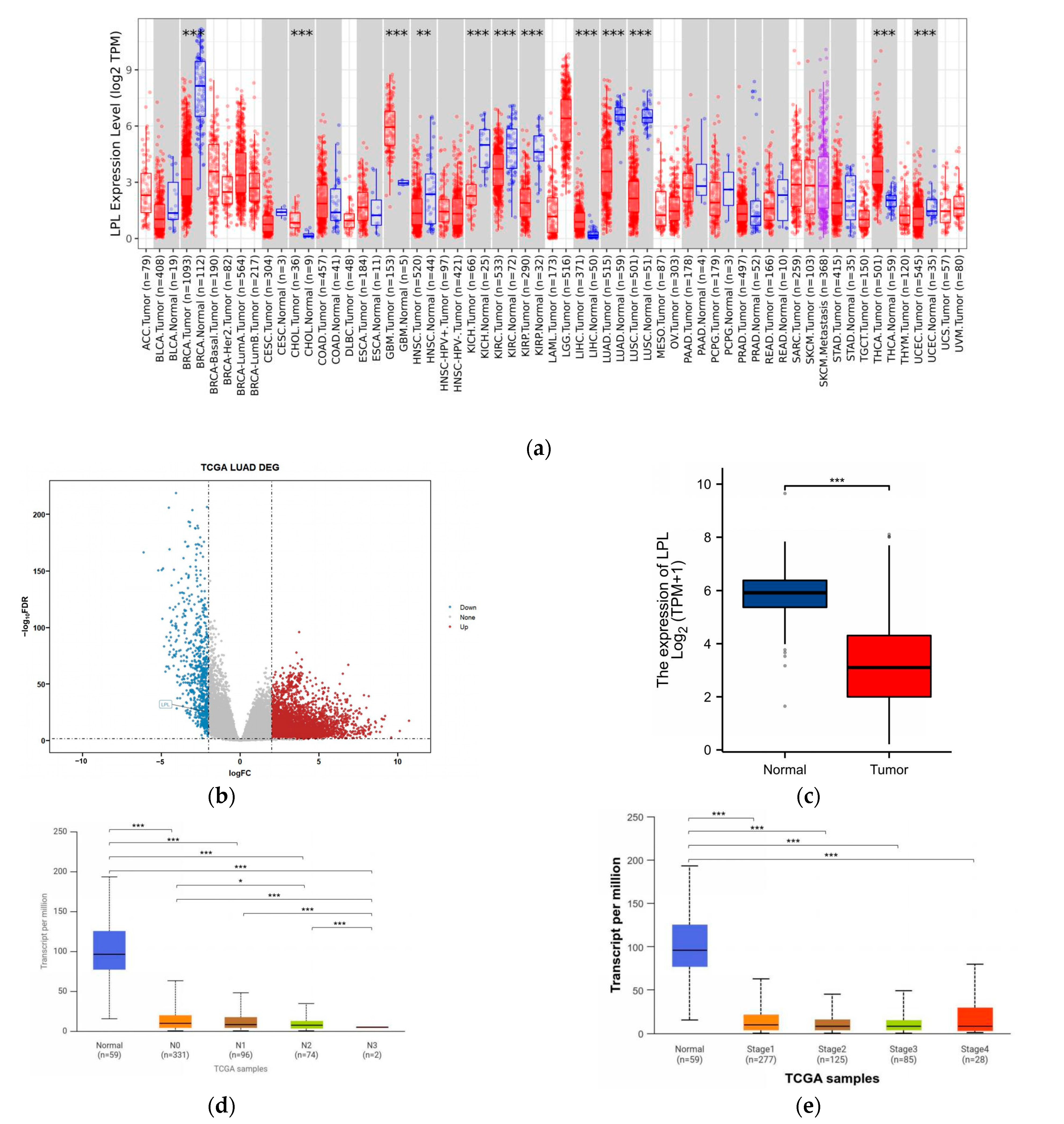
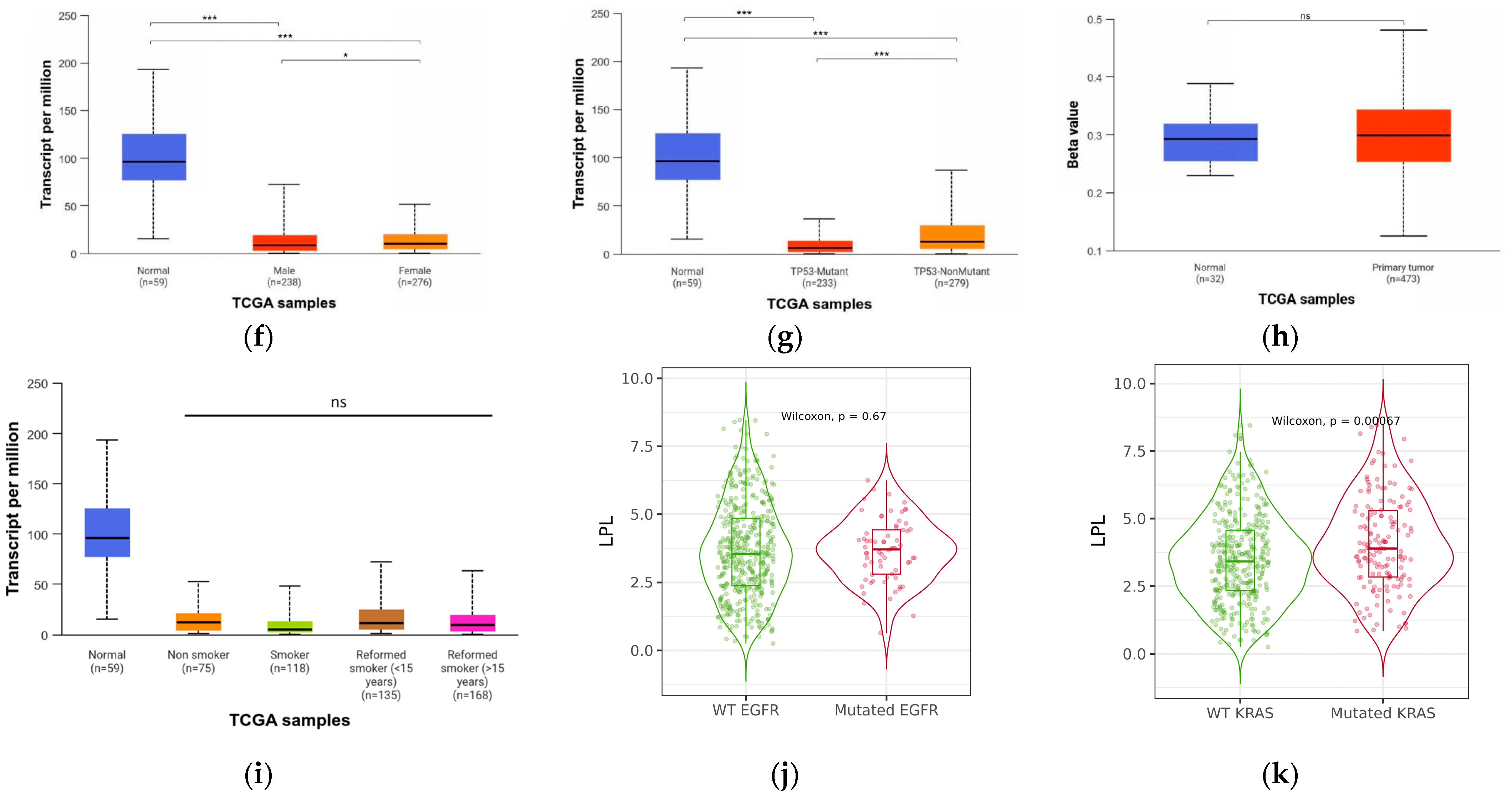
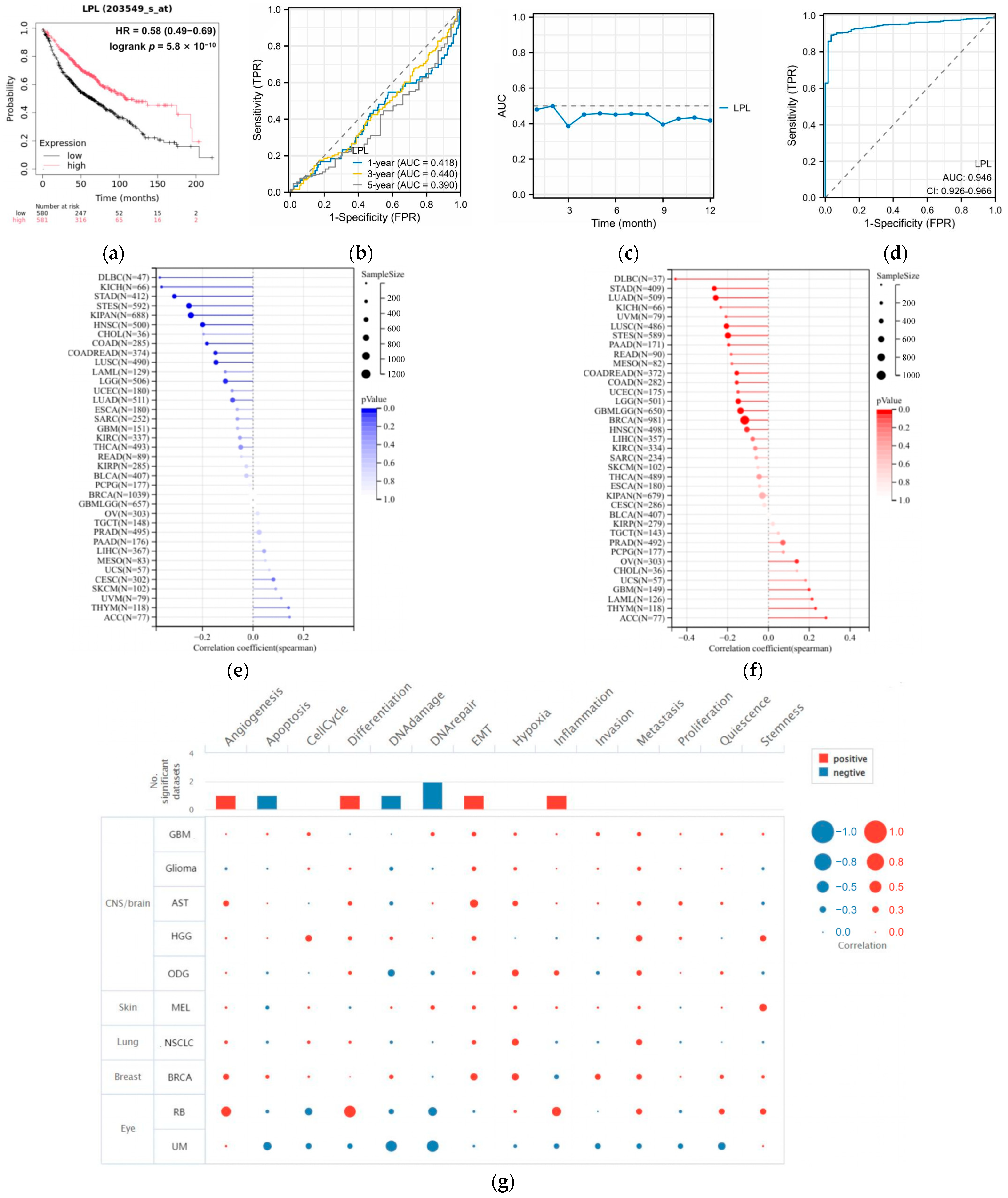
3.4. LPL Is Associated with Clinical Performance in LUAD
3.5. LPL Is a Diagnostic Marker for LUAD
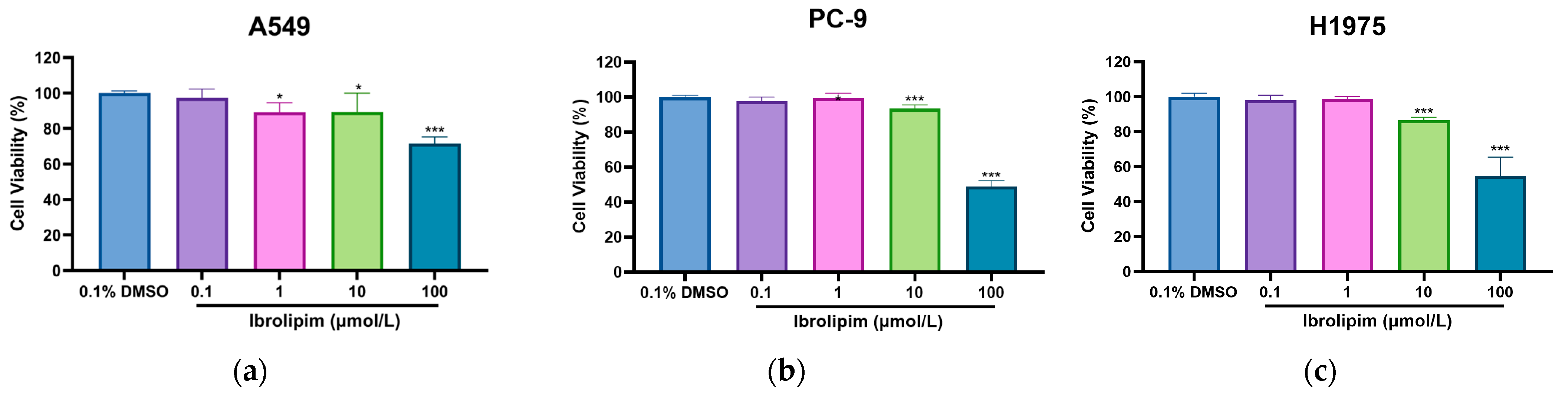
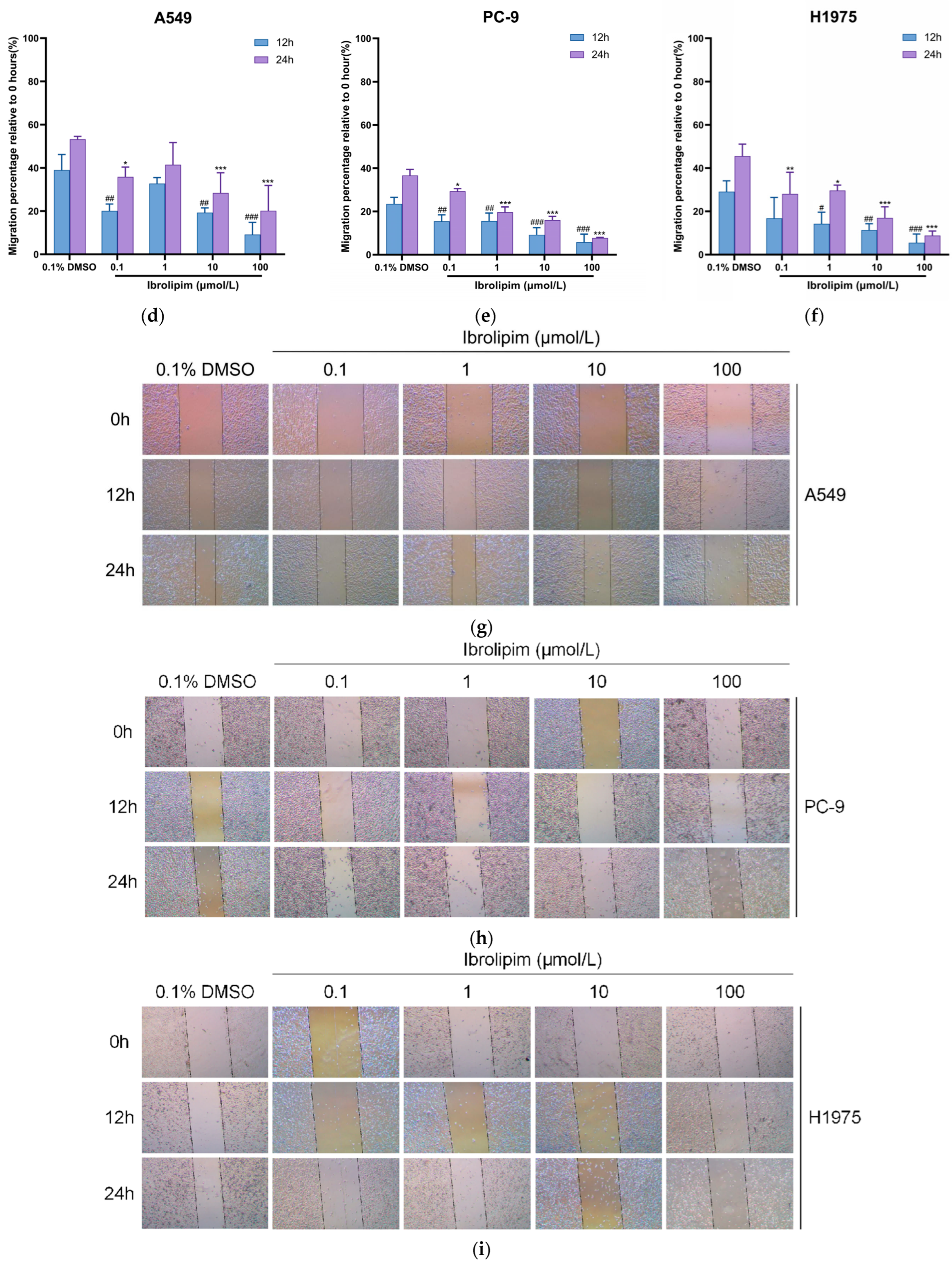
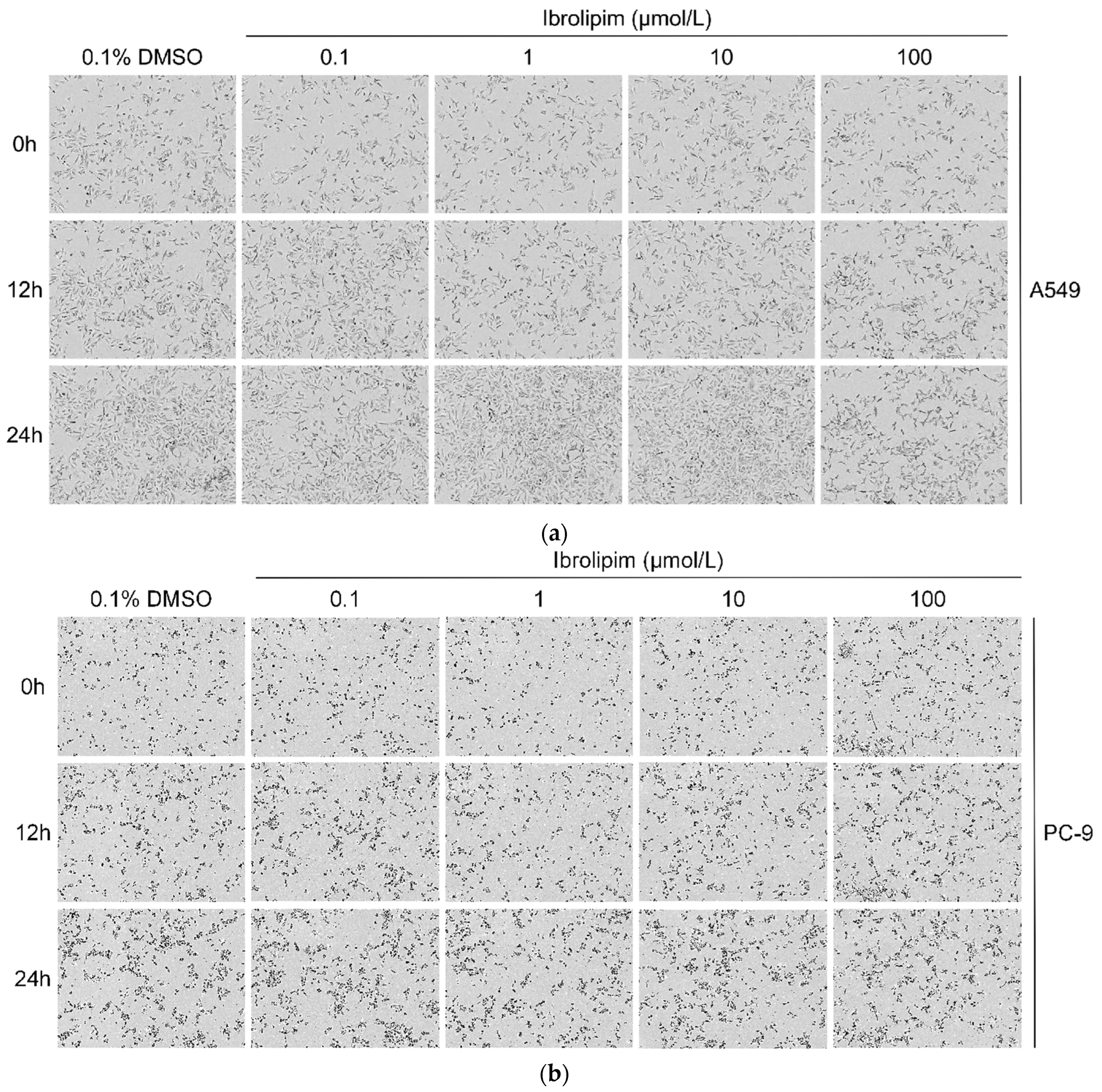
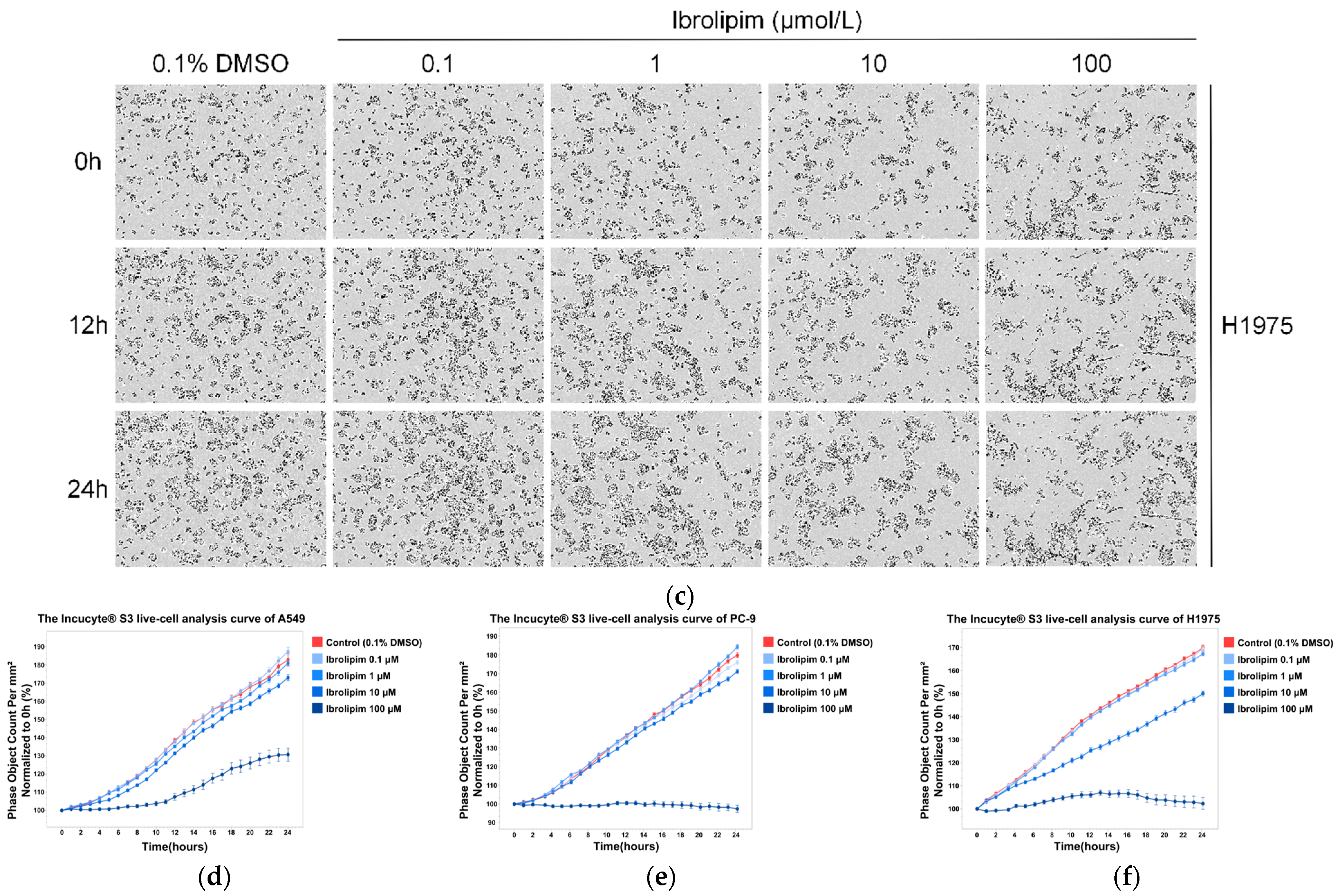
3.6. The LPL Activator Ibrolipim Inhibits the Proliferation and Migration of LUAD Cells
3.7. The Possible Regulatory and Functional Mechanisms of LPL in Lung Adenocarcinoma
3.8. LPL Is Associated with the Infiltration of Immune Cells in LUAD
3.9. LPL Exhibits a Correlation with the Efficacy of Immune Checkpoint Therapies
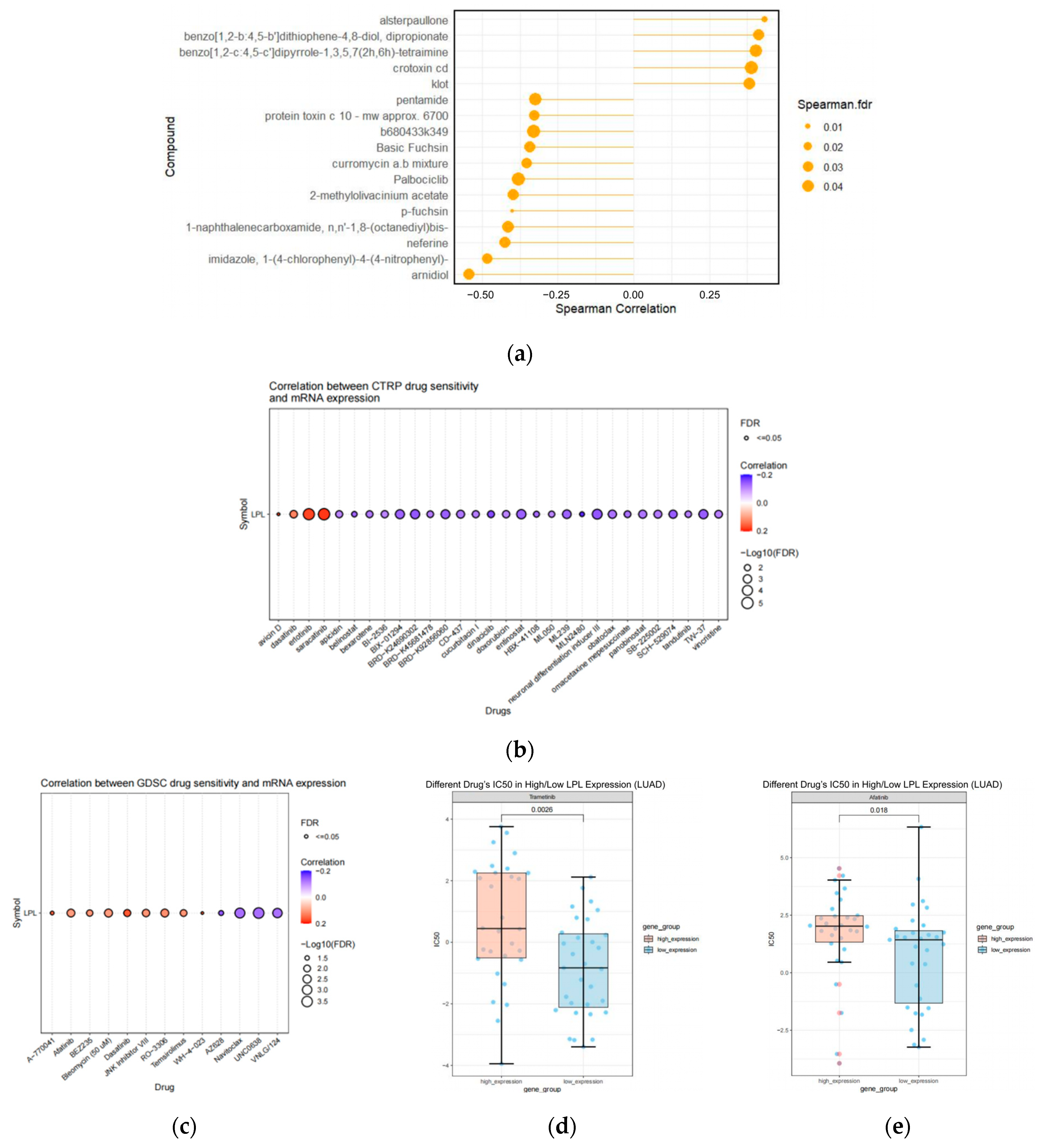
3.10. The Pharmacological Agents Targeting LPL
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALK | Anaplastic Lymphoma Kinase |
| AUC | Area Under Curve |
| CancerSEA | Cancer Single-Cell State Atlas |
| CCK-8 | Cell Counting Kit-8 |
| CPADS | Cancer Personalized Single-cell Atlas Data Server |
| CTRP | Cancer Therapeutics Response Portal |
| DEGs | Differentially Expressed Genes |
| DGIdb | Drug-Gene Interactions and the druggable genome |
| EGFR | Epidermal Growth Factor Receptor |
| eQTLs | Expression Quantitative Trait Loci |
| FBS | Fetal Bovine Serum |
| FDR | False Discovery Rate |
| GEO | Gene Expression Omnibus |
| GTEx | Genotype-Tissue Expression project |
| GWAS | Genome-Wide Association Study |
| HEIDI | Heterogeneity in Dependent Instruments |
| ICI | Immune Checkpoint Inhibitor |
| ILCCO | International Lung Cancer Consortium |
| IPS | Immunophenoscore |
| LPL | Lipoprotein Lipase |
| LUAD | Lung Adenocarcinoma |
| MSI | Microsatellite Instability |
| NCCN | National Comprehensive Cancer Network |
| NSCLC | Non-Small-Cell Lung Cancer |
| OR | Odds Ratio |
| PPH4 | Posterior Probability of Hypothesis 4 |
| ROC | Receiver Operating Characteristic |
| scRNA-seq | Single-cell RNA sequencing |
