Simple Summary
Prions are novel infectious agents that consist only of protein. It is known that in the same host species, different strains of prion can occur that differ by the cells that are infected and killed. The mechanism responsible for these observations is only beginning to be understood. Two main ideas, which are not mutually exclusive, may provide a framework to understand strain targeting. The first possibility is that as prions spread throughout the CNS they reach strain-specific populations of cells that trigger cell death. The second mechanism is that prions exist in two distinct forms, a replicative form that switches to the production of a toxic form that results in the destruction of cells and the eventual onset of clinical signs of disease. Both neurons and glia may participate in both of these possibilities. The relative contributions of each mechanism have yet to be determined.
Abstract
Prion diseases are caused by the disease-specific self-templating infectious conformation of the host-encoded prion protein, PrPSc. Prion strains are operationally defined as a heritable phenotype of disease under controlled conditions. One of the hallmark phenotypes of prion strain diversity is tropism within and between tissues. A defining feature of prion strains is the regional distribution of PrPSc in the CNS. Additionally, in both natural and experimental prion disease, stark differences in the tropism of prions in secondary lymphoreticular system tissues occur. The mechanism underlying prion tropism is unknown; however, several possible hypotheses have been proposed. Clinical target areas are prion strain-specific populations of neurons within the CNS that are susceptible to neurodegeneration following the replication of prions past a toxic threshold. Alternatively, the switch from a replicative to toxic form of PrPSc may drive prion tropism. The normal form of the prion protein, PrPC, is required for prion formation. More recent evidence suggests that it can mediate prion and prion-like disease neurodegeneration. In vitro systems for prion formation have indicated that cellular cofactors contribute to prion formation. Since these cofactors can be strain specific, this has led to the hypothesis that the distribution of prion formation cofactors can influence prion tropism. Overall, there is evidence to support several mechanisms of prion strain tropism; however, a unified theory has yet to emerge.
1. Introduction
Prion diseases are inevitably fatal neurodegenerative diseases of animals that affect several species [1]. Prion diseases of animals include bovine spongiform encephalopathy (BSE or ‘mad cow’ disease), scrapie of sheep and goats, chronic wasting disease (CWD) that affects captive and free-ranging cervids, camel prion disease and transmissible mink encephalopathy (TME) of ranch-raised mink [2,3]. Prion diseases of humans include Gerstmann–Sträussler–Scheinker (GSS) disease, fatal familial insomnia (FFI), and Creutzfeldt–Jakob disease (CJD) [4]. Prion diseases are unique in biology in that they can occur in three etiologies. First, prions can exist as a sporadic disease with no known genetic or environmental basis. Second, prion disease can occur in familial forms that correspond to mutations in the host-encoded prion protein (PrPC) [5]. Third, under natural or experimental conditions, prions can be transmitted between individuals via numerous routes of infection including per os, inhalation and scarification of the dermis [6,7]. Prions can be zoonotic, as evidenced by the interspecies transmission of BSE to several species including Sapiens [8,9].
The transmissible agent of prion diseases is unique in biology and is composed of only protein [10,11]. The enrichment of prion infectivity led to the identification of a protein, PrPSc, that was the major constituent of preparations that contained high levels of prion infectivity [12]. This protein, PrPSc, is resistant to degradation by proteases and insoluble in detergents. Subsequent studies determined that PrPSc was encoded by the host protein, PrPC, eliminating a viral origin [13]. These observations led to the prion hypothesis that PrPSc is the infectious agent of prion diseases [14]. Subsequent studies reconstituting infectious prions from minimal non-infectious components confirmed the prion hypothesis [15,16,17,18,19,20]. Prion formation occurs when PrPSc encounters PrPC and through an unknown mechanism can direct a global rearrangement of alpha helical structures present in PrPC to parallel in-register intermolecular beta sheet structures in PrPSc [21,22,23,24]. The fragmentation of the growing PrPSc fibril provides new free ends of PrPSc to engage in subsequent rounds of binding and converting PrPC to PrPSc [25]. This process can be recapitulated in vitro using protein misfolding cyclic amplification (PMCA), leading to the exponential propagation of PrPSc and prion infectivity [26,27].
Prion diseases exhibit strain diversity. Prion strains are operationally defined by heritable differences in the phenotype of disease under carefully controlled transmission conditions [28]. The phenotype of disease can include incubation period, clinical signs, distribution and intensity of neuropathology and tropism of PrPSc. Tropism can include strain-specific differences in tissues infected, and within a given tissue, strain-specific differences in region or cell type where prions are detected [29,30,31,32]. While strains are best characterized in rodents, prion strain diversity has been observed in natural host species including Sapiens [33,34]. While initially difficult for the prion hypothesis to provide a mechanism to explain prion strain diversity [35,36], a wealth of indirect evidence supports the hypothesis that strain-specific differences in the conformation of PrPSc encode prion strain diversity [37,38,39,40,41,42]. Recent evidence using cryo electron microscopy has revealed that PrPSc from two distinct murine prion strains, which share the same primary amino acid sequence of PrP, have distinct structures of PrPSc [43,44]. This direct evidence provides the most compelling evidence to date that the conformation of PrPSc encodes for prion strain diversity. The relationship between the strain-specific conformations of PrPSc and the phenotype of disease is, however, poorly understood.
The neuropathology of prion infection includes spongiform degeneration, reactive gliosis, and neuronal death in the absence of a cellular inflammatory infiltrate. Spongiform degeneration is caused by the development and merging of interneuronal vacuoles that is observed in nearly all prion diseases and may be a consequence of the unfolded protein response dysregulation phosphoinositide kinase PIKfyve [45]. However, in some instances, spongiform degeneration is not a prominent feature of prion infection [46,47]. Prion diseases are characterized by the activation of both astroglia and microglia [48,49,50]. Astrocytes can support prion formation, and the role of astrocytes in neurodegeneration is becoming increasingly clear [51]. The restriction of PrPC expression to astrocytes can result in prion formation in these cells; however, they do not undergo activation and neurodegeneration, and the development of clinical signs of prion infection is not observed [52]. These recent findings are consistent with previous work showing that brain grafts that express high levels of PrPC in a PrP−/− host undergo prion formation and the histological changes associated with prion infection, while PrPSc from the graft that entered the PrP−/− brain failed to cause neurodegeneration or glial activation [53]. Importantly, the pattern of reactive astrocytes is strain specific in a large cohort of murine-adapted prion strains [54]. The incubation period of prion infection corresponds to the degree of astrocyte activation and the fact that inhibition of the unfolded protein response in astrocytes can prevent neuronal loss [55,56]. Overall, these observations suggest that PrPC plays a critical role in neurodegeneration and that the interplay between neurons and glia is an important determinant in the outcome of disease.
2. Prion Strain Targeting
Neuropathological changes and PrPSc deposition patterns can vary based on prion strain, in part due to cellular targeting to a preferred neuronal subset. Different strains exhibit different cellular tropisms, evidenced by a multitude of studies. While many human prion strains cause a preferential and marked loss of PV+ neurons, fatal familial insomnia spares this subset within both the hippocampus and temporal cortex while also helping to mediate the loss within the frontal cortex [57]. PrPSc deposition patterns have been examined to differentiate between prion strains. Experiments on cultured organotypic cerebellar slices displayed strain-specific PrPSc deposition patterns [58]. In addition, patterns of PrPSc deposition from sheep infected with various prion strains have been used to reliably distinguish between various prion strains [59]. Genotype may also play a role in the degree of PrPSc deposition seen within these strain-specific patterns. Polymorphisms, associated with FFI, that confer longer disease duration tend to extend PrPSc deposition to areas of the brain outside of those affected in those with a shorter duration of disease [60]. Taken together, these experiments highlight the preferential cellular targeting of prion strains to certain areas within the brain, evidenced by strain-specific PrPSc deposition profiles.
