
Axonal Lysosomal Assays for Characterizing the Effects of LRRK2 G2019S

 ,
,  , , ,
, , ,  and
and
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Stem Cell Differentiation into Neurons
2.2. Treatment of Neurons with Compounds
2.3. Seeding Neurons into Microfluidic Chambers (MFCs)
2.4. Protein Extraction and Capillary Electrophoresis
2.5. Axotomy
2.6. Immunofluorescence
2.7. Imaging and Quantification
2.8. Live Cell Imaging
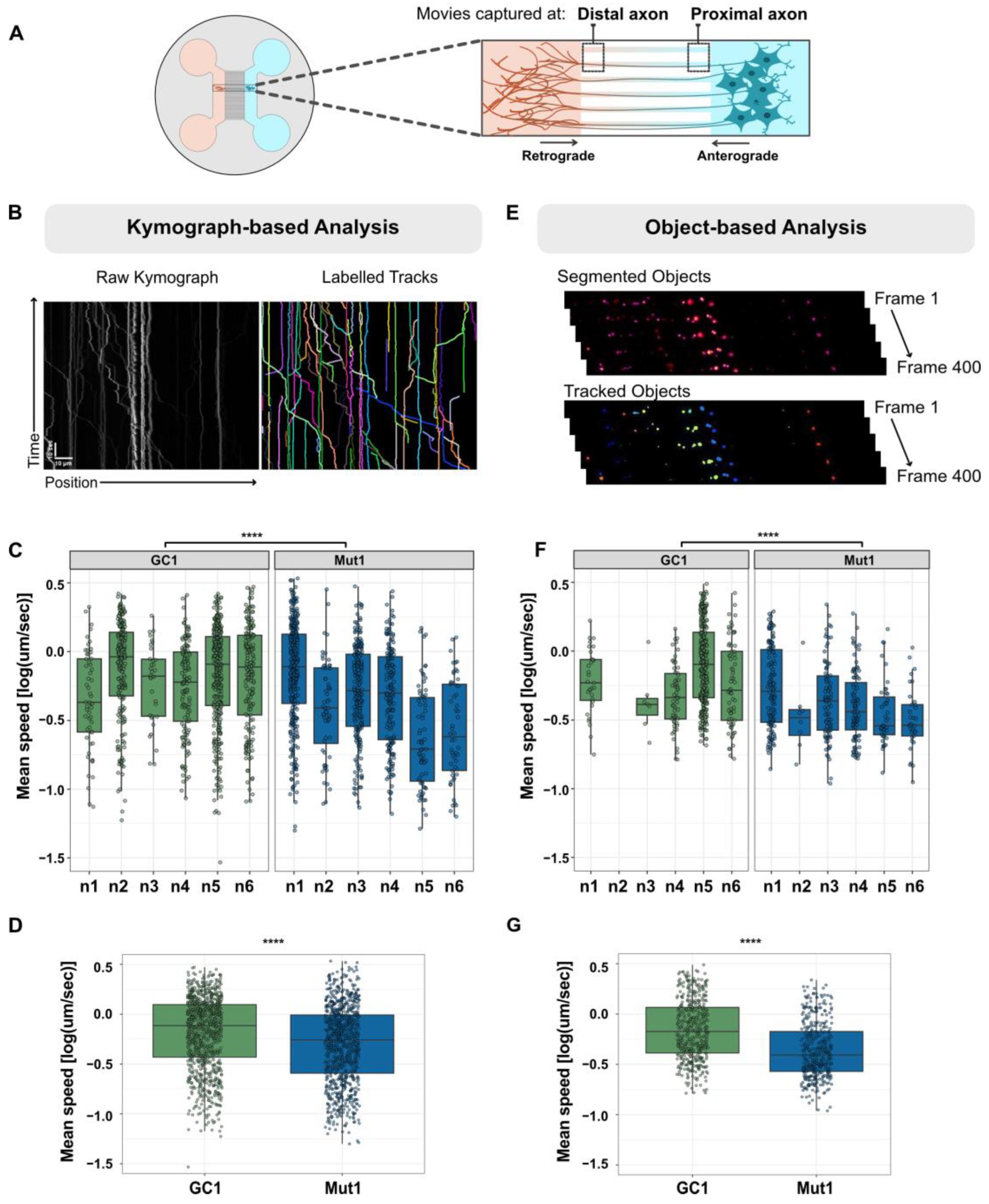
2.9. Quantification of Axonal Trafficking Using Kymographs
2.10. Quantification of Axonal Trafficking Using an Object-Based Method
2.11. Clinical and Biomarker Data Collection
3. Results
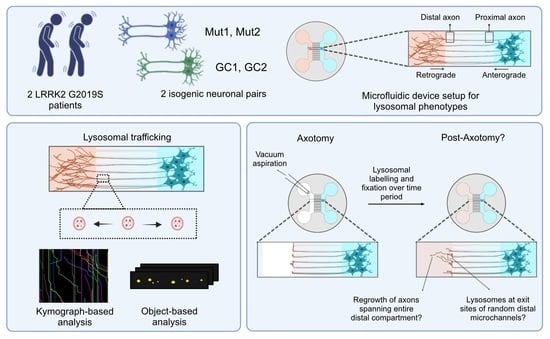
3.1. Establishing an Axonal Lysosomal Trafficking Assay
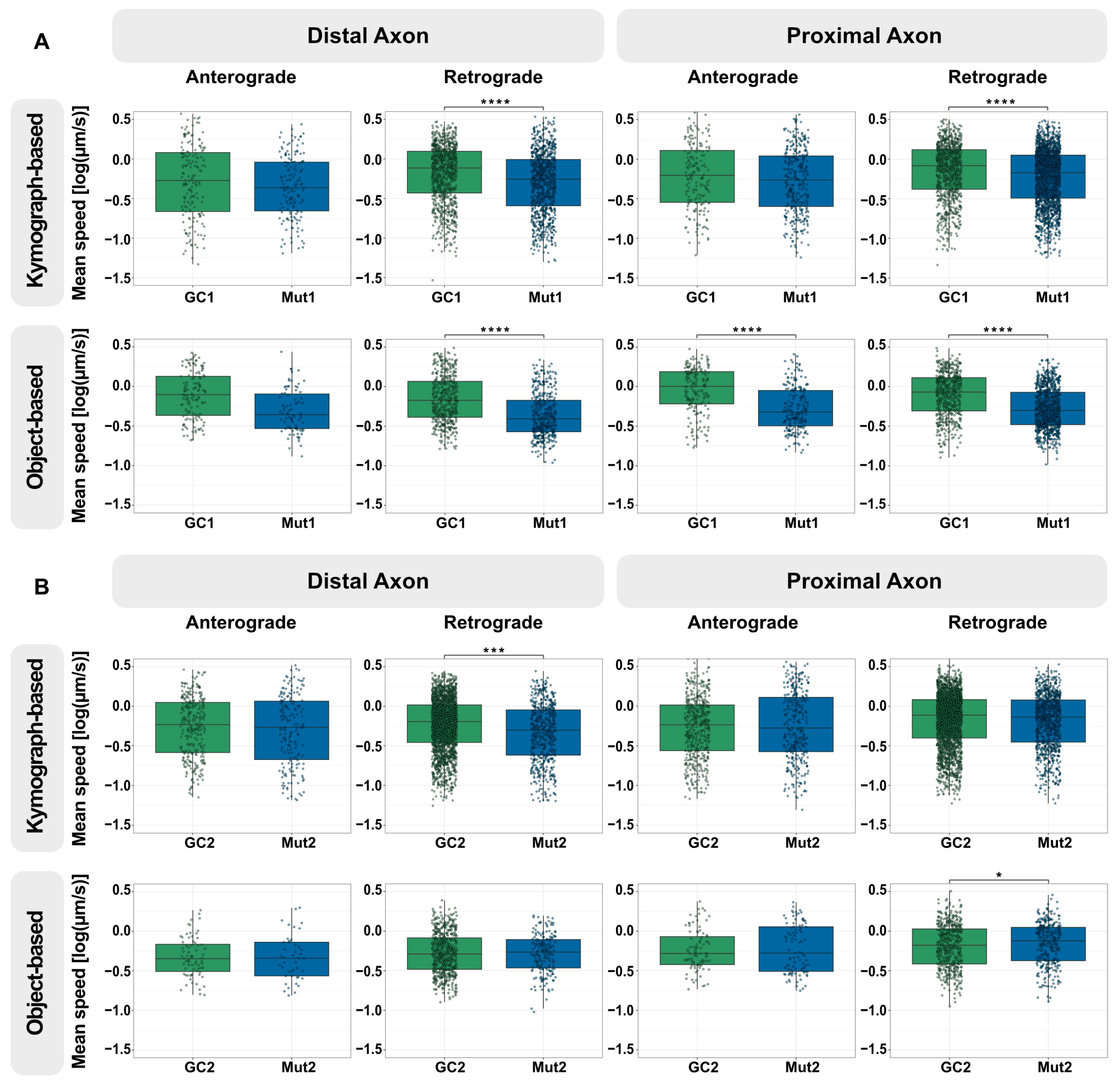
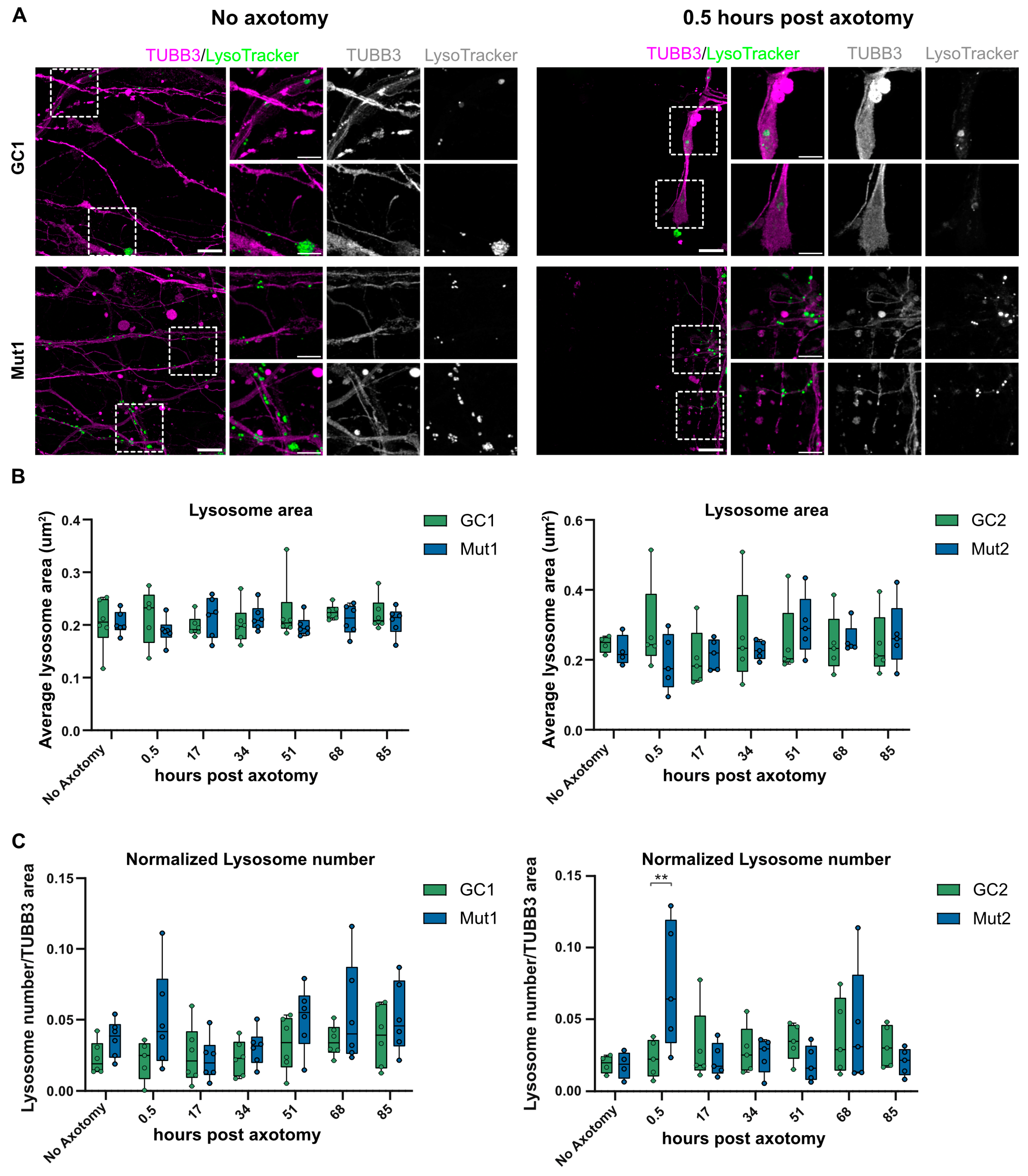
3.2. The Axonal Trafficking Assay Detects LRRK2 G2019S-Associated Changes in Lysosome Movement
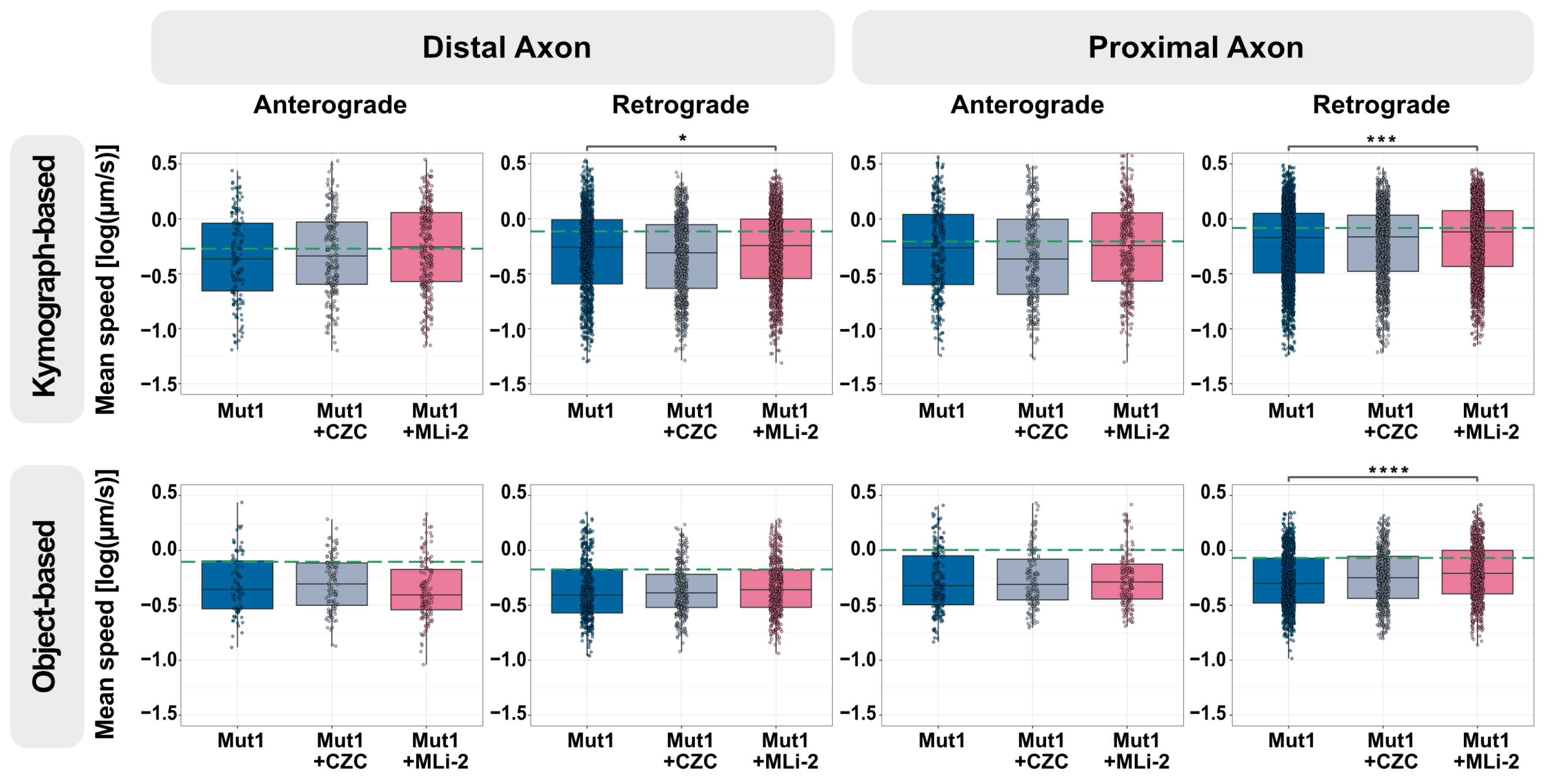
3.3. Axonal Trafficking Assay Detects Partial Rescue by a Small-Molecule LRRK2 Inhibitor
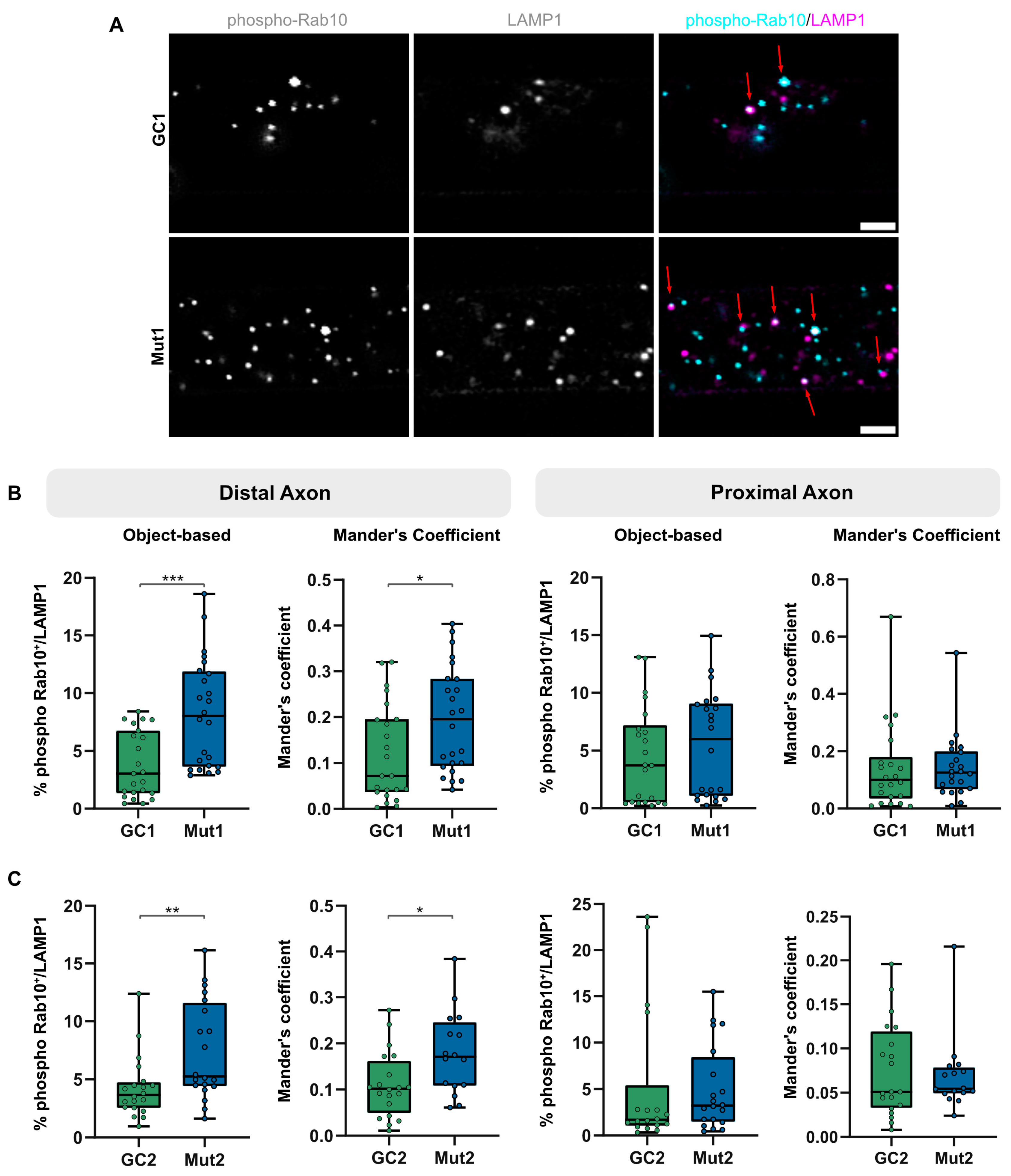
3.4. LRRK2 G2019S Is Associated with an Increase in Proportion of Lysosomes Colocalizing with Phosphorylated Rab10
3.5. Lysosomal Membrane Damage Increases LRRK2-Mediated Rab10 Phosphorylation
3.6. LRRK2 G2019S Is Not Associated with Consistent Effects on Long-Term Axonal Regrowth after Axotomy
3.7. LRRK2 G2019S Is Associated with Transient Accumulation of Lysosomes at Injury Site after Axotomy
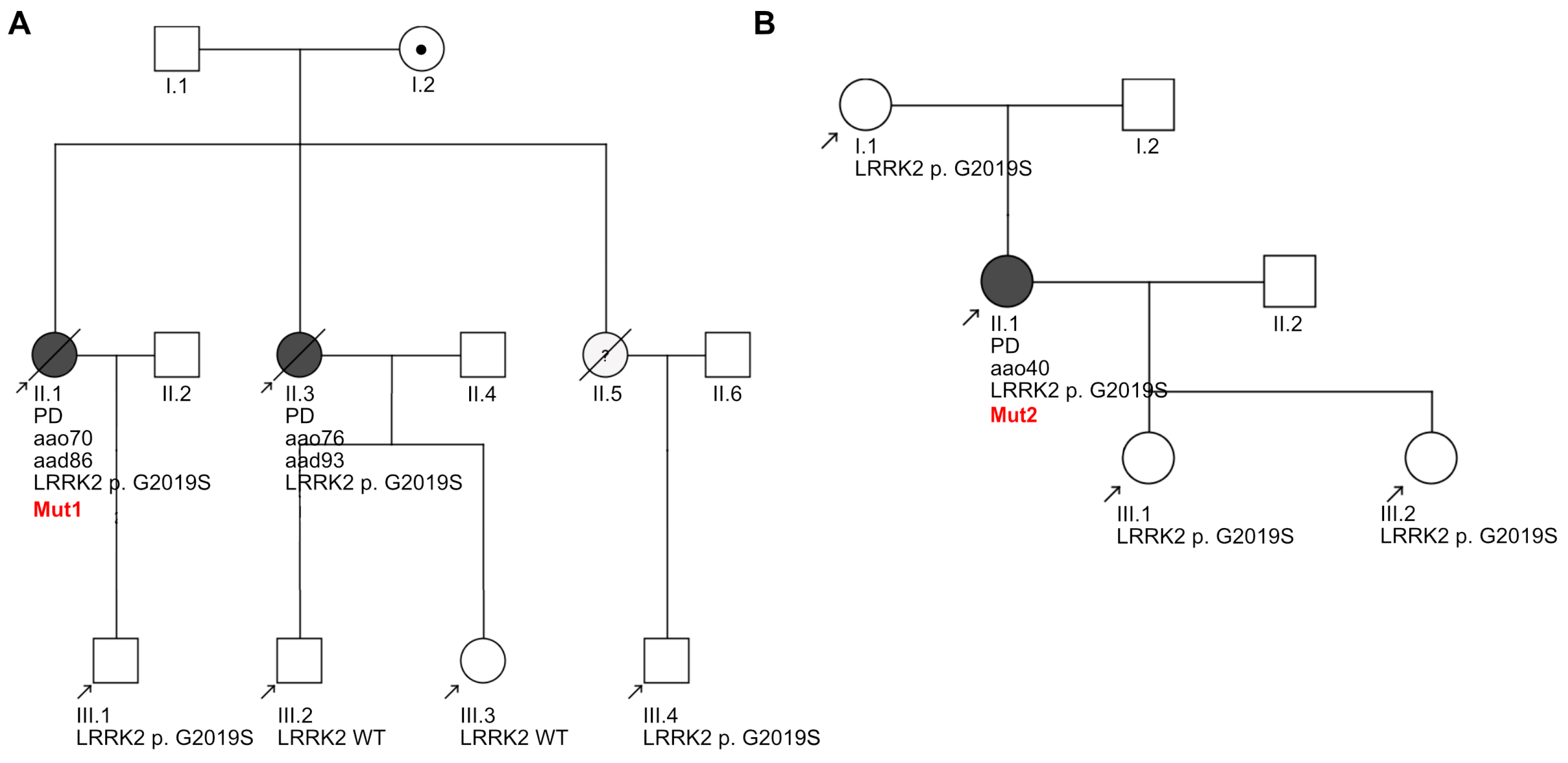
3.8. Clinical Heterogeneity within the Donating LRRK2 G2019S Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ou, Z.; Pan, J.; Tang, S.; Duan, D.; Yu, D.; Nong, H.; Wang, Z. Global Trends in the Incidence, Prevalence, and Years Lived with Disability of Parkinson’s Disease in 204 Countries/Territories from 1990 to 2019. Front. Public Health 2021, 9, 776847. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, J. An Essay on the Shaking Palsy; Whittingham and Rowland for Sherwood, Needly and Jones: London, UK, 1817. [Google Scholar]
- Goetz, C.G.; Lutge, W.; Tanner, C.M. Autonomic dysfunction in Parkinson’s disease. Neurology 1986, 36, 73. [Google Scholar] [CrossRef]
