
The Contribution of the Locus Coeruleus–Noradrenaline System Degeneration during the Progression of Alzheimer’s Disease
Abstract
Simple Summary
Abstract
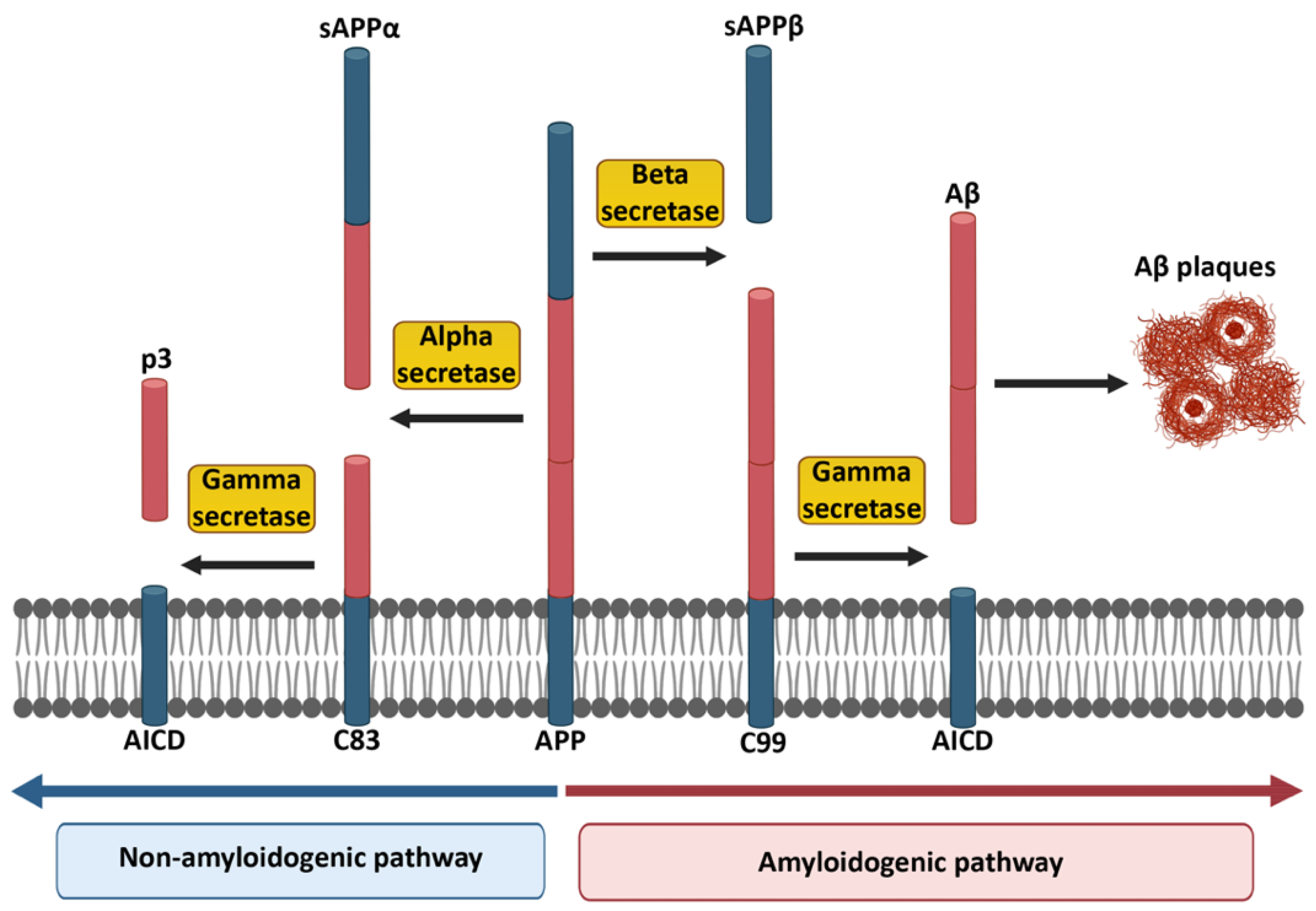
1. Alzheimer’s Disease
2. The Role of Microglia in Alzheimer’s Disease
3. The Locus Coeruleus
4. Noradrenaline
5. The Locus Coeruleus–Noradrenaline System in Cognitive Processes
6. Importance of the Locus Coeruleus in Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Model | Animal Species | LC Degeneration Model | References |
|---|---|---|---|
| Aggregated Aβ 1–42 injection | Rat | DSP4 | Heneka et al., 2002 [159] |
| Aggregated Aβ 1–42 and human recombinant IL-1β injection | Rat | DSP4 | Heneka et al., 2003 [160] |
| APP23 | Mouse | DSP4 | Heneka et al., 2006 [151] |
| TASTPM | Mouse | DSP4 | Pugh et al., 2007 [161] |
| H6 | Mouse | DSP4 | Kalinin et al., 2007 [162] |
| APP/PS1 | Mouse | DSP4 | Jardanhazi-Kurutz et al., 2010 [149] |
| APPV717I | Mouse | DSP4 | Heneka et al., 2010 [153] |
| APP/PS1 | Mouse | DSP4 | Jardanhazi-Kurutz et al., 2011 [163] |
| APP/PS1 | Mouse | DSP4 | Rey et al., 2012 [164] |
| P301S | Mouse | DSP4 | Chalermpalanupap et al., 2018 [155] |
| APP/PS1 | Mouse | DBH(−/−) | Hammerschmidt et al., 2013 [88] |
| P301S | Mouse | DBH(−/−) | Kang et al., 2020 [165] |
| APP/PS1 | Mouse | Ear2(−/−) | Kummer et al., 2014 [154] |
| APP/PS1 | Mouse | - | Liu et al., 2013 [166] |
| 5XFAD/BDNF KO * * Viral injection based local knocking out strategy for BDNF expression | Mouse | - | Braun et al., 2017 [167] |
| APP/PS1 | Mouse | - | Cao et al., 2021 [168] |
7. The Locus Coeruleus–Noradrenaline System and Microglia
8. Clinical Trials for Alzheimer’s Disease Based on the Locus Coeruleus–Noradrenaline System
9. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Alzheimer Reports. Available online: https://www.alz.co.uk/research/world-report (accessed on 20 December 2019).