Prion strain-specific differences in tissue tropism are observed. Prion conversion and deposition occurs in secondary lymphoreticular system tissues but has also been observed in other locations (e.g., skeletal muscle, fat) [61,62,63,64,65,66,67,68,69,70]. The extraneural distribution of PrPSc is not observed in all prion diseases and can differ based on prion strain. Sheep naturally infected with classic or Nor98 (atypical) scrapie have differences in the distribution of PrPSc in the spleen and lymph nodes [71,72]. A similar phenomenon is observed in hamsters infected with either the hyper (HY) or drowsy (DY) strains of hamster-adapted transmissible mink encephalopathy (TME) [37,73]. A widespread distribution of PrPSc is observed in HY TME-infected animals with the robust accumulation of PrPSc in the spleen, lymph nodes and skeletal muscle [62,74]. In contrast, prion infectivity and PrPSc is not detected in extraneural tissues in DY TME-infected animals [75]. This failure to establish infection is not due to a failure of prion transport as inoculum DY PrPSc can cross the epithelium and reach draining lymphatics following extranasal infection and the spleen following intraperitoneal infection [29]. Additionally, the spleen contains all the necessary components for DY TME formation as the spleen can support DY PrPSc formation in vitro during protein misfolding cyclic amplification [29]. Overall, strain-specific differences in prion tropism occur; however, the mechanism(s) are poorly described. The remainder of the review will explore potential mechanisms to explain prion tropism.
3. Clinical Target Areas
Clinical target areas (CTAs) are hypothesized to be areas of the brain that, when reached by PrPSc, lead to the development of clinical signs and ultimately the death of the host [76]. The time between the onset of prion replication within the brain and the development of clinical signs is known as the replication phase. The targeting of prions to CTAs determines the length of the replication phase within the brain; CTAs constitute a minority of the neurons within the brain and are strain specific as prion strains can differ in the clinical presentation of disease (Figure 1A). Altering the route of inoculation altered the length of the replication phase, suggesting that the pathway to a proposed CTA may vary in complexity [76,77].
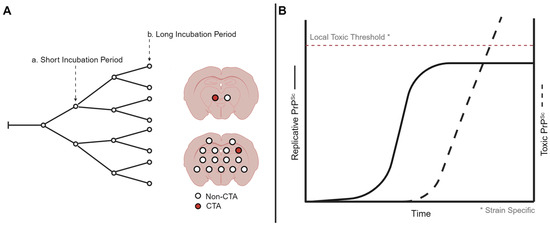
Figure 1.
Models of prion strain-specific targeting and neurodegeneration. (A) Strain-specific distribution of prion neuropathology may be due to differences in prion spread when population of neurons are reached and destroyed. Here, a short incubation period strain reaches the clinical target areas earlier compared to a long incubation period strain, resulting in regional differences in neuropathology. (B) At the cellular level, prion formation may start with a replicative form that plateaus, resulting in the production of a toxic prion species. The level needed for the toxic species to result in cell death may be strain and cell type specific. Created with BioRender.com.
Prions spread along defined neuroanatomical pathways following various inoculation routes independent of prion strain. Neuroinvasion, following peripheral routes of inoculation (e.g., intraperitoneal, per os), establish infection in follicular dendritic cells in the spleen and visceral lymph nodes and continued along autonomic nerves before reaching the midthoracic spinal cord [66,67,76,78,79,80,81,82,83,84,85,86]. Intralingual muscle inoculation provided the first evidence for retrograde axonal transport independent of prion strain, with spread retrograde axonal transport via the hypoglossal nerve to the hypoglossal nucleus and subsequent transsynaptic transport to the nucleus of the solitary tract [87]. Building upon these findings, the direct inoculation of the sciatic nerve with three different hamster-adapted prion strains indicated they all used retrograde axonal transport along the same four descending motor tracts: reticulospinal, vestibulospinal, rubrospinal and corticospinal tracts [88,89]. These observations suggest that prion strain targeting CTAs is not due to differences in prion transport in the CNS. The strain-specific differences in neuropathology and PrPSc deposition may be due to differences in the progression of the spread of PrPSc due to strain-specific differences when the CTAs are reached. Strain-independent retrograde axonal transport is also observed with other non-PrPSc prion-like diseases such as Parkinson’s disease and amyotrophic lateral sclerosis. Murine wild-type α synuclein (α Syn) fibrils and human E46K α Syn fibrils, associated with Parkinson’s disease, both underwent retrograde axonal transport along vestibulospinal and rubrospinal descending motor tracts following sciatic nerve inoculation [90]. The sciatic nerve inoculation of mutant SOD1 brain homogenates into transgenic mice expressing G58-SOD1:YFP resulted in the retrograde axonal transport of pathology along three descending motor tracts: the reticulospinal, vestibulospinal and rubrospinal tracts [91].
Differences in the duration of the asymptomatic replication phase of prions in the CNS are influenced by the route of infection. The prion replication phase in the CNS following intraperitoneal (i.p.) inoculation is shorter compared to the replication phase following intracerebral (i.c.) inoculation [92]. The shorter replication phase observed following i.p. inoculation suggests a more direct pathway to CTAs from the midthoracic cord compared to the pathways following i.c. inoculation. Consistent with this observation, the intraspinal inoculation of mice with 139A prions showed a shorter replication phase compared to i.c. inoculation, indicating a potential difference in the complexity of the neuronal pathway to the CTA available with a given inoculation site [77]. While the targeting and formation of PrPSc that results in neurodegeneration within the CTA determines the onset of clinical signs, prion spread to non-CTA regions is hypothesized to contribute to prion infectivity titers and the distribution of spongiform degeneration [76]. As spongiform degeneration precedes the onsets of clinical signs of disease, this means that either spongiform degeneration per se does not lead to clinical signs, or that spongiform degeneration in combination with another neurodegenerative event precipitates the onset of clinical signs of disease. Since it is hypothesized that the clinical signs associated with prion disease are a result of the replication and pathology only found within the CTAs, the route of infection thus may alter the pathways available to CTAs after neuroinvasion and therefore the duration of the replicative phase of disease.
4. Replicative vs. Toxic forms of PrPSc
Two distinct forms of PrPSc, a replicative form and a toxic form, are hypothesized to determine the onset of clinical signs of prion disease. The uncoupled replication and toxicity hypothesis describes two distinct phases of prion pathogenesis [93]. The first phase is characterized by the exponential accumulation of PrPSc that eventually plateaus. The plateau in PrPSc abundance is reached prior to the onset of clinical signs of prion infection. The second phase confers toxicity that is mediated by a toxic form of PrP, termed PrPL. The toxic effects associated with this form of PrP are thought to occur only after surpassing a toxic threshold (Figure 1B). Thus, the initial rapid propagation of replicative PrPSc is not responsible for toxicity, and toxicity occurs after the formation of PrPL to necessary threshold saturations.