- Poewe, W. Non-motor symptoms in Parkinson’s disease. Eur. J. Neurol. 2008, 15, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W. The Parkinson’s complex: Parkinsonism is just the tip of the iceberg. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2006, 59, 591–596. [Google Scholar] [CrossRef]
- Bryois, J.; Skene, N.G.; Hansen, T.F.; Kogelman, L.J.A.; Watson, H.J.; Liu, Z.; Brueggeman, L.; Breen, G.; Bulik, C.M.; Arenas, E.; et al. Genetic identification of cell types underlying brain complex traits yields insights into the etiology of Parkinson’s disease. Nat. Genet. 2020, 52, 482–493. [Google Scholar] [CrossRef]
- Adalbert, R.; Coleman, M.P. Review: Axon pathology in age-related neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 2013, 39, 90–108. [Google Scholar] [CrossRef]
- Cheng, H.-C.; Ulane, C.M.; Burke, R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Grosch, J.; Winkler, J.; Kohl, Z. Early Degeneration of Both Dopaminergic and Serotonergic Axons—A Common Mechanism in Parkinson’s Disease. Front. Cell. Neurosci. 2016, 10, 293. [Google Scholar] [CrossRef]
- Doppler, C.E.J.; Kinnerup, M.B.; Brune, C.; Farrher, E.; Betts, M.; Fedorova, T.D.; Schaldemose, J.L.; Knudsen, K.; Ismail, R.; Seger, A.D.; et al. Regional locus coeruleus degeneration is uncoupled from noradrenergic terminal loss in Parkinson’s disease. Brain 2021, 144, 2732–2744. [Google Scholar] [CrossRef]
- Lewy, F.H. Paralysis agitans. I. Pathologische Anatomie. In Handbuch der Neurologie, Vol. 3; Lewandowsky, M., Abelsdorff, G., Eds.; Springer: Berlin, Germany, 1912; pp. 920–933. [Google Scholar]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Latourelle, J.C.; Sun, M.; Lew, M.F.; Suchowersky, O.; Klein, C.; Golbe, L.I.; Mark, M.H.; Growdon, J.H.; Wooten, G.F.; Watts, R.L.; et al. The Gly2019Ser mutation in LRRK2 is not fully penetrant in familial Parkinson’s disease: The GenePD study. BMC Med. 2008, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Goldwurm, S.; Fonzo, A.D.; Simons, E.J.; Rohé, C.F.; Zini, M.; Canesi, M.; Tesei, S.; Zecchinelli, A.; Antonini, A.; Mariani, C.; et al. The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson’s disease and originates from a common ancestor. J. Med. Genet. 2005, 42, e65. [Google Scholar] [CrossRef] [PubMed]