| SMR | Summary-data-based Mendelian Randomization |
| SNPs | Single Nucleotide Polymorphisms |
| ssGSEA | single-sample Gene Set Enrichment Analysis |
| TCGA | The Cancer Genome Atlas |
| TCIA | The Cancer Immunome Atlas |
| TIMER | Tumor Immune Estimation Resource |
| TMB | Tumor Mutational Burden |
| TRICL | Transdisciplinary Research in Cancer of the Lung |
| UALCAN | University of Alabama Cancer Database |
References
- Brody, H. Lung Cancer. Nature 2020, 587, S7. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, L.E.L.; Remon, J.; Faivre-Finn, C.; Garassino, M.C.; Heymach, J.V.; Kerr, K.M.; Tan, D.S.W.; Veronesi, G.; Reck, M. Non-Small-Cell Lung Cancer. Nat. Rev. Dis. Primers 2024, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Juárez, M.; Serrano-Gómez, C.; Bote-de-Cabo, H.; Paz-Ares, L. Targeted Therapy for Lung Cancer: Beyond EGFR and ALK. Cancer 2023, 129, 1803–1820. [Google Scholar] [CrossRef]
- Succony, L.; Rassl, D.M.; Barker, A.P.; McCaughan, F.M.; Rintoul, R.C. Adenocarcinoma Spectrum Lesions of the Lung: Detection, Pathology and Treatment Strategies. Cancer Treat. Rev. 2021, 99, 102237. [Google Scholar] [CrossRef]
- Gillette, M.A.; Satpathy, S.; Cao, S.; Dhanasekaran, S.M.; Vasaikar, S.V.; Krug, K.; Petralia, F.; Li, Y.; Liang, W.-W.; Reva, B.; et al. Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 2020, 182, 200–225.e35. [Google Scholar] [CrossRef] [PubMed]
- Birney, E. Mendelian Randomization. Cold Spring Harb. Perspect. Med. 2022, 12, a041302. [Google Scholar] [CrossRef]
- Davies, N.M.; Holmes, M.V.; Davey Smith, G. Reading Mendelian Randomisation Studies: A Guide, Glossary, and Checklist for Clinicians. BMJ 2018, 362, k601. [Google Scholar] [CrossRef]
- Li, J.; Tang, M.; Gao, X.; Tian, S.; Liu, W. Mendelian Randomization Analyses Explore the Relationship between Cathepsins and Lung Cancer. Commun. Biol. 2023, 6, 1019. [Google Scholar] [CrossRef]