The formation of PrPL is hypothesized to be the result of off-target or intermediate forms of PrPSc. Compared to PrPSc, PrPL may have reduced self-templating activity, therefore requiring an increased time to accumulate to toxic levels. This hypothesis predicts that the rate of PrPL formation is influenced by the rate of PrPSc conversion and maturation [94]. With the increased conversion of PrPSc, more intermediates are generated, including PrPL. Fluctuations in isoforms occur in two phases, with synaptic alterations and neuropathological changes occurring after an increase in PK-sensitive isoforms generated in phase 2 [95]. The hypothesis of defined toxic thresholds is bolstered by the findings that similar levels of PrPSc must be reached for clinical disease to progress in phase 2, regardless of PrP expression [95]. PrPL may have a different conformation from PrPSc as both enriched PrPSc preparations and the sarkosyl treatment of RML-infected brain homogenates eliminated toxicity while maintaining prion infectivity. Collectively, these results are consistent with a toxic and replicative form of PrPSc.
Observations in animals during the subclinical phase of disease may provide support for the uncoupled replication and toxic forms of PrPSc hypothesis. Animals with subclinical prion disease replicate PrPSc and can live a normal life span in the absence of clinical signs, including ataxia [96]. Subclinical prion disease occurs in mice inoculated with the hamster strain Sc237 as these mice were able to replicate PrPSc to high levels but failed to exhibit clinical signs of disease [97]. In addition, inoculation with low-dose inoculums of mouse strains into mice overexpressing PrPC induced a subclinical disease state. Interestingly, in this study comparison of terminally ill high-titer inoculated and subclinical low-titer inoculated mice displayed similar levels of PrPSc within the brain stem [98]. If the rate of formation of PrPL is decreased in subclinical disease due to species barrier effect or slowed replication, a small amount of PrPL formed may undergo sufficient maturation into PrPSc, thus preventing clinical disease by eliminating the source of toxicity. In both instances, the propagation of PrPSc alone was not able to induce clinical symptoms, suggesting that another toxic mechanism is required to elicit irreversible clinical signs and terminal disease.
Factors, in addition to PrPL, may be involved in the development of clinical prion disease. A prediction of the replicative and toxic PrP hypothesis is that the initial sites of neuroinvasion would be the first areas of the CNS to reach a plateau in infectivity, followed by the accumulation of PrPL resulting in the onset of clinical signs of disease. This pattern, however, is not observed following extraneural routes of inoculation, where the initial site of neuroinvasion does not correspond with the development of clinical signs of disease [99]. For example, the inoculation of the sciatic nerve with HY TME prions results in the detection of PrPSc in ventral motor neurons (VMNs) in the lumbar spinal cord ipsilateral to the side of prion inoculation within 2 weeks postinfection [87,100]. From VMNs, PrPSc spreads along known neuroanatomical pathways until the onset of clinical signs 8 weeks later. The replicative vs. toxic PrP hypothesis would predict that hind limb motor deficits ipsilateral to the side of prion inoculation would be the first clinical sign of disease as VMNs are the first cell type infected and would first produce PrPL. What is observed, however, is clinical signs of hyperexcitability and cerebellar ataxia that is indistinguishable from other routes of infection [87,100]. A similar relationship between the initial deposition of PrPSc in VMNs and clinical disease is observed following the sciatic nerve inoculation of DY TME with animals developing clinical signs of progressive lethargy and not hind limb motor deficits. These observations constrain the properties of PrPL. First, as hind limb motor deficits are not observed, either PrPL is not produced in VMNs or PrPL is not toxic for VMNs. Second, as HY- and DY TME-infected animals have distinct clinical signs that occur independent of the route of infection, strain-specific forms of PrPL that affect different populations of neurons are required [7,62,75,101]. Additionally, synaptotoxic forms of PrPSc, presumably PrPL, are observed early during the pathogenesis of disease, prior to the plateauing of infectivity titers [102]. Overall, these observations suggest that host factors, in addition to PrPL reaching local toxic thresholds, contribute to prion disease development.
5. Role of PrPC in Neurotoxicity
PrPC is a cell surface protein that is anchored to the cell membrane by a glycosylphosphatidylinositol (GPI) anchor [13,103,104]. During prion formation, conversion occurs as PrPSc uses available PrPC as a substrate [22]. PrPSc serves as a template to misfold PrPC into the pathogenic and misfolded form, PrPSc. Two forms of PrPC exist, C1 and C2, resulting from the proteolytic cleavage of PrPC. C1 is the soluble, C terminal fragment of PrPC which has undergone α cleavage between amino acid residues 111/112, which eliminates the amyloidogenic region of residues 106–126 [105,106]. The C1 fragment is present in the brains of both healthy and prion-infected animals. While mice expressing C1 alone inoculated with RML failed to develop prion disease, mice co-expressing C1 and WT PrPC resulted in a prolonged incubation period and the slower accumulation of PrPSc [107]. These experiments provided evidence for a potential protective role of the C1 fragment, as it cannot be used as a substrate for PrPSc formation. In contrast, C2 is the insoluble form of PrP that is associated with β cleavage. C2 retains the PrP residues associated with the amyloidogenic region and is found only in the brains of prion-infected animals [105,106]. Therefore, while α cleavage may play a protective role in prion disease, β cleavage is associated with either PrPSc or PrPC and potentially contributes to disease.
The expression of PrPC is required for prion propagation. This is evidenced by both Prnp0/0 and Prnp0/+ mice inoculated with mouse adapted scrapie. While PrP−/− mice fail to develop prion disease and pathology, PrP−/+ mice have an increased incubation period when compared to WT mice [108]. Importantly, in PrP−/+ mice, an extended period of a prion infectivity plateau is observed, suggesting that PrPC plays a role in the onset of neurodegeneration and clinical signs of disease [108]. Conversely, the overexpression of PrPC shortens the incubation period of prion disease; however, it is unclear if this is due to an increased tempo of prion conversion or increased susceptibility to neurodegeneration [109,110,111,112].
Alterations in PrPC levels occur during prion infection. A downregulation in PrPC occurs prior to a plateau in infectivity in WT mice inoculated with RML prions [113,114]. This decrease in available substrate for conversion is hypothesized to result in a decrease in the rate of PrPSc formation. The plateau effect observed in PrPSc abundance was also observed in PMCA experiments, where it was shown that a higher quantity of PrPC and lower quantity of PrPSc leads to an increase in the replication rate of PrPSc, while the inverse is true when a higher quantity of PrPSc and lower quantity of PrPC is used [114]. Overall, alterations in the abundance of PrPC associated with prion disease may drive the plateau of infectivity seen in this proposed first phase of prion pathogenesis.
The expression of PrPC is required for the development of prion neuropathology. To investigate the role of PrPC expression and neurotoxicity, neural explants overexpressing PrPC were engrafted into the brains of PrP−/− mice. The intracerebral inoculation of these mice with RML prions did not result in the development of clinical signs of prion infection. It did, however, result in the development of prion formation and associated neuropathology in the graft expressing PrPC and not in the PrP−/− brain regions. Importantly, PrPSc produced in the engrafted brain migrated into the PrP−/− regions but did not result in observable neuropathological changes [53]. As neuropathology is observed in the engrafted PrPC-expressing brain regions, it should contain both the replicative vs. toxic forms of PrPSc; therefore, both forms should migrate into the PrP−/− brain region. However, neuropathological changes are absent in the PrP−/− brain region, leading to the inference that if PrPL is escaping from the explant, it is devoid of its toxic properties in the absence of PrPC. This experiment highlights the necessity of PrPC in neurodegeneration and its potential role as a mediator of neurotoxicity.