- Kawas, C.; Gray, S.; Brookmeyer, R.; Fozard, J.; Zonderman, A. Age-specific incidence rates of Alzheimer’s disease. Neurology 2000, 54, 2072–2077. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Jiang, Z.-F. Accumulated Amyloid-β Peptide and Hyperphosphorylated Tau Protein: Relationship and Links in Alzheimer’s Disease. J. Alzheimer’s Dis. 2009, 16, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Yankner, B.A.; Mesulam, M.-M. β-Amyloid and the Pathogenesis of Alzheimer’s Disease. N. Engl. J. Med. 1991, 325, 1849–1857. [Google Scholar]
- Roher, A.E.; Esh, C.L.; Kokjohn, T.A.; Castaño, E.M.; Van Vickle, G.D.; Kalback, W.M.; Patton, R.L.; Luehrs, D.C.; Daugs, I.D.; Kuo, Y.; et al. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimer’s Dement. 2009, 5, 18–29. [Google Scholar] [CrossRef]
- Ludewig, S.; Korte, M. Novel Insights into the Physiological Function of the APP (Gene) Family and Its Proteolytic Fragments in Synaptic Plasticity. Front. Mol. Neurosci. 2017, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Kögel, D.; Deller, T.; Behl, C. Roles of amyloid precursor protein family members in neuroprotection, stress signaling and aging. Exp. Brain Res. 2011, 217, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Mockett, B.G.; Richter, M.; Abraham, W.C.; Müller, U.C. Therapeutic Potential of Secreted Amyloid Precursor Protein APPsα. Front. Mol. Neurosci. 2017, 10, 30. [Google Scholar] [CrossRef]
- Sasmita, A.O. Current viral-mediated gene transfer research for treatment of Alzheimer’s disease. Biotechnol. Genet. Eng. Rev. 2018, 35, 26–45. [Google Scholar] [CrossRef]
- Polanco, J.C.; Li, C.; Bodea, L.-G.; Martinez-Marmol, R.; Meunier, F.A.; Gotz, J. Amyloid-β and tau complexity—Towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 2017, 14, 22–39. [Google Scholar] [CrossRef]
- Olsson, F.; Schmidt, S.; Althoff, V.; Munter, L.; Jin, S.; Rosqvist, S.; Lendahl, U.; Multhaup, G.; Lundkvist, J. Characterization of Intermediate Steps in Amyloid Beta (Aβ) Production under Near-native Conditions. J. Biol. Chem. 2014, 289, 1540–1550. [Google Scholar] [CrossRef]
- Hefter, D.; Kaiser, M.; Weyer, S.W.; Papageorgiou, I.E.; Both, M.; Kann, O.; Muller, U.C.; Draguhn, A. Amyloid Precursor Protein Protects Neuronal Network Function after Hypoxia via Control of Volt-age-Gated Calcium Channels. J. Neurosci. 2016, 36, 8356–8371. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.W.; Grosgen, S.; Rothhaar, T.L.; Burg, V.K.; Hundsdorfer, B.; Haupenthal, V.J.; Friess, P.; Muller, U.; Fassbender, K.; Riemenschneider, M.; et al. Intracellular APP Domain Regulates Serine-Palmitoyl-CoA Transferase Expression and Is Affected in Alzheimer’s Disease. Int. J. Alzheimer’s Dis. 2011, 2011, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-X.; Fuller, S.J.; Beyreuther, K.; Masters, C.L. The amyloid precursor protein of Alzheimer disease in human brain and blood. J. Leukoc. Biol. 1999, 66, 567–574. [Google Scholar] [CrossRef]
- Bali, J.; Gheinani, A.H.; Zurbriggen, S.; Rajendran, L. Role of genes linked to sporadic Alzheimer’s disease risk in the production of β-amyloid peptides. Proc. Natl. Acad. Sci. USA 2012, 109, 15307–15311. [Google Scholar] [CrossRef]
- Bird, T.D. Genetic aspects of Alzheimer disease. Anesthesia Analg. 2008, 10, 231–239. [Google Scholar] [CrossRef]
- Tanzi, R.E. The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef]
- Mandelkow, E.-M.; Mandelkow, E. Biochemistry and Cell Biology of Tau Protein in Neurofibrillary Degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef]
- Barron, M.; Gartlon, J.; Dawson, L.A.; Atkinson, P.J.; Pardon, M.-C. A state of delirium: Deciphering the effect of in-flammation on tau pathology in Alzheimer’s disease. Exp. Gerontol. 2017, 94, 103–107. [Google Scholar] [CrossRef]
- Niewiadomska, G.; Niewiadomski, W.; Steczkowska, M.; Gasiorowska, A. Tau Oligomers Neurotoxicity. Life 2021, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Cirrito, J.R.; Stewart, F.R.; Jiang, H.; Finn, M.B.; Holmes, B.; Binder, L.I.; Mandelkow, E.-M.; Diamond, M.I.; Lee, V.M.-Y.; et al. In Vivo Microdialysis Reveals Age-Dependent Decrease of Brain Interstitial Fluid Tau Levels in P301S Human Tau Transgenic Mice. J. Neurosci. 2011, 31, 13110–13117. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Holth, J.K.; Liao, F.; Stewart, F.R.; Mahan, T.; Jiang, H.; Cirrito, J.R.; Patel, T.K.; Hochgräfe, K.; Mandelkow, E.-M.; et al. Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 2014, 211, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.D.; Wassmer, T.; Fraser, G.; Smith, J.; Perkinton, M.; Billinton, A.; Livesey, F.J. Extracellular Monomeric and Aggregated Tau Efficiently Enter Human Neurons through Overlapping but Distinct Pathways. Cell Rep. 2018, 22, 3612–3624. [Google Scholar] [CrossRef]
- Ittner, L.M.; Fath, T.; Ke, Y.D.; Bi, M.; van Eersel, J.; Li, K.M.; Gunning, P.; Götz, J. Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2008, 105, 15997–16002. [Google Scholar] [CrossRef]
- Li, B.; Chohan, M.O.; Grundke-Iqbal, I.; Iqbal, K. Disruption of microtubule network by Alzheimer abnormally hyper-phosphorylated tau. Acta Neuropathol. 2007, 113, 501–511. [Google Scholar] [CrossRef]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.-L.; et al. Tau Mislocalization to Dendritic Spines Mediates Synaptic Dysfunction Independently of Neurodegen-eration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef]
- Walters, A.; Phillips, E.; Zheng, R.; Biju, M.; Kuruvilla, T. Evidence for neuroinflammation in Alzheimer’s disease: Neu-roinflammation in Alzheimer’s. Prog. Neurol. Psychiatry 2016, 20, 25–31. [Google Scholar] [CrossRef]
- Tuppo, E.E.; Arias, H.R. The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2005, 37, 289–305. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. The inflammatory response system of brain: Implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Rev. 1995, 21, 195–218. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 39, 383–421. [Google Scholar] [CrossRef] [PubMed]