- Hou, J.; Aerts, J.; den Hamer, B.; van Ijcken, W.; den Bakker, M.; Riegman, P.; van der Leest, C.; van der Spek, P.; Foekens, J.A.; Hoogsteden, H.C.; et al. Gene Expression-Based Classification of Non-Small Cell Lung Carcinomas and Survival Prediction. PLoS ONE 2010, 5, e10312. [Google Scholar] [CrossRef]
- Marwitz, S.; Depner, S.; Dvornikov, D.; Merkle, R.; Szczygieł, M.; Müller-Decker, K.; Lucarelli, P.; Wäsch, M.; Mairbäurl, H.; Rabe, K.F.; et al. Downregulation of the TGFβ Pseudoreceptor BAMBI in Non-Small Cell Lung Cancer Enhances TGFβ Signaling and Invasion. Cancer Res. 2016, 76, 3785–3801. [Google Scholar] [CrossRef]
- Moreno Leon, L.; Gautier, M.; Allan, R.; Ilié, M.; Nottet, N.; Pons, N.; Paquet, A.; Lebrigand, K.; Truchi, M.; Fassy, J.; et al. The Nuclear Hypoxia-Regulated NLUCAT1 Long Non-Coding RNA Contributes to an Aggressive Phenotype in Lung Adenocarcinoma through Regulation of Oxidative Stress. Oncogene 2019, 38, 7146–7165. [Google Scholar] [CrossRef]
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with Open Crowdsource Efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef] [PubMed]
- Finan, C.; Gaulton, A.; Kruger, F.A.; Lumbers, R.T.; Shah, T.; Engmann, J.; Galver, L.; Kelley, R.; Karlsson, A.; Santos, R.; et al. The Druggable Genome and Support for Target Identification and Validation in Drug Development. Sci. Transl. Med. 2017, 9, eaag1166. [Google Scholar] [CrossRef] [PubMed]
- The GTEx Consortium. The GTEx Consortium Atlas of Genetic Regulatory Effects across Human Tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- McKay, J.D.; Hung, R.J.; Han, Y.; Zong, X.; Carreras-Torres, R.; Christiani, D.C.; Caporaso, N.E.; Johansson, M.; Xiao, X.; Li, Y.; et al. Large-Scale Association Analysis Identifies New Lung Cancer Susceptibility Loci and Heterogeneity in Genetic Susceptibility across Histological Subtypes. Nat. Genet. 2017, 49, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; McKay, J.D.; Rafnar, T.; Wang, Z.; Timofeeva, M.N.; Broderick, P.; Zong, X.; Laplana, M.; Wei, Y.; Han, Y.; et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat. Genet. 2014, 46, 736–741. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of Summary Data from GWAS and eQTL Studies Predicts Complex Trait Gene Targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef]