Regions within PrPC may play a role in mediating prion neurodegeneration. The central region (CR) of PrP has been identified as a crucial region in the maintenance of the neuroprotective function of PrPC. In the absence of CR, residues 105–125, neurodegenerative phenotypes, are present in transgenic mice [115]. Deletions of the CR induce spontaneous ionic currents [116,117]. Experimentation with anti-PrP antibodies directed specifically towards the globular domain of PrPC found the majority to cause neurotoxicity in COCS. One such antibody, POM1, causes neuronal loss in vitro and in vivo [117]. ICSM18, a globular domain antibody that binds to an epitope of PrPC that overlaps the epitope of POM1, was also found to induce neurotoxicity in vivo [118]. The flexible N terminal region is hypothesized to play a role in the mediation of toxic effects associated with prion disease [117]. In experiments involving the comparison of hippocampal neuronal culture systems expressing PrPC with deletions in the N terminal region (Δ23–31) and WT PrPC, the toxic effects associated with PrPSc application were eliminated in the absence of this N terminal region [119]. Antibodies to the N terminal region, POM2, can neutralize the toxic effects associated with both RML prion disease as well as globular domain ligands in cerebellar organotypic culture slices [120]. The role of the N terminal region in toxicity is highlighted by cells expressing PrP(N)-EGFP-GPI, where spontaneous ionic currents are induced [121]. These experiments illustrate the interplay between key regions of PrPC that function in either neuroprotective or neurodegenerative capacities and the influence of PrPC and PrPSc interactions on these functions.
PrPC can act as a toxic mediator in prion-like diseases. PrPC functions as one of the receptors for oligomeric amyloid β, Aβo, which is associated with Alzheimer’s disease [122]. The binding of Aβo to PrPC initiates a toxic signaling pathway that culminates in neurodegeneration. The inhibition of long-term potentiation is blocked in both Prnp0/0 hippocampal slices treated with synthetic Aβo, as well as WT hippocampal slices pretreated with anti-PrP antibodies [122]. In vivo studies of Alzheimer’s disease transgenic mice revealed that while Aβ can still accumulate in mice lacking PrPC, axonal degeneration as well as impairments in memory and spatial learning are not observed [123]. The activation of Fyn, a Src family kinase, by the binding of Aβ to PrPC initiates a signaling pathway resulting in the synaptic dysfunction associated with NMDA receptors. With a specificity for Aβo, the binding of Aβo to PrPC activates Fyn, which in turn leads to an initial phosphorylation of a subunit of NMDA receptors, NR2B, which increases the amount of NMDA on the cell surface, ultimately leading to excitotoxicity [124]. This is not observed in neurons lacking the expression of PrPC, Fyn or in neurons treated with anti-PrP antibodies prior to Aβo exposure. Thus, this evidence collectively provides evidence for the necessity of PrPC expression in mediating the toxic effects of Aβo binding.
6. Cellular Cofactors and Prion Formation
The formation of PrPSc is aided by cellular co-factors. Prion formation is recapitulated by PMCA, leading to the production of infectious prions that maintain the strain properties of the initial input prion strain [27,125,126,127]. This system has been used to investigate the requirements for prion formation and has found that phosphatidylethanolamine (PE) and RNA facilitate prion formation in vitro. PMCA conversion of three prion strains that contained PE as the cofactor resulted in the three strains converging into a single strain that was indistinguishable from them and that also differed from the parental strain [128]. Since the information for prion strain diversity is encoded in the conformation of PrPSc, the cellular co-factors have been hypothesized to play a role in tropism. This system had identified strain-specific requirements in cellular co-factors for efficient in vitro formation. For example, differences in the effect of RNA in the formation of PrPSc correspond with prion strain, suggesting that cellular cofactors can influence the rate of prion formation [129,130,131]. As the rate of prion formation must exceed prion clearance, the distribution of cellular co-factors may influence prion tropism [29]. The role of strain-specific cofactors in PrPSc tropism in neurons and glia is unclear, as the currently identified co-factors may have a ubiquitous distribution [128,132,133,134,135].
7. Conclusions
Prion strain diversity is operationally defined by differences in tropism within and between tissues, with the regional distribution of PrPSc in the CNS being especially important (Figure 1). While the precise mechanism of prion tropism is unknown, the axonal transport of prions to clinical target areas may direct prions to populations of neurons and glia that contribute to disease pathology. Once there, the transition of a replicative form of PrPSc to that of a lethal form of PrPSc may trigger pathology. Additionally, strain-specific cellular cofactors may add further nuance to prion strain tropism.
Author Contributions
Conceptualization, S.M.S. and J.C.B.; writing—original draft preparation, S.M.S. and J.C.B.; writing—review and editing, S.M.S. and J.C.B.; funding acquisition, J.C.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institutes of Health National Institute of Neurological Disorders and Stroke R01NS103763, R01NS13305; the National Institute of Allergy and Infectious Disease 2P01 AI077774; and the Creutzfeldt Jacob disease foundation.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Prusiner, S.B. (Ed.) Prion Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017; p. 456. [Google Scholar]
- Wells, G.A.H.; Wilesmith, J.W. Bovine spongiform encephalopathy and related diseases. In Prion Biology and Diseases, 2nd ed.; Prusiner, S.B., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2004; pp. 595–628. [Google Scholar]
- Prusiner, S.B.; Williams, E.; Laplanche, J.-L.; Shinagawa, M. Scrapie, chronic wasting disease, and transmissible mink encephalopathy. In Prion Biology and Diseases, 2nd ed.; Prusiner, S.B., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2004; pp. 545–594. [Google Scholar]
- Will, R.G.; Ironside, J.W. Sporadic and infectious human prion diseases. In Prion Diseases; Prusiner, S.B., Ed.; Cold Spring Harbor Perspectives in Medicine; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017; pp. 73–93. [Google Scholar]