- Gilsbach, B.K.; Kortholt, A. Structural biology of the LRRK2 GTPase and kinase domains: Implications for regulation. Front. Mol. Neurosci. 2014, 7, 32. [Google Scholar] [CrossRef]
- West, A.B.; Moore, D.J.; Biskup, S.; Bugayenko, A.; Smith, W.W.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 16842–16847. [Google Scholar] [CrossRef]
- Civiero, L.; Vancraenenbroeck, R.; Belluzzi, E.; Beilina, A.; Lobbestael, E.; Reyniers, L.; Gao, F.; Micetic, I.; De Maeyer, M.; Bubacco, L.; et al. Biochemical characterization of highly purified leucine-rich repeat kinases 1 and 2 demonstrates formation of homodimers. PLoS ONE 2012, 7, e43472. [Google Scholar] [CrossRef]
- Greggio, E.; Zambrano, I.; Kaganovich, A.; Beilina, A.; Taymans, J.-M.; Daniëls, V.; Lewis, P.; Jain, S.; Ding, J.; Syed, A.; et al. The Parkinson Disease-associated Leucine-rich Repeat Kinase 2 (LRRK2) Is a Dimer that Undergoes Intramolecular Autophosphorylation. J. Biol. Chem. 2008, 283, 16906–16914. [Google Scholar] [CrossRef]
- Helton, L.G.; Soliman, A.; von Zweydorf, F.; Kentros, M.; Manschwetus, J.T.; Hall, S.; Gilsbach, B.; Ho, F.Y.; Athanasopoulos, P.S.; Singh, R.K.; et al. Allosteric Inhibition of Parkinson’s-Linked LRRK2 by Constrained Peptides. ACS Chem. Biol. 2021, 16, 2326–2338. [Google Scholar] [CrossRef]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 2016, 5, e12813. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Kuwahara, T.; Sakurai, M.; Komori, T.; Fujimoto, T.; Ito, G.; Yoshimura, S.-i.; Harada, A.; Fukuda, M.; Koike, M.; et al. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc. Natl. Acad. Sci. USA 2018, 115, E9115–E9124. [Google Scholar] [CrossRef]
- Herbst, S.; Campbell, P.; Harvey, J.; Bernard, E.M.; Papayannopoulos, V.; Wood, N.W.; Morris, H.R.; Gutierrez, M.G. LRRK2 activation controls the repair of damaged endomembranes in macrophages. EMBO J. 2020, 39, e104494. [Google Scholar] [CrossRef] [PubMed]