- Dansokho, C.; Heneka, M.T. Neuroinflammatory responses in Alzheimer’s disease. J. Neural. Transm. 2017, 125, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Hoeffel, G.; Ginhoux, F. Ontogeny of Tissue-Resident Macrophages. Front. Immunol. 2015, 6, 486. [Google Scholar] [CrossRef]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Aguzzi, A.; Barres, B.A.; Bennett, M.L. Microglia: Scapegoat, Saboteur, or Something Else? Science 2013, 339, 156–161. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Lenz, K.M.; McCarthy, M.M. A Starring Role for Microglia in Brain Sex Differences. Neuroscientist 2015, 21, 306–321. [Google Scholar] [CrossRef]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.P.; Chancey, J.; Enikolopov, G.; Wadiche, L.; Tsirka, S.E.; Maletic-Savatic, M. Microglia Shape Adult Hippocampal Neurogenesis through Apoptosis-Coupled Phagocytosis. Cell. Stem. Cell. 2010, 7, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting Microglia Directly Monitor the Functional State of Synapses In Vivo and Determine the Fate of Ischemic Terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Hristovska, I.; Pascual, O. Deciphering Resting Microglial Morphology and Process Motility from a Synaptic Prospect. Front. Integr. Neurosci. 2016, 9, 73. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Tremblay, M.-È.; Stevens, B.; Sierra, A.; Wake, H.; Bessis, A.; Nimmerjahn, A. The Role of Microglia in the Healthy Brain. J. Neurosci. 2011, 31, 16064–16069. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Paren-chyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Bolmont, T.; Haiss, F.; Eicke, D.; Radde, R.; Mathis, C.A.; Klunk, W.; Kohsaka, S.; Jucker, M.; Calhoun, M.E. Dynamics of the Microglial/Amyloid Interaction Indicate a Role in Plaque Maintenance. J. Neurosci. 2008, 28, 4283–4292. [Google Scholar] [CrossRef]
- Graeber, M.B.; Tetzlaff, W.; Streit, W.J.; Kreutzberg, G.W. Microglial cells but not astrocytes undergo mitosis following rat facial nerve axotomy. Neurosci. Lett. 1988, 85, 317–321. [Google Scholar] [CrossRef]
- Mrak, R.E. Microglia in Alzheimer Brain: A Neuropathological Perspective. Int. J. Alzheimer’s Dis. 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Czeh, M.; Gressens, P.; Kaindl, A.M. The Yin and Yang of Microglia. Dev. Neurosci. 2011, 33, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Serpente, M.; Bonsi, R.; Scarpini, E.; Galimberti, D. Innate Immune System and Inflammation in Alzheimer’s Disease: From Pathogenesis to Treatment. Neuroimmunomodulation 2014, 21, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Monje, M.L.; Toda, H.; Palmer, T.D. Inflammatory Blockade Restores Adult Hippocampal Neurogenesis. Science 2003, 302, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- Waisman, A.; Liblau, R.S.; Becher, B. Innate and adaptive immune responses in the CNS. Lancet Neurol. 2015, 14, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef]
- Baik, S.H.; Kang, S.; Son, S.M.; Mook-Jung, I. Microglia contributes to plaque growth by cell death due to uptake of am-yloid β in the brain of Alzheimer’s disease mouse model: Aβ Plaque Formation and Microglial Activation. Glia 2016, 64, 2274–2290. [Google Scholar] [CrossRef]
- Stalder, M.; Phinney, A.; Probst, A.; Sommer, B.; Staufenbiel, M.; Jucker, M. Association of Microglia with Amyloid Plaques in Brains of APP23 Transgenic Mice. Am. J. Pathol. 1999, 154, 1673–1684. [Google Scholar] [CrossRef]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.-L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; et al. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 2000, 6, 916–919. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial Dysfunction and Defective β-Amyloid Clearance Pathways in Aging Alzheimer’s Disease Mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.P.; Vogl, T.; Axt, D.; Griep, A.; Vieira-Saecker, A.; Jessen, F.; Gelpi, E.; Roth, J.; Heneka, M.T. Mrp14 Deficiency Ameliorates Amyloid Burden by Increasing Microglial Phagocytosis and Modula-tion of Amyloid Precursor Protein Processing. J. Neurosci. 2012, 32, 17824–17829. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Gabriela Sanchez, M.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gomez-Nicola, D.; Luheshi, G.; Vallieres, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states: A New Microglial Phenotype. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Martini, A.C.; Helman, A.M.; Mccarty, K.L.; Lott, I.T.; Doran, E.; Schmitt, F.A.; Head, E. Distribution of microglial phenotypes as a function of age and Alzheimer’s disease neuropathology in the brains of people with Down syndrome. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12113. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef]
- Chen, Y.; Colonna, M. Microglia in Alzheimer’s disease at single-cell level. Are there common patterns in humans and mice? J. Exp. Med. 2021, 218, e20202717. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.-E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Patel, N.S.; Paris, D.; Mathura, V.; Quadros, A.N.; Crawford, F.C.; Mullan, M.J. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J. Neuroinflammation 2005, 2, 9–10. [Google Scholar] [CrossRef]
- Lue, L.-F.; Rydel, R.; Brigham, E.F.; Yang, L.-B.; Hampel, H.; Murphy, G.M.; Brachova, L.; Yan, S.-D.; Walker, D.G.; Shen, Y.; et al. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia 2001, 35, 72–79. [Google Scholar] [CrossRef]
- Prieto, G.A.; Tong, L.; Smith, E.D.; Cotman, C.W. TNFα and IL-1β but not IL-18 Suppresses Hippocampal Long-Term Potentiation Directly at the Synapse. Neurochem. Res. 2018, 44, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Quan, N. Microglia and CNS Interleukin-1: Beyond Immunological Concepts. Front. Neurol. 2018, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Hewett, S.J.; Csernansky, C.A.; Choi, D.W. Selective potentiation of NMDA-induced neuronal injury following induc-tion of astrocytic iNOS. Neuron 1994, 13, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Amaral, D.G.; Sinnamon, H.M. The locus coeruleus: Neurobiology of a central noradrenergic nucleus. Prog. Neurobiol. 1977, 9, 147–196. [Google Scholar] [CrossRef]
- Berridge, C.W.; Waterhouse, B.D. The locus coeruleus–noradrenergic system: Modulation of behavioral state and state-dependent cognitive processes. Brain Res. Rev. 2003, 42, 33–84. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Tanaka, C. Morphological organization of catecholamine terminals in the diencephalon of the rhesus monkey. Brain Res. 1977, 119, 43–55. [Google Scholar] [CrossRef]
- Jones, B.E.; Yang, T.-Z. The efferent projections from the reticular formation and the locus coeruleus studied by antero-grade and retrograde axonal transport in the rat. J. Comp. Neurol. 1985, 242, 56–92. [Google Scholar] [CrossRef]
- Osaka, T.; Matsumura, H. Noradrenergic inputs to sleep-related neurons in the preoptic area from the locus coeruleus and the ventrolateral medulla in the rat. Neurosci. Res. 1994, 19, 39–50. [Google Scholar] [CrossRef]
- Modirrousta, M.; Mainville, L.; Jones, B.E. Gabaergic neurons with α2-adrenergic receptors in basal forebrain and preop-tic area express c-Fos during sleep. Neuroscience 2004, 129, 803–810. [Google Scholar] [CrossRef]