- Hsiao, K.; Baker, H.F.; Crow, T.J.; Poulter, M.; Owen, F.; Terwilliger, J.D.; Westaway, D.; Ott, J.; Prusiner, S.B. Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature 1989, 338, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Stamp, J.T.; Brotherston, J.G.; Zlotnik, I.; Mackay, J.M.K.; Smith, W. Further studies on scrapie. J. Comp. Pathol. 1959, 69, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, A.E.; Bartz, J.C. The nasal cavity is a route for prion infection in hamsters. J. Virol. 2007, 81, 4482–4491. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.E.; Will, R.G.; Ironside, J.W.; McConnell, I.; Drummond, D.; Suttie, A.; McCardle, L.; Chree, A.; Hope, J.; Birkett, C.; et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent [see comments]. Nature 1997, 389, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Sidle, K.C.L.; Meads, J.; Ironside, J.; Hill, A.F. Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature 1996, 383, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J. Self-replication and scrapie. Nature 1967, 215, 1043–1044. [Google Scholar] [CrossRef]
- Bolton, D.; McKinley, M.; Prusiner, S. Identification of a protein that purifies with the scrapie prion. Science 1982, 218, 1309–1311. [Google Scholar] [CrossRef]
- Oesch, B.; Westaway, D.; Wälchli, M.; McKinley, M.P.; Kent, S.B.H.; Aebersold, R.; Barry, R.A.; Tempst, P.; Teplow, D.B.; Hood, L.E.; et al. A cellular gene encodes scrapie PrP 27–30 protein. Cell 1985, 40, 735–746. [Google Scholar] [CrossRef]
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef]
- Deleault, N.R.; Harris, B.T.; Rees, J.R.; Supattapone, S. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. USA 2007, 104, 9741–9746. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, X.; Yuan, C.G.; Ma, J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010, 327, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.W.; Giles, K.; Legname, G.; Wille, H.; Baskakov, I.V.; DeArmond, S.J.; Prusiner, S.B. Design and construction of diverse mammalian prion strains. Proc. Natl. Acad. Sci. USA 2009, 106, 20417–20422. [Google Scholar] [CrossRef] [PubMed]
- Erana, H.; Diaz-Dominguez, C.M.; Charco, J.M.; Vidal, E.; Gonzalez-Miranda, E.; Perez-Castro, M.A.; Pineiro, P.; Lopez-Moreno, R.; Sampedro-Torres-Quevedo, C.; Fernandez-Veiga, L.; et al. Understanding the key features of the spontaneous formation of bona fide prions through a novel methodology that enables their swift and consistent generation. Acta Neuropathol. Commun. 2023, 11, 145. [Google Scholar] [CrossRef]
- Makarava, N.; Kovacs, G.G.; Bocharova, O.; Savtchenko, R.; Alexeeva, I.; Budka, H.; Rohwer, R.G.; Baskakov, I.V. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010, 119, 177–187. [Google Scholar] [CrossRef]
- Block, A.J.; Shikiya, R.A.; Eckland, T.E.; Kincaid, A.E.; Walters, R.W.; Ma, J.; Bartz, J.C. Efficient interspecies transmission of synthetic prions. PLoS Pathog. 2021, 17, e1009765. [Google Scholar] [CrossRef]
- Kraus, A.; Hoyt, F.; Schwartz, C.L.; Hansen, B.; Artikis, E.; Hughson, A.G.; Raymond, G.J.; Race, B.; Baron, G.S.; Caughey, B. High-resolution structure and strain comparison of infectious mammalian prions. Mol. Cell 2021, 81, 4540–4551.e6. [Google Scholar] [CrossRef]
- Caughey, B.; Raymond, G.J. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J. Biol. Chem. 1991, 266, 18217–18223. [Google Scholar] [CrossRef]
- Riek, R.; Hornemann, S.; Wider, G.; Glockshuber, R.; Wuthrich, K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231). FEBS Lett. 1997, 413, 282–288. [Google Scholar] [CrossRef]
- Riek, R.; Hornemann, S.; Wider, G.; Billeter, M.; Glockshuber, R.; Wuthrich, K. NMR structure of the mouse prion protein domain PrP(121-321). Nature 1996, 382, 180–182. [Google Scholar] [CrossRef]
- Sun, Y.; Jack, K.; Ercolani, T.; Sangar, D.; Hosszu, L.; Collinge, J.; Bieschke, J. Direct Observation of Competing Prion Protein Fibril Populations with Distinct Structures and Kinetics. ACS Nano 2023, 17, 6575–6588. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Saa, P.; Hetz, C.; Soto, C. In vitro generation of infectious scrapie prions. Cell 2005, 121, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001, 411, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Bartz, J.C. Prion Strain Diversity. Cold Spring Harb. Perspect. Med. 2016, 6, a024349. [Google Scholar] [CrossRef] [PubMed]
- Shikiya, R.A.; Langenfeld, K.A.; Eckland, T.E.; Trinh, J.; Holec, S.A.M.; Mathiason, C.K.; Kincaid, A.E.; Bartz, J.C. PrPSc formation and clearance as determinants of prion tropism. PLoS Pathog. 2017, 13, e1006298. [Google Scholar] [CrossRef] [PubMed]
- Taraboulos, A.; Jendroska, K.; Serban, D.; Yang, S.; DeArmond, S.; Prusiner, S. Regional mapping of prion proteins in brain. Proc. Natl. Acad. Sci. USA 1992, 89, 7620–7624. [Google Scholar] [CrossRef]
- Sisó, S.; Jeffrey, M.; Martin, S.; Chianini, F.; Dagleish, M.P.; Gonzalez, L. Characterization of strains of ovine transmissible spongiform encephalopathy with a short PrPd profiling method. J. Comp. Pathol. 2010, 142, 300–310. [Google Scholar] [CrossRef]
- Jeffrey, M.; Mcgovern, G.; Goodsir, C.M.; González, L. Strain-Associated Variations in Abnormal PrP Trafficking of Sheep Scrapie. Brain Pathol. 2009, 19, 1–11. [Google Scholar] [CrossRef]
- Parchi, P.; Capellari, S.; Chen, S.G.; Petersen, R.B.; Gambetti, P.; Kopp, N.; Brown, P.; Kitamoto, T.; Tateishi, J.; Giese, A.; et al. Typing prion isoforms. Nature 1997, 386, 232–234. [Google Scholar] [CrossRef]
- Parchi, P.; Zou, W.; Wang, W.; Brown, P.; Capellari, S.; Ghetti, B.; Kopp, N.; Schulz-Schaeffer, W.J.; Kretzschmar, H.A.; Head, M.W.; et al. Genetic influence on the structural variations of the abnormal prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 10168–10172. [Google Scholar] [CrossRef]
- Bruce, M.E.; Dickinson, A.G. Biological evidence that the scrapie agent has an independent genome. J. Gen. Virol. 1987, 68, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, A.; Outram, G.; Taylor, D.M.; Foster, J.D. Further evidence that scrapie agent has an independent genome. In Unconventional Virus Diseases of the Central Nervous System; Court, L., Dormont, D., Brown, P., Kingsbury, D., Eds.; Commissariat a l’Energie Atomique: Paris, France, 1986; pp. 446–460. [Google Scholar]
- Bessen, R.; Marsh, R. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J. Virol. 1992, 66, 2096–2101. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have PrP(Sc) molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Telling, G.C.; Parchi, P.; DeArmond, S.J.; Cortelli, P.; Montagna, P.; Gabizon, R.; Mastrianni, J.; Lugaresi, E.; Gambetti, P.; Prusiner, S.B. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996, 274, 2079–2082. [Google Scholar] [CrossRef] [PubMed]
- Ayers, J.I.; Schutt, C.R.; Shikiya, R.A.; Aguzzi, A.; Kincaid, A.E.; Bartz, J.C. The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog. 2011, 7, e1001317. [Google Scholar] [CrossRef] [PubMed]