- Bonet-Ponce, L.; Beilina, A.; Williamson, C.D.; Lindberg, E.; Kluss, J.H.; Saez-Atienzar, S.; Landeck, N.; Kumaran, R.; Mamais, A.; Bleck, C.K.E.; et al. LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci. Adv. 2020, 6, eabb2454. [Google Scholar] [CrossRef] [PubMed]
- Schapansky, J.; Khasnavis, S.; DeAndrade, M.P.; Nardozzi, J.D.; Falkson, S.R.; Boyd, J.D.; Sanderson, J.B.; Bartels, T.; Melrose, H.L.; LaVoie, M.J. Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble α-synuclein in neurons. Neurobiol. Dis. 2018, 111, 26–35. [Google Scholar] [CrossRef]
- Bhatia, P. Developing Assays to Characterize the Effects of LRRK2 G2019S on Axonal Lysosomes. Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany, 2023. [Google Scholar]
- Reinhardt, L. LRRK2 G2019S Induces Axonal Dysfunction in Human iPSC-Derived Neurons That Can Be Linked to α-Synuclein, Tau and Clinical Severity. Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany, 2018. [Google Scholar]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic Correction of a LRRK2 Mutation in Human iPSCs Links Parkinsonian Neurodegeneration to ERK-Dependent Changes in Gene Expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Glatza, M.; Hemmer, K.; Tsytsyura, Y.; Thiel, C.S.; Hoing, S.; Moritz, S.; Parga, J.A.; Wagner, L.; Bruder, J.M.; et al. Derivation and Expansion Using Only Small Molecules of Human Neural Progenitors for Neurodegenerative Disease Modeling. PLoS ONE 2013, 8, e59252. [Google Scholar] [CrossRef]
- Taylor, A.M.; Blurton-Jones, M.; Rhee, S.W.; Cribbs, D.H.; Cotman, C.W.; Jeon, N.L. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods 2005, 2, 599–605. [Google Scholar] [CrossRef]
- Bolte, S.; CordeliÈRes, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Otsu, N. A Threshold Selection Method from Gray-Level Histograms. IEEE Trans. Syst. Man. Cybern. 1979, 9, 62–66. [Google Scholar] [CrossRef]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Günther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Jakobs, M.A.H.; Dimitracopoulos, A.; Franze, K. KymoButler, a deep learning software for automated kymograph analysis. eLife 2019, 8, e42288. [Google Scholar] [CrossRef] [PubMed]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. KNIME: The Konstanz Information Miner. In Data Analysis, Machine Learning and Applications; Springer: Berlin/Heidelberg, Germany, 2008; pp. 319–326. [Google Scholar]