- Swanson, L.W.; E Sawchenko, P. Hypothalamic Integration: Organization of the Paraventricular and Supraoptic Nuclei. Annu. Rev. Neurosci. 1983, 6, 269–324. [Google Scholar] [CrossRef]
- Proudfit, H.; Clark, F. The projections of locus coeruleus neurons to the spinal cord. Prog. Brain Res. 1991, 88, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Olson, L.; Fuxe, K. On the projections from the locus coeruleus noradrealine neurons: The cerebellar innervation. Brain Res. 1971, 28, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Chandler, D.J.; Waterhouse, B.D.P.D. Evidence for Broad Versus Segregated Projections from Cholinergic and Noradrenergic Nuclei to Functionally and Anatomically Discrete Subregions of Prefrontal Cortex. Front. Behav. Neurosci. 2012, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Chandler, D.J.; Lamperski, C.S.; Waterhouse, B.D. Identification and distribution of projections from monoaminergic and cholinergic nuclei to functionally differentiated subregions of prefrontal cortex. Brain Res. 2013, 1522, 38–58. [Google Scholar] [CrossRef]
- Chandler, D.J.; Gao, W.-J.; Waterhouse, B.D. Heterogeneous organization of the locus coeruleus projections to prefrontal and motor cortices. Proc. Natl. Acad. Sci. USA 2014, 111, 6816–6821. [Google Scholar] [CrossRef]
- Breton-Provencher, V.; Drummond, G.T.; Sur, M. Locus Coeruleus Norepinephrine in Learned Behavior: Anatomical Modularity and Spatiotemporal Integration in Targets. Front. Neural Circuits 2021, 15, 46. [Google Scholar] [CrossRef]
- Hammerschmidt, T.; Kummer, M.P.; Terwel, D.; Martinez, A.; Gorji, A.; Pape, H.-C.; Rommelfanger, K.S.; Schroeder, J.P.; Stoll, M.; Schultze, J.; et al. Selective Loss of Noradrenaline Exacerbates Early Cognitive Dysfunction and Synaptic Deficits in APP/PS1 Mice. Biol. Psychiatry 2013, 73, 454–463. [Google Scholar] [CrossRef]
- Sara, S.J. The locus coeruleus and noradrenergic modulation of cognition. Nat. Rev. Neurosci. 2009, 10, 211–223. [Google Scholar] [CrossRef]
- Sara, S.J.; Bouret, S. Orienting and Reorienting: The Locus Coeruleus Mediates Cognition through Arousal. Neuron 2012, 76, 130–141. [Google Scholar] [CrossRef]
- Mason, S.T.; Fibiger, H.C. Regional topography within noradrenergic locus coeruleus as revealed by retrograde transport of horseradish peroxidase. J. Comp. Neurol. 1979, 187, 703–724. [Google Scholar] [CrossRef]
- Waterhouse, B.D.; Lin, C.S.; Burne, R.A.; Woodward, D.J. The distribution of neocortical projection neurons in the locus coeruleus. J. Comp. Neurol. 1983, 217, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, L.A.; Luo, L. Organization of the Locus Coeruleus-Norepinephrine System. Curr. Biol. 2015, 25, R1051–R1056. [Google Scholar] [CrossRef] [PubMed]
- Atzori, M.; Cuevas-Olguin, R.; Esquivel-Rendon, E.; Garcia-Oscos, F.; Salgado-Delgado, R.C.; Saderi, N.; Miranda-Morales, M.; Trevino, M.; Pineda, J.C.; Salgado, H. Locus Ceruleus Norepinephrine Release: A Central Regulator of CNS Spatio-Temporal Activation? Front. Synaptic Neurosci. 2016, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Aston-Jones, G.; Bloom, F. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J. Neurosci. 1981, 1, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Aston-Jones, G.; Bloom, F.E. Norepinephrine-containing locus coeruleus neurons in behaving rats exhibit pronounced responses to non-noxious environmental stimuli. J. Neurosci. 1981, 1, 887–900. [Google Scholar] [CrossRef]
- Hobson, J.A.; McCarley, R.W.; Wyzinski, P.W. Sleep Cycle Oscillation: Reciprocal Discharge by Two Brainstem Neuronal Groups. Science 1975, 189, 55–58. [Google Scholar] [CrossRef]
- Foote, S.L.; Aston-Jones, G.; Bloom, F.E. Impulse activity of locus coeruleus neurons in awake rats and monkeys is a function of sensory stimulation and arousal. Proc. Natl. Acad. Sci. USA 1980, 77, 3033–3037. [Google Scholar] [CrossRef]
- Iijima, K. An immunocytochemical study on the GABA-ergic and serotonin-ergic neurons in rat locus ceruleus with special reference to possible existence of the masked indoleamine cells. Acta Histochem. 1989, 87, 43–57. [Google Scholar] [CrossRef]
- McLean, J.H.; Shipley, M.T.; Nickell, W.T.; Aston-Jones, G.; Reyher, C.K.H. Chemoanatomical organization of the nor-adrenergic input from locus coeruleus to the olfactory bulb of the adult rat. J. Comp. Neurol. 1989, 285, 339–349. [Google Scholar] [CrossRef]
- Olpe, H.-R.; Steinmann, M. Responses of locus coeruleus neurons to neuropeptides. Prog. Brain Res. 1991, 88, 241–248. [Google Scholar] [CrossRef]
- Simpson, K.; Waterhouse, B.; Lin, R. Origin, distribution, and morphology of galaninergic fibers in the rodent trigeminal system. J. Comp. Neurol. 1999, 411, 524–534. [Google Scholar] [CrossRef]
- Bylund, D.; Bylund, K. Norepinephrine. In Encyclopedia of the Neurological Sciences; Elsevier: Amsterdam, The Netherlands, 2014; pp. 614–616. [Google Scholar] [CrossRef]
- Wassall, R.; Teramoto, N.; Cunnane, T. Noradrenaline. In Encyclopedia of Neuroscience; Squire, L.R., Ed.; Academic Press: Cambridge, MA, USA, 2009; pp. 1221–1230. [Google Scholar] [CrossRef]
- Benarroch, E.E. The locus ceruleus norepinephrine system: Functional organization and potential clinical significance. Neurology 2009, 73, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A. The Catecholaminergic Innervation of Primate Prefrontal Cortex. In Advances in Neuroscience and Schizophrenia; Tuma, A.H., Ed.; Springer: Berlin/Heidelberg, Germany, 1992. [Google Scholar]
- Foote, S.L.; Morrison, J.H. Extrathalamic modulation of cortical function. Annu. Rev. Neurosci. 1987, 10, 67–95. [Google Scholar] [CrossRef] [PubMed]
- Ramos, B.P.; Arnsten, A.F. Adrenergic pharmacology and cognition: Focus on the prefrontal cortex. Pharmacol. Ther. 2007, 113, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, B.D.; Devilbiss, D.; Fleischer, D.; Sessler, F.M.; Simpson, K.L. New Perspectives on the Functional Organi-zation and Postsynaptic Influences of the Locus Ceruleus Efferent Projection System. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 1997; Volume 42, pp. 749–754. [Google Scholar]
- Sirviö, J.; MacDonald, E. Central α1-adrenoceptors: Their role in the modulation of attention and memory formation. Pharmacol. Ther. 1999, 83, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Goldman-Rakic, P.; Lidow, M.; Gallager, D. Overlap of dopaminergic, adrenergic, and serotoninergic receptors and com-plementarity of their subtypes in primate prefrontal cortex. J. Neurosci. 1990, 10, 2125–2138. [Google Scholar] [CrossRef]