- Giambartolomei, C.; Vukcevic, D.; Schadt, E.E.; Franke, L.; Hingorani, A.D.; Wallace, C.; Plagnol, V. Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. PLoS Genet. 2014, 10, e1004383. [Google Scholar] [CrossRef]
- Yuan, H.; Yan, M.; Zhang, G.; Liu, W.; Deng, C.; Liao, G.; Xu, L.; Luo, T.; Yan, H.; Long, Z.; et al. CancerSEA: A Cancer Single-Cell State Atlas. Nucleic Acids Res. 2019, 47, D900–D908. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for Analysis of Tumor-Infiltrating Immune Cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An Update to the Integrated Cancer Data Analysis Platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Győrffy, B. Transcriptome-Level Discovery of Survival-Associated Biomarkers and Therapy Targets in Non-Small-Cell Lung Cancer. Br. J. Pharmacol. 2024, 181, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An Open-Source Package for R and S+ to Analyze and Compare ROC Curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Chen, D.; Xu, L.; Xing, H.; Shen, W.; Song, Z.; Li, H.; Zhu, X.; Li, X.; Wu, L.; Jiao, H.; et al. Sangerbox 2: Enhanced functionalities and update for a comprehensive clinical bioinformatics data analysis platform. iMeta 2024, 3, e238. [Google Scholar] [CrossRef]
- Jin, M.; O’Nuallain, B.; Hong, W.; Boyd, J.; Lagomarsino, V.N.; O’Malley, T.T.; Liu, W.; Vanderburg, C.R.; Frosch, M.P.; Young-Pearse, T.; et al. An in Vitro Paradigm to Assess Potential Anti-Aβ Antibodies for Alzheimer’s Disease. Nat. Commun. 2018, 9, 2676. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An Enhanced Web Server for Large-Scale Expression Profiling and Interactive Analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-Cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]