- Tixador, P.; Herzog, L.; Reine, F.; Jaumain, E.; Chapuis, J.; Le Dur, A.; Laude, H.; Beringue, V. The physical relationship between infectivity and prion protein aggregates is strain-dependent. PLoS Pathog. 2010, 6, e1000859. [Google Scholar] [CrossRef] [PubMed]
- Cortez, L.M.; Nemani, S.K.; Duque Velasquez, C.; Sriraman, A.; Wang, Y.; Wille, H.; McKenzie, D.; Sim, V.L. Asymmetric-flow field-flow fractionation of prions reveals a strain-specific continuum of quaternary structures with protease resistance developing at a hydrodynamic radius of 15 nm. PLoS Pathog. 2021, 17, e1009703. [Google Scholar] [CrossRef]
- Manka, S.W.; Wenborn, A.; Betts, J.; Joiner, S.; Saibil, H.R.; Collinge, J.; Wadsworth, J.D.F. A structural basis for prion strain diversity. Nat. Chem. Biol. 2023, 19, 607–613. [Google Scholar] [CrossRef]
- Manka, S.W.; Wenborn, A.; Collinge, J.; Wadsworth, J.D.F. Prion strains viewed through the lens of cryo-EM. Cell Tissue Res. 2022, 392, 167–178. [Google Scholar] [CrossRef]
- Lakkaraju, A.K.K.; Frontzek, K.; Lemes, E.; Herrmann, U.; Losa, M.; Marpakwar, R.; Aguzzi, A. Loss of PIKfyve drives the spongiform degeneration in prion diseases. EMBO Mol. Med. 2021, 13, e14714. [Google Scholar] [CrossRef]
- Begara-McGorum, I.; Gonzalez, L.; Simmons, M.; Hunter, N.; Houston, F.; Jeffrey, M. Vacuolar lesion profile in sheep scrapie: Factors influencing its variation and relationship to disease-specific PrP accumulation. J. Comp. Pathol. 2002, 127, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Marsh, R.; Sipe, J.; Morse, S.; Hanson, R. Transmissible mink encephalopathy. Reduced spongiform degeneration in aged mink of the Chediak-Higashi genotype. Lab. Investig. 1976, 34, 381–386. [Google Scholar] [PubMed]
- Brown, D.; Schmidt, B.; Kretzschmar, H. Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature 1996, 380, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Marsh, R. Studies on the Reactive Astrocyte in Mink Encepalopathy. Master’s Thesis, 1966; pp. 1–60. [Google Scholar]
- Barcikowska, M.; Liberski, P.P.; Boellaard, J.W.; Brown, P.; Gajdusek, D.C.; Budka, H. Microglia is a component of the prion protein amyloid plaque in the Gerstmann-Straussler-Scheinker syndrome. Acta Neuropathol. 1993, 85, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; Goodsir, C.M.; Race, R.E.; Chesebro, B. Scrapie-specific neuronal lesions are independent of neuronal PrP expression. Ann. Neurol. 2004, 55, 781–792. [Google Scholar] [CrossRef]
- Lakkaraju, A.K.K.; Sorce, S.; Senatore, A.; Nuvolone, M.; Guo, J.; Schwarz, P.; Moos, R.; Pelczar, P.; Aguzzi, A. Glial activation in prion diseases is selectively triggered by neuronal PrP(Sc). Brain Pathol. 2022, 32, e13056. [Google Scholar] [CrossRef]
- Brandner, S.; Isenmann, S.; Raeber, A.; Fischer, M.; Sailer, A.; Kobayashi, Y.; Marino, S.; Weissmann, C.; Aguzzi, A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996, 379, 339–343. [Google Scholar] [CrossRef]
- Bradford, B.M.; Wijaya, C.A.W.; Mabbott, N.A. Discrimination of Prion Strain Targeting in the Central Nervous System via Reactive Astrocyte Heterogeneity in CD44 Expression. Front. Cell Neurosci. 2019, 13, 411. [Google Scholar] [CrossRef]
- Makarava, N.; Mychko, O.; Chang, J.C.; Molesworth, K.; Baskakov, I.V. The degree of astrocyte activation is predictive of the incubation time to prion disease. Acta Neuropathol. Commun. 2021, 9, 87. [Google Scholar] [CrossRef]
- Smith, H.L.; Freeman, O.J.; Butcher, A.J.; Holmqvist, S.; Humoud, I.; Schatzl, T.; Hughes, D.T.; Verity, N.C.; Swinden, D.P.; Hayes, J.; et al. Astrocyte Unfolded Protein Response Induces a Specific Reactivity State that Causes Non-Cell-Autonomous Neuronal Degeneration. Neuron 2020, 105, 855–866.e5. [Google Scholar] [CrossRef]
- Guentchev, M.; Wanschitz, J.; Voigtländer, T.; Flicker, H.; Budka, H. Selective neuronal vulnerability in human prion diseases. Fatal familial insomnia differs from other types of prion diseases. Am. J. Pathol. 1999, 155, 1453–1457. [Google Scholar] [CrossRef] [PubMed]
- Falsig, J.; Sonati, T.; Herrmann, U.S.; Saban, D.; Li, B.; Arroyo, K.; Ballmer, B.; Liberski, P.P.; Aguzzi, A. Prion pathogenesis is faithfully reproduced in cerebellar organotypic slice cultures. PLoS Pathog. 2012, 8, e1002985. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.; Martin, S.; Jeffrey, M. Distinct profiles of PrP(d) immunoreactivity in the brain of scrapie- and BSE-infected sheep: Implications for differential cell targeting and PrP processing. J. Gen. Virol. 2003, 84, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Parchi, P.; Castellani, R.; Cortelli, P.; Montagna, P.; Chen, S.G.; Petersen, R.B.; Manetto, V.; Vnencak-Jones, C.L.; McLean, M.J.; Sheller, J.R.; et al. Regional distribution of protease-resistant prion protein in fatal familial insomnia. Ann. Neurol. 1995, 38, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Race, B.; Meade-White, K.; Oldstone, M.; Race, R.; Chesebro, B. Detection of prion infectivity in fat tissues of scrapie-infected mice. PLoS Pathog. 2008, 4, e1000232. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, E.R.; Bartz, J.C.; Kincaid, A.E.; Bessen, R.A. Prion infection of skeletal muscle cells and papillae in the tongue. J. Virol. 2004, 78, 6792–6798. [Google Scholar] [CrossRef][Green Version]
- Angers, R.C.; Browning, S.R.; Seward, T.S.; Sigurdson, C.J.; Miller, M.W.; Hoover, E.A.; Telling, G.C. Prions in skeletal muscles of deer with chronic wasting disease. Science 2006, 311, 1117. [Google Scholar] [CrossRef]
- Bosque, P.J.; Ryou, C.; Telling, G.; Peretz, D.; Legname, G.; DeArmond, S.J.; Prusiner, S.B. Prions in skeletal muscle. Proc. Natl. Acad. Sci. USA 2002, 99, 3812–3817. [Google Scholar] [CrossRef]
- Bosque, P.J.; Telling, G.C.; Cayetano, J.; DeArmond, S.J.; Prusiner, S.B. Evidence for prion replication in skeletal muscle. Ann. Neurol. 1997, 42, 986. [Google Scholar]
- McCulloch, L.; Brown, K.L.; Bradford, B.M.; Hopkins, J.; Bailey, M.; Rajewsky, K.; Manson, J.C.; Mabbott, N.A. Follicular dendritic cell-specific prion protein (PrP) expression alone is sufficient to sustain prion infection in the spleen. PLoS Pathog. 2011, 7, e1002402. [Google Scholar] [CrossRef]
- Brown, K.; Stewart, K.; Ritchie, D.; Mabbott, N.; Williams, A.; Fraser, H.; Morrison, W.; Bruce, M. Scrapie replication in lymphoid tissues depends on prion protein- expressing follicular dendritic cells. Nat. Med. 1999, 5, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.C.; Haig, D.A. Multiplication of scrapie agent in mouse spleen. Res. Vet. Sci. 1971, 12, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.; Dickinson, A.G. Pathogenesis of scrapie in the mouse: The role of the spleen. Nature 1970, 226, 462–463. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, A.G.; Fraser, H. Genetical control of the concentration of ME7 scrapie agent in mouse spleen. J. Comp. Pathol. 1969, 79, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Benestad, S.L.; Sarradin, P.; Thu, B.; Schönheit, J.; Tranulis, M.A.; Bratberg, B. Cases of scrapie with unusual features in Norway and designation of a new type, Nor98. Vet. Rec. 2003, 153, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Andreoletti, O.; Orge, L.; Benestad, S.L.; Beringue, V.; Litaise, C.; Simon, S.; Le Dur, A.; Laude, H.; Simmons, H.; Lugan, S.; et al. Atypical/Nor98 scrapie infectivity in sheep peripheral tissues. PLoS Pathog. 2011, 7, e1001285. [Google Scholar] [CrossRef] [PubMed]