- Stirling, D.R.; Swain-Bowden, M.J.; Lucas, A.M.; Carpenter, A.E.; Cimini, B.A.; Goodman, A. CellProfiler 4: Improvements in speed, utility and usability. BMC Bioinform. 2021, 22, 433. [Google Scholar] [CrossRef] [PubMed]
- Mölder, F.; Jablonski, K.; Letcher, B.; Hall, M.; Tomkins-Tinch, C.; Sochat, V.; Forster, J.; Lee, S.; Twardziok, S.; Kanitz, A.; et al. Sustainable data analysis with Snakemake [version 1; peer review: 1 approved, 1 approved with reservations]. F1000Research 2021, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Litvan, I.; Bhatia, K.P.; Burn, D.J.; Goetz, C.G.; Lang, A.E.; McKeith, I.; Quinn, N.; Sethi, K.D.; Shults, C.; Wenning, G.K. SIC Task Force appraisal of clinical diagnostic criteria for parkinsonian disorders. Mov. Disord. 2003, 18, 467–486. [Google Scholar] [CrossRef] [PubMed]
- Goetz, C.G.; Tilley, B.C.; Shaftman, S.R.; Stebbins, G.T.; Fahn, S.; Martinez-Martin, P.; Poewe, W.; Sampaio, C.; Stern, M.B.; Dodel, R.; et al. Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Scale presentation and clinimetric testing results. Mov. Disord. 2008, 23, 2129–2170. [Google Scholar] [CrossRef]
- Goetz, C.G.; Poewe, W.; Rascol, O.; Sampaio, C.; Stebbins, G.T.; Counsell, C.; Giladi, N.; Holloway, R.G.; Moore, C.G.; Wenning, G.K.; et al. Movement Disorder Society Task Force report on the Hoehn and Yahr staging scale: Status and recommendations The Movement Disorder Society Task Force on rating scales for Parkinson’s disease. Mov. Disord. 2004, 19, 1020–1028. [Google Scholar] [CrossRef]
- Hoops, S.; Nazem, S.; Siderowf, A.D.; Duda, J.E.; Xie, S.X.; Stern, M.B.; Weintraub, D. Validity of the MoCA and MMSE in the detection of MCI and dementia in Parkinson disease. Neurology 2009, 73, 1738–1745. [Google Scholar] [CrossRef]