- Huang, C.; Liu, Z.; Guo, Y.; Wang, W.; Yuan, Z.; Guan, Y.; Pan, D.; Hu, Z.; Sun, L.; Fu, Z.; et al. scCancerExplorer: A Comprehensive Database for Interactively Exploring Single-Cell Multi-Omics Data of Human Pan-Cancer. Nucleic Acids Res. 2025, 53, D1526–D1535. [Google Scholar] [CrossRef]
- Dong, Q.; Li, F.; Xu, Y.; Xiao, J.; Xu, Y.; Shang, D.; Zhang, C.; Yang, H.; Tian, Z.; Mi, K.; et al. RNAactDrug: A comprehensive database of RNAs associated with drug sensitivity from multi-omics data. Brief. Bioinform. 2020, 21, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A Resource for Therapeutic Biomarker Discovery in Cancer Cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Cokol, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef]
- Li, K.; Yang, H.; Lin, A.; Xie, J.; Wang, H.; Zhou, J.; Carr, S.R.; Liu, Z.; Li, X.; Zhang, J.; et al. CPADS: A web tool for comprehensive pancancer analysis of drug sensitivity. Brief. Bioinform. 2024, 25, bbae237. [Google Scholar] [CrossRef]
- Riely, G.J.; Wood, D.E.; Ettinger, D.S.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; et al. Non-Small Cell Lung Cancer, Version 4.2024, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2024, 22, 249–274. [Google Scholar] [CrossRef]
- Podgornik, H.; Sok, M.; Kern, I.; Marc, J.; Cerne, D. Lipoprotein Lipase in Non-Small Cell Lung Cancer Tissue Is Highly Expressed in a Subpopulation of Tumor-Associated Macrophages. Pathol.-Res. Pract. 2013, 209, 516–520. [Google Scholar] [CrossRef]
- Xiao, J.; Cao, S.; Wang, J.; Li, P.; Cheng, Q.; Zhou, X.; Dong, J.; Li, Y.; Zhao, X.; Xu, Z.; et al. Leptin-mediated suppression of lipoprotein lipase cleavage enhances lipid uptake and facilitates lymph node metastasis in gastric cancer. Cancer Commun. 2024, 44, 855–878. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.M.; Kraynie, A.M.; Roggli, V.L. Immunostaining in Lung Cancer for the Clinician. Commonly Used Markers for Differentiating Primary and Metastatic Pulmonary Tumors. Ann. Am. Thorac. Soc. 2015, 12, 429–435. [Google Scholar] [CrossRef]