- Bessen, R.; Marsh, R. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J. Gen. Virol. 1992, 73, 329–334. [Google Scholar] [CrossRef]
- Bartz, J.C.; Aiken, J.M.; Bessen, R.A. Delay in onset of prion disease for the HY strain of transmissible mink encephalopathy as a result of prior peripheral inoculation with the replication-deficient DY strain. J. Gen. Virol. 2004, 85, 265–273. [Google Scholar] [CrossRef]
- Bartz, J.C.; Dejoia, C.; Tucker, T.; Kincaid, A.E.; Bessen, R.A. Extraneural prion neuroinvasion without lymphoreticular system infection. J. Virol. 2005, 79, 11858–11863. [Google Scholar] [CrossRef]
- Kimberlin, R.; Walker, C.A. Transport, targeting and replication of scrapie in the CNS. In Unconventional Virus Diseases of the Central Nervous System, 1st ed.; Court, L., Dormont, D., Brown, P., Kingsbury, D., Eds.; Commissariat a l’Energie Atomique: Paris, France, 1986; Volume 1, pp. 547–562. [Google Scholar]
- Kimberlin, R.; Cole, S.; Walker, C. Pathogenesis of scrapie is faster when infection is intraspinal instead of intracerebral. Microb. Pathog. 1987, 2, 405–415. [Google Scholar] [CrossRef]
- Clarke, M.; Kimberlin, R. Pathogenesis of mouse scrapie: Distribution of agent in the pulp and stroma of infected spleens. Vet. Microbiol. 1984, 9, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Walker, C.A. Pathogenesis of mouse scrapie: Patterns of agent replication in different parts of the CNS following intraperitoneal infection. J. R. Soc. Med. 1982, 75, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Walker, C.A. Pathogenesis of mouse scrapie: Evidence for neural spread of infection to the CNS. J. Gen. Virol. 1980, 51, 183–187. [Google Scholar] [CrossRef]
- Kimberlin, R.; Walker, C. Pathogenesis of mouse scrapie: Dynamics of agent replication in spleen, spinal cord and brain after infection by different routes. J. Comp. Pathol. 1979, 89, 551–562. [Google Scholar] [CrossRef] [PubMed]
- van Keulen, L.J.M.; Bossers, A.; van Zijderveld, F. TSE pathogenesis in cattle and sheep. Vet. Res. 2008, 39, 24. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Heikenwalder, M.; Junt, T.; Schwarz, P.; Glatzel, M.; Heppner, F.L.; Fu, Y.-X.; Lipp, M.; Aguzzi, A. Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 2003, 425, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, D.S.; Kobayashi, A.; Ohno, H.; Yagita, H.; Williams, I.R.; Mabbott, N.A. M cell-depletion blocks oral prion disease pathogenesis. Mucosal Immunol. 2012, 5, 216–225. [Google Scholar] [CrossRef]
- Mohan, J.; Bruce, M.E.; Mabbott, N.A. Follicular dendritic cell dedifferentiation reduces scrapie susceptibility following inoculation via the skin. Immunology 2005, 114, 225–234. [Google Scholar] [CrossRef]
- Mabbott, N.A.; Mackay, F.; Minns, F.; Bruce, M.E. Temporary inactivation of follicular dendritic cells delays neuroinvasion of scrapie. Nat. Med. 2000, 6, 719–720. [Google Scholar] [CrossRef]
- Bartz, J.C.; Kincaid, A.E.; Bessen, R.A. Retrograde transport of transmissible mink encephalopathy within descending motor tracts. J. Virol. 2002, 76, 5759–5768. [Google Scholar] [CrossRef]
- Langenfeld, K.A.; Shikiya, R.A.; Kincaid, A.E.; Bartz, J.C. Incongruity between Prion Conversion and Incubation Period following Coinfection. J. Virol. 2016, 90, 5715–5723. [Google Scholar] [CrossRef] [PubMed]
- Ayers, J.I.; Kincaid, A.E.; Bartz, J.C. Prion strain targeting independent of strain-specific neuronal tropism. J. Virol. 2009, 83, 81–87. [Google Scholar] [CrossRef]
- Ayers, J.I.; Riffe, C.J.; Sorrentino, Z.A.; Diamond, J.; Fagerli, E.; Brooks, M.; Galaleldeen, A.; Hart, P.J.; Giasson, B.I. Localized induction of wild-type and mutant alpha-synuclein aggregation reveals propagation along neuroanatomical tracts. J. Virol. 2018, 92, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Ayers, J.I.; Fromholt, S.E.; O’Neal, V.M.; Diamond, J.H. Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol. 2016, 131, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.; Walker, C. Pathogenesis of scrapie (strain 263K) in hamsters infected intracerebrally, intraperitoneally or intraocularly. J. Gen. Virol. 1986, 67, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.K.; Al-Doujaily, H.; Sharps, B.; Clarke, A.R.; Collinge, J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011, 470, 540–542. [Google Scholar] [CrossRef]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef]
- Sandberg, M.K.; Al-Doujaily, H.; Sharps, B.; De Oliveira, M.W.; Schmidt, C.; Richard-Londt, A.; Lyall, S.; Linehan, J.M.; Brandner, S.; Wadsworth, J.D.F.; et al. Prion neuropathology follows the accumulation of alternate prion protein isoforms after infective titre has peaked. Nat. Commun. 2014, 5, 4347. [Google Scholar] [CrossRef]
- Dickinson, A.; Fraser, H.; Outram, G. Scrapie incubation time can exceed natural lifespan. Nature 1975, 256, 732–733. [Google Scholar] [CrossRef]
- Hill, A.F.; Joiner, S.; Linehan, J.; Desbruslais, M.; Lantos, P.L.; Collinge, J. Species-barrier-independent prion replication in apparently resistant species. Proc. Natl. Acad. Sci. USA 2000, 97, 10248–10253. [Google Scholar] [CrossRef]
- Thackray, A.M.; Klein, M.A.; Aguzzi, A.; Bujdoso, R. Chronic subclinical prion disease induced by low-dose inoculum. J. Virol. 2002, 76, 2510–2517. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Hall, S.M.; Walker, C.A. Pathogenesis of mouse scrapie. Evidence for direct neural spread of infection to the CNS after injection of sciatic nerve. J. Neurol. Sci. 1983, 61, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Bartz, J.C.; Kramer, M.L.; Sheehan, M.H.; Hutter, J.A.; Ayers, J.I.; Bessen, R.A.; Kincaid, A.E. Prion interference is due to a reduction in strain-specific PrPSc levels. J. Virol. 2007, 81, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Bartz, J.C.; Kincaid, A.E.; Bessen, R.A. Rapid prion neuroinvasion following tongue infection. J. Virol. 2003, 77, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Foliaki, S.T.; Lewis, V.; Islam, A.M.T.; Ellett, L.J.; Senesi, M.; Finkelstein, D.I.; Roberts, B.; Lawson, V.A.; Adlard, P.A.; Collins, S.J. Early existence and biochemical evolution characterise acutely synaptotoxic PrPSc. PLoS Pathog. 2019, 15, e1007712. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, M.A.; Stahl, N.; Hecker, R.; Pan, K.-M.; Burlingame, A.L.; Prusiner, S.B. Glycosylinositol phospholipid anchors of prion proteins. In Prion Diseases of Humans and Animals; Prusiner, S.B., Collinge, J., Powell, J., Anderton, B., Eds.; Ellis Horwood: London, UK, 1992; pp. 380–397. [Google Scholar]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef]