- Maetzler, W.; Schmid, S.P.; Wurster, I.; Liepelt, I.; Gaenslen, A.; Gasser, T.; Berg, D. Reduced but not oxidized cerebrospinal fluid glutathione levels are lowered in Lewy body diseases. Mov. Disord. 2011, 26, 176–181. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Faghri, F.; Pihlstrom, L.; Geiger, J.T.; Elbaz, A.; Lesage, S.; Corvol, J.C.; May, P.; Nicolas, A.; Abramzon, Y.; et al. NeuroChip, an updated version of the NeuroX genotyping platform to rapidly screen for variants associated with neurological diseases. Neurobiol. Aging 2017, 57, 247.e9–247.e13. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Glaß, H.; Naumann, M.; Kreiter, N.; Japtok, J.; Sczech, R.; Hermann, A. High content organelle trafficking enables disease state profiling as powerful tool for disease modelling. Sci. Data 2018, 5, 180241. [Google Scholar] [CrossRef]
- Glaß, H.; Neumann, P.; Pal, A.; Reinhardt, P.; Storch, A.; Sterneckert, J.; Hermann, A. Combined Dendritic and Axonal Deterioration Are Responsible for Motoneuronopathy in Patient-Derived Neuronal Cell Models of Chorea-Acanthocytosis. Int. J. Mol. Sci. 2020, 21, 1797. [Google Scholar] [CrossRef] [PubMed]
- Hollenbeck, P.J. Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J. Cell Biol. 1993, 121, 305–315. [Google Scholar] [CrossRef]
- Ramsden, N.; Perrin, J.; Ren, Z.; Lee, B.D.; Zinn, N.; Dawson, V.L.; Tam, D.; Bova, M.; Lang, M.; Drewes, G.; et al. Chemoproteomics-based design of potent LRRK2-selective lead compounds that attenuate Parkinson’s disease-related toxicity in human neurons. ACS Chem. Biol. 2011, 6, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Fell, M.J.; Mirescu, C.; Basu, K.; Cheewatrakoolpong, B.; DeMong, D.E.; Ellis, J.M.; Hyde, L.A.; Lin, Y.; Markgraf, C.G.; Mei, H.; et al. MLi-2, a Potent, Selective, and Centrally Active Compound for Exploring the Therapeutic Potential and Safety of LRRK2 Kinase Inhibition. J. Pharmacol. Exp. Ther. 2015, 355, 397–409. [Google Scholar] [CrossRef]
- Kelly, K.; Wang, S.; Boddu, R.; Liu, Z.; Moukha-Chafiq, O.; Augelli-Szafran, C.; West, A.B. The G2019S mutation in LRRK2 imparts resiliency to kinase inhibition. Exp. Neurol. 2018, 309, 1–13. [Google Scholar] [CrossRef]
- Thiele, D.L.; Lipsky, P.E. Mechanism of L-leucyl-L-leucine methyl ester-mediated killing of cytotoxic lymphocytes: Dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc. Natl. Acad. Sci. USA 1990, 87, 83–87. [Google Scholar] [CrossRef]