- Minneman, K.P.; Pittman, R.N.; Molinoff, P.B. Beta-Adrenergic Receptor Subtypes: Properties, Distribution, and Regulation. Annu. Rev. Neurosci. 1981, 4, 419–461. [Google Scholar] [CrossRef]
- Tully, K.; Bolshakov, V.Y. Emotional enhancement of memory: How norepinephrine enables synaptic plasticity. Mol. Brain 2010, 3, 15. [Google Scholar] [CrossRef]
- Murugaiah, K.; O’Donnell, J. Beta adrenergic receptors facilitate norepinephrine release from rat hypothalamic and hip-pocampal slices. Res. Commun. Mol. Pathol. Pharmacol. 1995, 90, 179–190. [Google Scholar]
- Samuels, E.R.; Szabadi, E.R.S.A.E. Functional Neuroanatomy of the Noradrenergic Locus Coeruleus: Its Roles in the Regulation of Arousal and Autonomic Function Part I: Principles of Functional Organisation. Curr. Neuropharmacol. 2008, 6, 235–253. [Google Scholar] [CrossRef]
- Sara, S.J. Locus Coeruleus in time with the making of memories. Curr. Opin. Neurobiol. 2015, 35, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.W. Norepinephrine and dopamine as learning signals. Neural Plast. 2004, 11, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Roozendaal, B.; Mcgaugh, J. Memory Modulation. Behav. Neurosci. 2011, 125, 797–824. [Google Scholar] [CrossRef] [PubMed]
- Ferry, B.; Parrot, S.; Marien, M.; Lazarus, C.; Cassel, J.-C.; McGaugh, J.L. Noradrenergic influences in the basolateral amygdala on inhibitory avoidance memory are mediated by an action on α2-adrenoceptors. Psychoneuroendocrinology 2014, 51, 68–79. [Google Scholar] [CrossRef]
- Tanaka, T.; Yokoo, H.; Mizoguchi, K.; Yoshida, M.; Tsuda, A.; Tanaka, M. Noradrenaline release in the rat amygdala is increased by stress: Studies with intracerebral microdialysis. Brain Res. 1991, 544, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Galvez, R.; Mesches, M.H.; Mcgaugh, J.L. Norepinephrine Release in the Amygdala in Response to Footshock Stimula-tion. Neurobiol. Learn. Mem. 1996, 66, 253–257. [Google Scholar] [CrossRef]
- McIntyre, C.K.; Hatfield, T.; McGaugh, J.L. Amygdala norepinephrine levels after training predict inhibitory avoidance retention performance in rats: Amygdala norepinephrine predicts long-term memory. Eur. J. Neurosci. 2002, 16, 1223–1226. [Google Scholar] [CrossRef]
- McGaugh, J.L.; Introini-Collison, I.B.; Cahill, L.F.; Castellano, C.; Dalmaz, C.; Parent, M.B.; Williams, C.L. Neuromodulatory systems and memory storage: Role of the amygdala. Behav. Brain Res. 1993, 58, 81–90. [Google Scholar] [CrossRef]
- Van Stegeren, A.H. The role of the noradrenergic system in emotional memory. Acta Psychologica 2008, 127, 532–541. [Google Scholar] [CrossRef]
- McGaugh, J.L. The Amygdala Modulates the Consolidation of Memories of Emotionally Arousing Experiences. Annu. Rev. Neurosci. 2004, 27, 1–28. [Google Scholar] [CrossRef]
- Stanton, P.; Sarvey, J. Depletion of norepinephrine, but not serotonin, reduces long-term potentiation in the dentate gy-rus of rat hippocampal slices. J. Neurosci. 1985, 5, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, W.; Johnston, D. Frequency-dependent noradrenergic modulation of long-term potentiation in the hippocam-pus. Science 1984, 226, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, W.F.; Johnston, D. Noradrenergic enhancement of long-term potentiation at mossy fiber synapses in the hip-pocampus. J. Neurophysiol. 1988, 59, 667–687. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, T.J.; Connor, S.A.; Gelinas, J.N.; Nguyen, P.V. Viagra for your synapses: Enhancement of hippocampal long-term potentiation by activation of beta-adrenergic receptors. Cell. Signal. 2010, 22, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Salgado, H.; Treviño, M.; Atzori, M. Layer- and area-specific actions of norepinephrine on cortical synaptic transmission. Brain Res. 2016, 1641, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Ito, M. Long-Term Depression: Cerebellum. In Encyclopedia of Neuroscience; Squire, L.R., Ed.; Academic Press: Cambridge, MA, USA, 2009; pp. 533–539. [Google Scholar] [CrossRef]
- Trevino, M.; Frey, S.; Kohr, G. Alpha-1 Adrenergic Receptors Gate Rapid Orientation-Specific Reduction in Visual Dis-crimination. Cerebral Cortex 2012, 22, 2529–2541. [Google Scholar] [CrossRef]
- Scheiderer, C.L.; Dobrunz, L.E.; McMahon, L.L. Novel Form of Long-Term Synaptic Depression in Rat Hippocampus Induced by Activation of α1 Adrenergic Receptors. J. Neurophysiol. 2004, 91, 1071–1077. [Google Scholar] [CrossRef]
- Marien, M.R.; Colpaert, F.C.; Rosenquist, A.C. Noradrenergic mechanisms in neurodegenerative diseases: A theory. Brain Res. Rev. 2004, 45, 38–78. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2010, 121, 171–181. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Alzheimer’s pathogenesis: Is there neuron-to-neuron propagation? Acta Neuropathol. 2011, 121, 589–595. [Google Scholar] [CrossRef]
- Grudzien, A.; Shaw, P.; Weintraub, S.; Bigio, E.; Mash, D.C.; Mesulam, M.M. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alz-heimer’s disease. Neurobiol. Aging 2007, 28, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Bondareff, W.; Mountjoy, C.Q.; Roth, M.; Rossor, M.N.; Iversen, L.L.; Reynolds, G.P.; Hauser, D.L. Neuronal Degeneration in Locus Ceruleus and Cortical Correlates of Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 1987, 1, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Matthews, K.L.; Chen, C.P.-H.; Esiri, M.M.; Keene, J.; Minger, S.L.; Francis, P.T. Noradrenergic changes, aggressive behavior, and cognition in patients with dementia. Biol. Psychiatry 2002, 51, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M.; Wilcock, G.K.; Esiri, M.M.; Francis, P.T.; Bowen, D.M. Monoaminergic innervation of the frontal and temporal lobes in Alzheimer’s disease. Brain Res. 1987, 401, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M.A.; Lincoln, J.; Yates, P.O.; Stamp, J.E.; Toper, S. Changes in the Monoamine Containing Neurones of the Human Cns in Senile Dementia. Br. J. Psychiatry 1980, 136, 533–541. [Google Scholar] [CrossRef]
- Adolfsson, R.; Gottfries, C.G.; Roos, B.E.; Winblad, B.; Gartner, J.; Langford, A.; O’Brien, A.; Hosang, G.M.; Fisher, H.L.; Hodgson, K.; et al. Changes in the Brain Catecholamines in Patients with Dementia of Alzheimer Type. Br. J. Psychiatry 1979, 135, 216–223. [Google Scholar] [CrossRef]
- Šimic, G.; Leko, M.B.; Wray, S.; Harrington, C.R.; Delalle, I.; Jovanov-Milosevic, N.; Bavzadona, D.; Buee, L.; De Silva, R.; Di Giovanni, G.; et al. Monoaminergic neuropathology in Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 101–138. [Google Scholar] [CrossRef]