- Fatima, N.; Cohen, C.; Lawson, D.; Siddiqui, M.T. TTF-1 and Napsin A Double Stain: A Useful Marker for Diagnosing Lung Adenocarcinoma on Fine-Needle Aspiration Cell Blocks. Cancer Cytopathol. 2011, 119, 127–133. [Google Scholar] [CrossRef]
- Gurda, G.T.; Zhang, L.; Wang, Y.; Chen, L.; Geddes, S.; Cho, W.C.; Askin, F.; Gabrielson, E.; Li, Q.K. Utility of Five Commonly Used Immunohistochemical Markers TTF-1, Napsin A, CK7, CK5/6 and P63 in Primary and Metastatic Adenocarcinoma and Squamous Cell Carcinoma of the Lung: A Retrospective Study of 246 Fine Needle Aspiration Cases. Clin. Trans. Med. 2015, 4, 16. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Inoue, Y.; Hagi, A.; Murase, T. The Novel Compound NO-1886 Elevates Plasma High-Density Lipoprotein Cholesterol Levels in Hamsters and Rabbits by Increasing Lipoprotein Lipase without Any Effect on Cholesteryl Ester Transfer Protein Activity. Metabolism 1997, 46, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Tsutsumi, K. Lipoprotein Lipase Activator NO-1886. Cardiovasc. Drug Rev. 2003, 21, 133–142. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Inoue, Y.; Shima, A.; Iwasaki, K.; Kawamura, M.; Murase, T. The Novel Compound NO-1886 Increases Lipoprotein Lipase Activity with Resulting Elevation of High Density Lipoprotein Cholesterol, and Long-Term Administration Inhibits Atherogenesis in the Coronary Arteries of Rats with Experimental Atherosclerosis. J. Clin. Investig. 1993, 92, 411–417. [Google Scholar] [CrossRef]
- Gidda, A.K.; Chittaranjan, S.; Gorski, S.M. Real-Time IncuCyte® Assay for the Dynamic Assessment of Live and Dead Cells in 2D Cultures. Bio Protoc. 2025, 15, e5209. [Google Scholar] [CrossRef] [PubMed]
- Marrone, G.; Urciuoli, S.; Candi, E.; Bernini, R.; Vanni, G.; Masci, C.; Guerriero, C.; Mancini, M.; De Lorenzo, A.; Vignolini, P.; et al. Biological Activities of Molecules Derived from Olea europaea L. Tested In Vitro. Life 2023, 14, 49. [Google Scholar] [CrossRef]