- Gerasimenko, J.V.; Gerasimenko, O.V.; Petersen, O.H. Membrane repair: Ca2+-elicited lysosomal exocytosis. Curr. Biol. 2001, 11, R971–R974. [Google Scholar] [CrossRef]
- Andrews, N.W. Membrane resealing: Synaptotagmin VII keeps running the show. Sci. STKE 2005, 2005, pe19. [Google Scholar] [CrossRef]
- Encarnação, M.; Espada, L.; Escrevente, C.; Mateus, D.; Ramalho, J.; Michelet, X.; Santarino, I.; Hsu, V.W.; Brenner, M.B.; Barral, D.C.; et al. A Rab3a-dependent complex essential for lysosome positioning and plasma membrane repair. J. Cell Biol. 2016, 213, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Ostrowski, P.; Jaumouillé, V.; Grinstein, S. The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 2016, 212, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Zhang, Y.; Pan, H.; Xu, Q.; Guo, J.; Tang, B.; Sun, Q. Clinical Heterogeneity Among LRRK2 Variants in Parkinson’s Disease: A Meta-Analysis. Front. Aging Neurosci. 2018, 10, 283. [Google Scholar] [CrossRef]
- Kwon, E.H.; Tennagels, S.; Gold, R.; Gerwert, K.; Beyer, L.; Tönges, L. Update on CSF Biomarkers in Parkinson’s Disease. Biomolecules 2022, 12, 329. [Google Scholar] [CrossRef] [PubMed]
- Saijo, E.; Groveman, B.R.; Kraus, A.; Metrick, M.; Orrù, C.D.; Hughson, A.G.; Caughey, B. Ultrasensitive RT-QuIC Seed Amplification Assays for Disease-Associated Tau, α-Synuclein, and Prion Aggregates. In Protein Misfolding Diseases: Methods and Protocols; Gomes, C.M., Ed.; Springer: New York, NY, USA, 2019; pp. 19–37. [Google Scholar]
- Vermunt, L.; Otte, M.; Verberk, I.M.W.; Killestein, J.; Lemstra, A.W.; van der Flier, W.M.; Pijnenburg, Y.A.L.; Vijverberg, E.G.B.; Bouwman, F.H.; Gravesteijn, G.; et al. Age- and disease-specific reference values for neurofilament light presented in an online interactive support interface. Ann. Clin. Transl. Neurol. 2022, 9, 1832–1837. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Rodríguez, A.; Martínez-Rodríguez, I.; Sánchez-Juan, P.; Sierra, M.; González-Aramburu, I.; Rivera-Sánchez, M.; Andrés-Pacheco, J.; Gutierrez-González, Á.; García-Hernández, A.; Madera, J.; et al. Serial DaT-SPECT imaging in asymptomatic carriers of LRRK2 G2019S mutation: 8 years’ follow-up. Eur. J. Neurol. 2021, 28, 4204–4208. [Google Scholar] [CrossRef] [PubMed]