- Manaye, K.F.; McIntire, D.D.; Mann, D.M.A.; German, D.C. Locus coeruleus cell loss in the aging human brain: A non-random process. J. Comp. Neurol. 1995, 358, 79–87. [Google Scholar] [CrossRef]
- Gulyás, B.; Brockschnieder, D.; Nag, S.; Pavlova, E.; Kasa, P.; Beliczai, Z.; Legradi, A.; Gulya, K.; Thiele, A.; Dyrks, T.; et al. The norepinephrine transporter (NET) radioligand (S,S)-[18F]FMeNER-D2 shows significant decreases in NET density in the human brain in Alzheimer’s disease: A post-mortem autoradiographic study. Neurochem. Int. 2010, 56, 789–798. [Google Scholar] [CrossRef]
- Gannon, M.; Wang, Q. Complex noradrenergic dysfunction in Alzheimer’s disease: Low norepinephrine input is not always to blame. Brain Res. 2019, 1702, 12–16. [Google Scholar] [CrossRef]
- Vincent, S. Norepinephrine Transporter. In Encyclopedia of Endocrine Diseases; Martini, L., Ed.; Elsevier: Amsterdam, The Netherlands, 2004; pp. 382–386. [Google Scholar] [CrossRef]
- Zhou, J. Norepinephrine transporter inhibitors and their therapeutic potential. Drugs Future 2004, 29, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Jardanhazi-Kurutz, D.; Kummer, M.; Terwel, D.; Vogel, K.; Dyrks, T.; Thiele, A.; Heneka, M.T. Induced LC degeneration in APP/PS1 transgenic mice accelerates early cerebral amyloidosis and cognitive deficits. Neurochem. Int. 2010, 57, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.B.; Stenfors, C. DSP4, a Selective Neurotoxin for the Locus Coeruleus Noradrenergic System. A Review of Its Mode of Action. Neurotox. Res. 2014, 27, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T. Locus Ceruleus Degeneration Promotes Alzheimer Pathogenesis in Amyloid Precursor Protein 23 Trans-genic Mice. J. Neurosci. 2006, 26, 1343–1354. [Google Scholar] [CrossRef]
- Kummer, M.P.; Hermes, M.; Delekarte, A.; Hammerschmidt, T.; Kumar, S.; Terwel, D.; Walter, J.; Pape, H.-C.; König, S.; Roeber, S.; et al. Nitration of Tyrosine 10 Critically Enhances Amyloid β Aggregation and Plaque Formation. Neuron 2011, 71, 833–844. [Google Scholar] [CrossRef]
- Heneka, M.T.; Nadrigny, F.; Regen, T.; Martinez-Hernandez, A.; Dumitrescu-Ozimek, L.; Terwel, D.; Jardanhazi-Kurutz, D.; Walter, J.; Kirchhoff, F.; Hanisch, U.-K.; et al. Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proc. Natl. Acad. Sci. USA 2010, 107, 6058–6063. [Google Scholar] [CrossRef]
- Kummer, M.P.; Hammerschmidt, T.; Martinez, A.; Terwel, D.; Eichele, G.; Witten, A.; Figura, S.; Stoll, M.; Schwartz, S.; Pape, H.-C.; et al. Ear2 Deletion Causes Early Memory and Learning Deficits in APP/PS1 Mice. J. Neurosci. 2014, 34, 8845–8854. [Google Scholar] [CrossRef]
- Chalermpalanupap, T.; Schroeder, J.P.; Rorabaugh, J.M.; Liles, L.C.; Lah, J.J.; Levey, A.I.; Weinshenker, D. Locus Coeruleus Ablation Exacerbates Cognitive Deficits, Neuropathology, and Lethality in P301S Tau Transgenic Mice. J. Neurosci. 2017, 38, 74–92. [Google Scholar] [CrossRef]
- Szot, P.; Miguelez, C.; White, S.; Franklin, A.; Sikkema, C.; Wilkinson, C.; Ugedo, L.; Raskind, M. A comprehensive analysis of the effect of DSP4 on the locus coeruleus noradrenergic system in the rat. Neuroscience 2010, 166, 279–291. [Google Scholar] [CrossRef]
- Weinshenker, D.; Miller, N.S.; Blizinsky, K.; Laughlin, M.L.; Palmiter, R.D. Mice with chronic norepinephrine deficiency resemble amphetamine-sensitized animals. Proc. Natl. Acad. Sci. USA 2002, 99, 13873–13877. [Google Scholar] [CrossRef]
- Warnecke, M.; Oster, H.; Revelli, J.-P.; Alvarez-Bolado, G.; Eichele, G. Abnormal development of the locus coeruleus in Ear2(Nr2f6)-deficient mice impairs the functionality of the forebrain clock and affects nociception. Genes Dev. 2005, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Dumitrescu-Ozimek, L.; Klockgether, T.; Feinstein, D.L.; Galea, E.; Gavrilyak, V. Noradrenergic depletion potentiates beta -amyloid-induced cortical inflammation: Implications for Alzheimer’s disease. J. Neurosci. 2002, 22, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Gavrilyuk, V.; Landreth, G.E.; O’Banion, M.K.; Weinberg, G.; Feinstein, D.L. Noradrenergic depletion increases inflammatory responses in brain: Effects on IκB and HSP70 expres-sion: PPARγ agonist reduce brain inflammatory responses. J. Neurochem. 2003, 85, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Pugh, P.L.; Vidgeon-Hart, M.P.; Ashmeade, T.; Culbert, A.A.; Seymour, Z.; Perren, M.J.; Joyce, F.; Bate, S.T.; Babin, A.; Virley, D.J.; et al. Repeated administration of the noradrenergic neurotoxin N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4) modulates neuroinflammation and amyloid plaque load in mice bearing amyloid precursor protein and prese-nilin-1 mutant transgenes. J. Neuroinflammation 2007, 4, 8. [Google Scholar] [CrossRef]
- Kalinin, S.; Gavrilyuk, V.; Polak, P.E.; Vasser, R.; Zhao, J.; Heneka, M.T.; Feinstein, D.L. Noradrenaline deficiency in brain increases β-amyloid plaque burden in an animal model of Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1206–1214. [Google Scholar] [CrossRef]
- Jardanhazi-Kurutz, D.; Kummer, M.P.; Terwel, D.; Vogel, K.; Thiele, A.; Heneka, M.T. Distinct adrenergic system changes and neuroinflammation in response to induced locus ce-ruleus degeneration in APP/PS1 transgenic mice. Neuroscience 2011, 176, 396–407. [Google Scholar] [CrossRef]
- Rey, N.L.; Jardanhazi-Kurutz, D.; Terwel, D.; Kummer, M.P.; Jourdan, F.; Didier, A.; Heneka, M.T. Locus coeruleus degeneration exacerbates olfactory deficits in APP/PS1 transgenic mice. Neurobiol. Aging 2012, 33, 426.e1–426.e11. [Google Scholar] [CrossRef]
- Kang, S.S.; Liu, X.; Ahn, E.H.; Xiang, J.; Manfredsson, F.P.; Yang, X.; Luo, H.R.; Liles, L.C.; Weinshenker, D.; Ye, K. Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. J. Clin. Investig. 2019, 130, 422–437. [Google Scholar] [CrossRef]
- Liu, L.; Luo, S.; Zeng, L.; Wang, W.; Yuan, L.; Jian, X. Degenerative alterations in noradrenergic neurons of the locus coeruleus in Alzheimer’s disease. Neural Regen. Res. 2013, 8, 2249–2255. [Google Scholar] [CrossRef]
- Braun, D.J.; Kalinin, S.; Feinstein, D.L. Conditional Depletion of Hippocampal Brain-Derived Neurotrophic Factor Exac-erbates Neuropathology in a Mouse Model of Alzheimer’s Disease. ASN Neuro 2017, 9, 1–14. [Google Scholar] [CrossRef]