- Liam, C.-K.; Andarini, S.; Lee, P.; Ho, J.C.-M.; Chau, N.Q.; Tscheikuna, J. Lung Cancer Staging Now and in the Future. Respirology 2015, 20, 526–534. [Google Scholar] [CrossRef]
- Spella, M.; Stathopoulos, G.T. Immune Resistance in Lung Adenocarcinoma. Cancers 2021, 13, 384. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Suda, K.; Hamada, A.; Tsutani, Y. Recent Advances in Perioperative Immunotherapies in Lung Cancer. Biomolecules 2023, 13, 1377. [Google Scholar] [CrossRef]
- Yu, Y.; Li, L.; Luo, B.; Chen, D.; Yin, C.; Jian, C.; You, Q.; Wang, J.; Fang, L.; Cai, D.; et al. Predicting Potential Therapeutic Targets and Small Molecule Drugs for Early-Stage Lung Adenocarcinoma. Biomed. Pharmacother. 2024, 174, 116528. [Google Scholar] [CrossRef]
- Perrier, A.; Didelot, A.; Laurent-Puig, P.; Blons, H.; Garinet, S. Epigenetic Mechanisms of Resistance to Immune Checkpoint Inhibitors. Biomolecules 2020, 10, 1061. [Google Scholar] [CrossRef]
- Wu, S.A.; Kersten, S.; Qi, L. Lipoprotein Lipase and Its Regulators: An Unfolding Story. Trends Endocrinol. Metab. 2021, 32, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Mead, J.R.; Irvine, S.A.; Ramji, D.P. Lipoprotein Lipase: Structure, Function, Regulation, and Role in Disease. J. Mol. Med. 2002, 80, 753–769. [Google Scholar] [CrossRef]
- Gehrisch, S. Common Mutations of the Lipoprotein Lipase Gene and Their Clinical Significance. Curr. Atheroscler. Rep. 1999, 1, 70–78. [Google Scholar] [CrossRef]
- Hayden, M.R.; Kastelein, J.J.; Funke, H.; Brunzell, J.D.; Ma, Y. Phenotypic Variation of Mutations in the Human Lipoprotein-Lipase Gene. Biochem. Soc. Trans. 1993, 21, 506–509. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Liu, X.; Zhang, Y.; Tao, Y.; Zhao, L.; Abusalamah, H.; Huffman, C.; Harbison, R.A.; Puram, S.V.; Wang, Y.; et al. Tumor Extracellular Vesicle–Derived PD-L1 Promotes T Cell Senescence through Lipid Metabolism Reprogramming. Sci. Transl. Med. 2025, 17, eadm7269. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Metzger, T.; Bailey-Bucktrout, S. Tumor Infiltrating T Cells Have Abnormal Lipid Metabolism That Can Be Modulated by PD-L1 Blockade. J. Immunol. 2016, 196, 144.21. [Google Scholar] [CrossRef]