- Boecker, C.A.; Goldsmith, J.; Dou, D.; Cajka, G.G.; Holzbaur, E.L.F. Increased LRRK2 kinase activity alters neuronal autophagy by disrupting the axonal transport of autophagosomes. Curr. Biol. 2021, 31, 2140–2154.e2146. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef]
- Bolam, J.P.; Pissadaki, E.K. Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov. Disord. 2012, 27, 1478–1483. [Google Scholar] [CrossRef]
- Pal, A.; Kretner, B.; Abo-Rady, M.; Glaβ, H.; Dash, B.P.; Naumann, M.; Japtok, J.; Kreiter, N.; Dhingra, A.; Heutink, P.; et al. Concomitant gain and loss of function pathomechanisms in C9ORF72 amyotrophic lateral sclerosis. Life Sci. Alliance 2021, 4, e202000764. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhatia, P.; Bickle, M.; Agrawal, A.A.; Truss, B.; Nikolaidi, A.; Brockmann, K.; Reinhardt, L.; Vogel, S.; Szegoe, E.M.; Pal, A.; et al. Axonal Lysosomal Assays for Characterizing the Effects of LRRK2 G2019S. Biology 2024, 13, 58. https://doi.org/10.3390/biology13010058
Bhatia P, Bickle M, Agrawal AA, Truss B, Nikolaidi A, Brockmann K, Reinhardt L, Vogel S, Szegoe EM, Pal A, et al. Axonal Lysosomal Assays for Characterizing the Effects of LRRK2 G2019S. Biology. 2024; 13(1):58. https://doi.org/10.3390/biology13010058
Chicago/Turabian StyleBhatia, Priyanka, Marc Bickle, Amay A. Agrawal, Buster Truss, Aikaterina Nikolaidi, Kathrin Brockmann, Lydia Reinhardt, Stefanie Vogel, Eva M. Szegoe, Arun Pal, and et al. 2024. "Axonal Lysosomal Assays for Characterizing the Effects of LRRK2 G2019S" Biology 13, no. 1: 58. https://doi.org/10.3390/biology13010058
APA StyleBhatia, P., Bickle, M., Agrawal, A. A., Truss, B., Nikolaidi, A., Brockmann, K., Reinhardt, L., Vogel, S., Szegoe, E. M., Pal, A., Hermann, A., Mikicic, I., Yun, M., Falkenburger, B., & Sterneckert, J. (2024). Axonal Lysosomal Assays for Characterizing the Effects of LRRK2 G2019S. Biology, 13(1), 58. https://doi.org/10.3390/biology13010058