- Cao, S.; Fisher, D.W.; Rodriguez, G.; Yu, T.; Dong, H. Comparisons of neuroinflammation, microglial activation, and degeneration of the locus coeruleus-norepinephrine system in APP/PS1 and aging mice. J. Neuroinflammation 2021, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rho, H.-J.; Kim, J.-H.; Lee, S.-H. Function of Selective Neuromodulatory Projections in the Mammalian Cerebral Cortex: Comparison Between Cholinergic and Noradrenergic Systems. Front. Neural Circuits 2018, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.; Zeppenfeld, D.; McConnell, E.; Pena, S.; Nedergaard, M. Norepinephrine: A Neuromodulator That Boosts the Function of Multiple Cell Types to Optimize CNS Performance. Neurochem. Res. 2012, 37, 2496–2512. [Google Scholar] [CrossRef]
- Feinstein, D.L.; Galea, E.; Reis, D.J. Norepinephrine Suppresses Inducible Nitric Oxide Synthase Activity in Rat Astroglial Cultures. J. Neurochem. 1993, 60, 1945–1948. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.; Madrigal, J.L.M.; Feinstein, U.L. Noradrenergic Regulation of Glial Activation: Molecular Mechanisms and Therapeutic Implications. Curr. Neuropharmacol. 2014, 12, 342–352. [Google Scholar] [CrossRef]
- Russo, C.D.; Boullerne, A.I.; Gavrilyuk, V.; Feinstein, D.L. Inhibition of microglial inflammatory responses by norepinephrine: Effects on nitric oxide and interleukin-1β production. J. Neuroinflammation 2004, 1, 9. [Google Scholar] [CrossRef][Green Version]
- Feinstein, D.L.; Heneka, M.T.; Gavrilyuk, V.; Russo, C.D.; Weinberg, G.; Galea, E. Noradrenergic regulation of inflammatory gene expression in brain. Neurochem. Int. 2002, 41, 357–365. [Google Scholar] [CrossRef]
- Mori, K.; Ozaki, E.; Zhang, B.; Yang, L.; Yokoyama, A.; Takeda, I.; Maeda, N.; Sakanaka, M.; Tanaka, J. Effects of norepinephrine on rat cultured microglial cells that express α1, α2, β1 and β2 adrenergic receptors. Neuropharmacology 2002, 43, 1026–1034. [Google Scholar] [CrossRef]
- Madrigal, J.L.; Dello Russo, C.; Gavrilyuk, V.; Feinstein, D.L. Effects of Noradrenaline on Neuronal NOS2 Expression and Viability. Antioxid. Redox Signal. 2006, 8, 885–892. [Google Scholar] [CrossRef]
- Evans, A.K.; Ardestani, P.M.; Yi, B.; Park, H.H.; Lam, R.K.; Shamloo, M. Beta-adrenergic receptor antagonism is proinflammatory and exacerbates neuroinflammation in a mouse model of Alzheimer’s Disease. Neurobiol. Dis. 2020, 146, 105089. [Google Scholar] [CrossRef]
- Liu, Y.U.; Ying, Y.; Li, Y.; Eyo, U.B.; Chen, T.; Zheng, J.; Umpierre, A.D.; Zhu, J.; Bosco, D.B.; Dong, H.; et al. Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nat. Neurosci. 2019, 22, 1771–1781. [Google Scholar] [CrossRef] [PubMed]
- Stowell, R.D.; Sipe, G.O.; Dawes, R.P.; Batchelor, H.N.; Lordy, K.A.; Whitelaw, B.S.; Stoessel, M.B.; Bidlack, J.M.; Brown, E.; Sur, M.; et al. Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nat. Neurosci. 2019, 22, 1782–1792. [Google Scholar] [CrossRef] [PubMed]
- Mercan, D.; Heneka, M.T. Norepinephrine as a modulator of microglial dynamics. Nat. Neurosci. 2019, 22, 1745–1746. [Google Scholar] [CrossRef] [PubMed]
- David, M.C.B.; Del Giovane, M.; Liu, K.Y.; Gostick, B.; Rowe, J.B.; Oboh, I.; Howard, R.; Malhotra, P. Cognitive and neuropsychiatric effects of noradrenergic treatment in Alzheimer’s disease: Systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2022, 93, 1080–1090. [Google Scholar] [CrossRef]
- Al-Majed, A.; Bakheit, A.H.; Alharbi, R.M.; Abdel Aziz, H.A. Chapter Two—Mirtazapine. In Profiles of Drug Substances, Excipients and Related Methodology; Brittain, H.G., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 43, pp. 209–254. [Google Scholar]
- Correia, A.S.; Vale, N. Antidepressants in Alzheimer’s Disease: A Focus on the Role of Mirtazapine. Pharmaceuticals 2021, 14, 930. [Google Scholar] [CrossRef]
- Raji, M.A.; Brady, S.R. Mirtazapine for Treatment of Depression and Comorbidities in Alzheimer Disease. Ann. Pharmacother. 2001, 35, 1024–1027. [Google Scholar] [CrossRef]
- Cakir, S.; Kulaksizoglu, I.B. The efficacy of mirtazapine in agitated patients with Alzheimer’s disease: A 12-week open-label pilot study. Neuropsychiatr. Dis. Treat. 2008, 4, 963–966. [Google Scholar] [CrossRef]
- Banerjee, S.; Hellier, J.; Dewey, M.; Romeo, R.; Ballard, C.; Baldwin, R.; Bentham, P.; Fox, C.; Holmes, C.; Katona, C.; et al. Sertraline or mirtazapine for depression in dementia (HTA-SADD): A randomised, multicentre, double-blind, placebo-controlled trial. Lancet 2011, 378, 403–411. [Google Scholar] [CrossRef]
- Banerjee, S.; Banerjee, S.; High, J.; Stirling, S.; Shepstone, L.; Swart, A.M.; Telling, T.; Henderson, C.; Ballard, C.; Bentham, P.; et al. Study of mirtazapine for agitated behaviours in dementia (SYMBAD): A randomised, double-blind, place-bo-controlled trial. Lancet 2021, 398, 1487–1497. [Google Scholar] [CrossRef]
- Manzoor, S.; Hoda, N. A comprehensive review of monoamine oxidase inhibitors as Anti-Alzheimer’s disease agents: A review. Eur. J. Med. Chem. 2020, 206, 112787. [Google Scholar] [CrossRef]
- Emilsson, L.; Saetre, P.; Balciuniene, J.; Castensson, A.; Cairns, N.; Jazin, E.E. Increased monoamine oxidase messenger RNA expression levels in frontal cortex of Alzheimer’s disease patients. Neurosci. Lett. 2002, 326, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, D.; Sehgal, A.; Singh, S.; Sharma, N.; Zengin, G.; Andronie-Cioara, F.L.; Toma, M.M.; Bungau, S.; Bumbu, A.G.; et al. Role of Monoamine Oxidase Activity in Alzheimer’s Disease: An Insight into the Therapeutic Potential of In-hibitors. Molecules 2021, 26, 3724. [Google Scholar] [CrossRef] [PubMed]
- Gulyás, B.; Pavlova, E.; Kasa, P.; Gulya, K.; Bakota, L.; Varszegi, S.; Keller, E.; Horvath, M.C.; Nag, S.; Hermecz, I.; et al. Activated MAO-B in the brain of Alzheimer patients, demonstrated by [11C]-l-deprenyl using whole hem-isphere autoradiography. Neurochem. Int. 2011, 58, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E.; et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef]
- Magni, G.; Meibach, R. Lazabemide for the long-term treatment of Alzheimer’s disease. Eur. Neuropsychopharmacol. 1999, 9, 142. [Google Scholar] [CrossRef]
- Piccinin, G.L.; Finali, G.; Piccirilli, M. Neuropsychological effects of L-deprenyl in Alzheimer’s type dementia. Clin Neuropharmacol 1990, 13, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Bönisch, H.; Brüss, M. The Noradrenaline Transporter of the Neuronal Plasma Membranea. Ann. N. Y. Acad. Sci. 1994, 733, 193–202. [Google Scholar] [CrossRef]
- Wong, D.T.; Threlkeld, P.G.; Best, K.L.; Bymaster, F.P. A new inhibitor of norepinephrine uptake devoid of affinity for receptors in rat brain. J. Pharmacol. Exp. Ther. 1982, 222, 61–65. [Google Scholar]