- Chen, S.; Teplow, D.; Parchi, P.; Teller, J.; Gambetti, P.; Autilio-Gambetti, L. Truncated forms of the human prion protein in normal brain and in prion diseases. J. Biol. Chem. 1995, 270, 19173–19180. [Google Scholar] [CrossRef]
- Mange, A.; Beranger, F.; Peoc’h, K.; Onodera, T.; Frobert, Y.; Lehmann, S. Alpha- and beta- cleavages of the amino-terminus of the cellular prion protein. Biol. Cell 2004, 96, 125–132. [Google Scholar] [CrossRef]
- Westergard, L.; Turnbaugh, J.A.; Harris, D.A. A naturally occurring C-terminal fragment of the prion protein (PrP) delays disease and acts as a dominant-negative inhibitor of PrPSc formation. J. Biol. Chem. 2011, 286, 44234–44242. [Google Scholar] [CrossRef]
- Bueler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef]
- Fischer, M.; Rülicke, T.; Raeber, A.; Sailer, A.; Moser, M.; Oesch, B.; Brandner, S.; Aguzzi, A.; Weissmann, C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996, 15, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Le Dur, A.; Laï, T.L.; Stinnakre, M.-G.; Laisné, A.; Chenais, N.; Rakotobe, S.; Passet, B.; Reine, F.; Soulier, S.; Herzog, L.; et al. Divergent prion strain evolution driven by PrP(C) expression level in transgenic mice. Nat. Commun. 2017, 8, 14170. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.; Clarke, A.; McBride, P.; McConnell, I.; Hope, J. PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration 1994, 3, 331–340. [Google Scholar] [PubMed]
- Westaway, D.; Mirenda, C.A.; Foster, D.; Zebarjadian, Y.; Scott, M.; Torchia, M.; Yang, S.-L.; Serban, H.; DeArmond, S.J.; Ebeling, C.; et al. Paradoxical shortening of scrapie incubation times by expression of prion protein transgenes derived from long incubation period mice. Neuron 1991, 7, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; van der Merwe, J.; Kim, C.; Haldiman, T.; McKenzie, D.; Safar, J.G.; Westaway, D. Prion infectivity plateaus and conversion to symptomatic disease originate from falling precursor levels and increased oligomeric PrPSc species. J. Virol. 2015, 89, 12418–12426. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; Kim, C.; Haldiman, T.; van der Merwe, J.; Lau, A.; Yang, J.; Grams, J.; Di Bari, M.A.; Nonno, R.; Telling, G.C.; et al. Prion disease tempo determined by host-dependent substrate reduction. J. Clin. Investig. 2014, 124, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Christensen, H.M.; Stewart, L.R.; Roth, K.A.; Chiesa, R.; Harris, D.A. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 2007, 26, 548–558. [Google Scholar] [CrossRef]
- Solomon, I.H.; Khatri, N.; Biasini, E.; Massignan, T.; Huettner, J.E.; Harris, D.A. An N-terminal polybasic domain and cell surface localization are required for mutant prion protein toxicity. J. Biol. Chem. 2011, 286, 14724–14736. [Google Scholar] [CrossRef]
- Sonati, T.; Reimann, R.R.; Falsig, J.; Baral, P.K.; O’connor, T.; Hornemann, S.; Yaganoglu, S.; Li, B.; Herrmann, U.S.; Wieland, B.; et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 2013, 501, 102–106. [Google Scholar] [CrossRef]
- Reimann, R.R.; Sonati, T.; Hornemann, S.; Herrmann, U.S.; Arand, M.; Hawke, S.; Aguzzi, A. Differential Toxicity of Antibodies to the Prion Protein. PLoS Pathog. 2016, 12, e1005401. [Google Scholar] [CrossRef]
- Fang, C.; Imberdis, T.; Garza, M.C.; Wille, H.; Harris, D.A. A Neuronal Culture System to Detect Prion Synaptotoxicity. PLoS Pathog. 2016, 12, e1005623. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, U.S.; Sonati, T.; Falsig, J.; Reimann, R.R.; Dametto, P.; O’Connor, T.; Li, B.; Lau, A.; Hornemann, S.; Sorce, S.; et al. Prion infections and anti-PrP antibodies trigger converging neurotoxic pathways. PLoS Pathog. 2015, 11, e1004662. [Google Scholar] [CrossRef]
- Wu, B.; McDonald, A.J.; Markham, K.; Rich, C.B.; McHugh, K.P.; Tatzelt, J.; Colby, D.W.; Millhauser, G.L.; Harris, D.A. The N-terminus of the prion protein is a toxic effector regulated by the C-terminus. Elife 2017, 6, e23473. [Google Scholar] [CrossRef] [PubMed]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Gimbel, D.A.; Nygaard, H.B.; Coffey, E.E.; Gunther, E.C.; Lauren, J.; Gimbel, Z.A.; Strittmatter, S.M. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 2010, 30, 6367–6374. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef]
- Castilla, J.; Morales, R.; Saa, P.; Barria, M.; Gambetti, P.; Soto, C. Cell-free propagation of prion strains. EMBO J. 2008, 27, 2557–2566. [Google Scholar] [CrossRef]
- Eckland, T.E.; Shikiya, R.A.; Bartz, J.C. Independent amplification of co-infected long incubation period low conversion efficiency prion strains. PLoS Pathog. 2018, 14, e1007323. [Google Scholar] [CrossRef]
- Lucassen, R.; Nishina, K.; Supattapone, S. In vitro amplification of protease-resistant prion protein requires free sulfhydryl groups. Biochemistry 2003, 42, 4127–4135. [Google Scholar] [CrossRef]
- Deleault, N.R.; Walsh, D.J.; Piro, J.R.; Wang, F.; Wang, X.; Ma, J.; Rees, J.R.; Supattapone, S. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. USA 2012, 109, E1938–E1946. [Google Scholar] [CrossRef]
- Deleault, N.R.; Kascsak, R.; Geoghegan, J.C.; Supattapone, S. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 2010, 49, 3928–3934. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Montalban, N.; Lee, Y.J.; Makarava, N.; Savtchenko, R.; Baskakov, I.V. Changes in prion replication environment cause prion strain mutation. FASEB J. 2013, 27, 3702–3710. [Google Scholar] [CrossRef] [PubMed]
- Saá, P.; Sferrazza, G.F.; Ottenberg, G.; Oelschlegel, A.M.; Dorsey, K.; Lasmézas, C.I. Strain-Specific Role of RNAs in Prion Replication. J. Virol. 2012, 86, 10494–10504. [Google Scholar] [CrossRef] [PubMed]
- Supattapone, S. Elucidating the role of cofactors in mammalian prion propagation. Prion 2014, 8, 100–105. [Google Scholar] [CrossRef]
- Supattapone, S. Synthesis of High Titer Infectious Prions with Cofactor Molecules. J. Biol. Chem. 2014, 289, 19850–19854. [Google Scholar] [CrossRef]
- Miller, M.B.; Wang, D.W.; Wang, F.; Noble, G.P.; Ma, J.; Woods, V.L.; Li, S.; Supattapone, S. Cofactor Molecules Induce Structural Transformation during Infectious Prion Formation. Structure 2013, 21, 2061–2068. [Google Scholar] [CrossRef]
- Deleault, N.R.; Piro, J.R.; Walsh, D.J.; Wang, F.; Ma, J.; Geoghegan, J.C.; Supattapone, S. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. USA 2012, 109, 8546–8551. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).