- Shan, Y.; Xie, T.; Sun, Y.; Lu, Z.; Topatana, W.; Juengpanich, S.; Chen, T.; Han, Y.; Cao, J.; Hu, J.; et al. Lipid Metabolism in Tumor-Infiltrating Regulatory T Cells: Perspective to Precision Immunotherapy. Biomark. Res. 2024, 12, 41. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, W.; Wei, M.; Huang, Y.; Qin, J.; Liu, M.; Liu, N.; He, Y.; Chen, C.; Huang, Y.; Yin, H.; et al. Integrated Bioinformatics Analysis and Cellular Experimental Validation Identify Lipoprotein Lipase Gene as a Novel Biomarker for Tumorigenesis and Prognosis in Lung Adenocarcinoma. Biology 2025, 14, 566. https://doi.org/10.3390/biology14050566
He W, Wei M, Huang Y, Qin J, Liu M, Liu N, He Y, Chen C, Huang Y, Yin H, et al. Integrated Bioinformatics Analysis and Cellular Experimental Validation Identify Lipoprotein Lipase Gene as a Novel Biomarker for Tumorigenesis and Prognosis in Lung Adenocarcinoma. Biology. 2025; 14(5):566. https://doi.org/10.3390/biology14050566
Chicago/Turabian StyleHe, Wanwan, Meilian Wei, Yan Huang, Junsen Qin, Meng Liu, Na Liu, Yanli He, Chuanbing Chen, Yali Huang, Heng Yin, and et al. 2025. "Integrated Bioinformatics Analysis and Cellular Experimental Validation Identify Lipoprotein Lipase Gene as a Novel Biomarker for Tumorigenesis and Prognosis in Lung Adenocarcinoma" Biology 14, no. 5: 566. https://doi.org/10.3390/biology14050566
APA StyleHe, W., Wei, M., Huang, Y., Qin, J., Liu, M., Liu, N., He, Y., Chen, C., Huang, Y., Yin, H., & Zhang, R. (2025). Integrated Bioinformatics Analysis and Cellular Experimental Validation Identify Lipoprotein Lipase Gene as a Novel Biomarker for Tumorigenesis and Prognosis in Lung Adenocarcinoma. Biology, 14(5), 566. https://doi.org/10.3390/biology14050566