- Bymaster, F.P.; Katner, J.S.; Nelson, D.L.; Hemrick-Luecke, S.K.; Threlkeld, P.G.; Heiligenstein, J.H.; Morin, S.M.; Gehlert, D.R.; Perry, K.W. Atomoxetine Increases Extracellular Levels of Norepinephrine and Dopamine in Prefrontal Cortex of Rat a Potential Mechanism for Efficacy in Attention Deficit/Hyperactivity Disorder. Neuropsychopharmacology 2002, 27, 699–711. [Google Scholar] [CrossRef]
- Kielbasa, W.; Kalvass, J.C.; Stratford, R. Microdialysis Evaluation of Atomoxetine Brain Penetration and Central Nervous System Pharmacokinetics in Rats. Drug Metab. Dispos. 2008, 37, 137–142. [Google Scholar] [CrossRef]
- Mohs, R.C.; Shiovitz, T.M.; Tariot, P.N.; Porsteinsson, A.P.; Baker, K.D.; Feldman, P.D. Atomoxetine Augmentation of Cholinesterase Inhibitor Therapy in Patients With Alzheimer Disease: 6-Month, Randomized, Double-Blind, Placebo-Controlled, Parallel-Trial Study. Am. J. Geriatr. Psychiatry 2009, 17, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.I.; Qiu, D.; Zhao, L.; Hu, W.T.; Duong, D.M.; Higginbotham, L.; Dammer, E.B.; Seyfried, N.T.; Wingo, T.S.; Hales, C.M.; et al. A phase II study repurposing atomoxetine for neuroprotection in mild cognitive impairment. Brain 2022, 145, 1924–1938. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Caballero, M.; Warming, H.; Walker, R.; Holmes, C.; Cruickshank, G.; Patel, B. Vagus Nerve Stimulation as a Potential Therapy in Early Alzheimer’s Disease: A Review. Front. Hum. Neurosci. 2022, 16, e866434. [Google Scholar] [CrossRef] [PubMed]
- Kenny, B.J.; Bordoni, B. Neuroanatomy, Cranial Nerve 10 (Vagus Nerve). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Rosas-Ballina, M.; Olofsson, P.S.; Ochani, M.; Valdés-Ferrer, S.I.; Levine, Y.A.; Reardon, C.; Tusche, M.W.; Pavlov, V.A.; Andersson, U.; Chavan, S.; et al. Acetylcholine-Synthesizing T Cells Relay Neural Signals in a Vagus Nerve Circuit. Science 2011, 334, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus Nerve as Modulator of the Brain-Gut Axis in Psychiatric and Inflammatory Disorders. Front Psychiatry 2018, 9, 44. [Google Scholar] [CrossRef]
- Hulsey, D.R.; Riley, J.R.; Loerwald, K.W.; Rennaker II, R.L.; Kilgard, M.P.; Hays, S.A. Parametric characterization of neural activity in the locus coeruleus in response to vagus nerve stimula-tion. Exp. Neurol. 2017, 289, 21–30. [Google Scholar] [CrossRef]
- Pisapia, J.; Baltuch, G. Vagus nerve stimulation. In Neuromodulation in Psychiatry; John Wiley: Hoboken, NJ, USA, 2015; pp. 325–334. [Google Scholar] [CrossRef]
- Roosevelt, R.W.; Smith, D.C.; Clough, R.W.; Jensen, R.A.; Browning, R.A. Increased extracellular concentrations of norepinephrine in cortex and hippocampus following vagus nerve stimulation in the rat. Brain Res. 2006, 1119, 124–132. [Google Scholar] [CrossRef]
- Manta, S.; Dong, J.; Debonnel, G.; Blier, P. Enhancement of the function of rat serotonin and norepinephrine neurons by sustained vagus nerve stimulation. J. Psychiatry Neurosci. 2009, 34, 272–280. [Google Scholar]
- Follesa, P.; Biggio, F.; Gorini, G.; Caria, S.; Talani, G.; Dazzi, L.; Puligheddu, M.; Marrosu, F.; Biggio, G. Vagus nerve stimulation increases norepinephrine concentration and the gene expression of BDNF and bFGF in the rat brain. Brain Res. 2007, 1179, 28–34. [Google Scholar] [CrossRef]
- Sun, L.; Peräkylä, J.; Holm, K.; Haapasalo, J.; Lehtimäki, K.; Ogawa, K.H.; Peltola, J.; Hartikainen, K.M. Vagus nerve stimulation improves working memory performance. J. Clin. Exp. Neuropsychol. 2017, 39, 954–964. [Google Scholar] [CrossRef]
- Jacobs, H.I.L.; Priovoulos, N.; Riphagen, J.M.; Poser, B.A.; Napadow, V.; Uludag, K.; Sclocco, R.; Ivanov, D.; Verhey, F.R.J. Transcutaneous vagus nerve stimulation increases locus coeruleus function and memory performance in older individuals: Featured research and focused topic sessions: Interventions targeting the noradrenergic system in Alzheimer’s and neurodegenerative disease. Alzheimer’s Dement. 2020, 16, e044766. [Google Scholar]
- Sjögren, M.J.C. Cognition-Enhancing Effect of Vagus Nerve Stimulation in Patients with Alzheimer’s Disease: A Pilot Study. J. Clin. Psychiatry 2002, 63, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Merrill, C.A.; Jonsson, M.A.G.; Minthon, L.; Ejnell, H.; Silander, H.C.-s.; Blennow, K.; Karlsson, M.; Nordlund, A.; Rolstad, S.; Warkentin, S.; et al. Vagus Nerve Stimulation in Patients with Alzheimer’s Disease: Additional Follow-Up Results of a Pilot Study Through 1 Year. J. Clin. Psychiatry 2006, 67, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Hoang, K.; Watt, H.; Golemme, M.; Perry, R.J.; Ritchie, C.; Wilson, D.; Pickett, J.; Fox, C.; Howard, R.; Malhotra, P.A. Noradrenergic Add-on Therapy with Extended-Release Guanfacine in Alzheimer’s Disease (NorAD): Study protocol for a randomised clinical trial and COVID-19 amendments. Trials 2022, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Noradrenergic Add-on Therapy with Guanfacine (NorAD). Available online: https://clinicaltrials.gov/ct2/show/NCT03116126 (accessed on 14 November 2022).
- David, M.; Malhotra, P.A. New approaches for the quantification and targeting of noradrenergic dysfunction in Alz-heimer’s disease. Ann. Clin. Transl. Neurol. 2022, 9, 582–596. [Google Scholar] [CrossRef] [PubMed]
- Implantable Vagus Nerve Stimulation Modulation of Coeruleus-Norepinephrine Network for Mild-Moderate AD Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT05575271 (accessed on 14 November 2022).
- Modulating the Locus Coeruleus Function. Available online: https://clinicaltrials.gov/ct2/show/NCT04877782 (accessed on 14 November 2022).
- The Wandering Nerve: Gateway to Boost Alzheimer’s Disease Related Cognitive Performance (WALLe). Available online: https://clinicaltrials.gov/ct2/show/NCT04908358 (accessed on 14 November 2022).





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mercan, D.; Heneka, M.T. The Contribution of the Locus Coeruleus–Noradrenaline System Degeneration during the Progression of Alzheimer’s Disease. Biology 2022, 11, 1822. https://doi.org/10.3390/biology11121822
Mercan D, Heneka MT. The Contribution of the Locus Coeruleus–Noradrenaline System Degeneration during the Progression of Alzheimer’s Disease. Biology. 2022; 11(12):1822. https://doi.org/10.3390/biology11121822
Chicago/Turabian StyleMercan, Dilek, and Michael Thomas Heneka. 2022. "The Contribution of the Locus Coeruleus–Noradrenaline System Degeneration during the Progression of Alzheimer’s Disease" Biology 11, no. 12: 1822. https://doi.org/10.3390/biology11121822
APA StyleMercan, D., & Heneka, M. T. (2022). The Contribution of the Locus Coeruleus–Noradrenaline System Degeneration during the Progression of Alzheimer’s Disease. Biology, 11(12), 1822. https://doi.org/10.3390/biology11121822