
Nlrp3 Deficiency Alleviates Lipopolysaccharide-Induced Acute Kidney Injury via Suppressing Renal Inflammation and Ferroptosis in Mice

 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
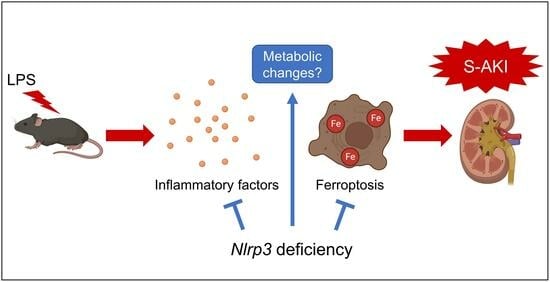
1. Introduction
2. Materials and Methods
2.1. Human Kidney Samples
2.2. Animals and S-AKI Model
2.3. Histological Assessment
2.4. Prussian Blue Staining
2.5. Immunohistochemistry (IHC)
2.6. Real-Time Quantitative PCR (qPCR)
2.7. Laboratory Examination
2.8. RNA Sequencing and Bioinformatics Analysis
2.9. Statistical Analysis
3. Results
3.1. Patients with S-AKI Have an Elevated Level of NLRP3 Expression in the Kidneys
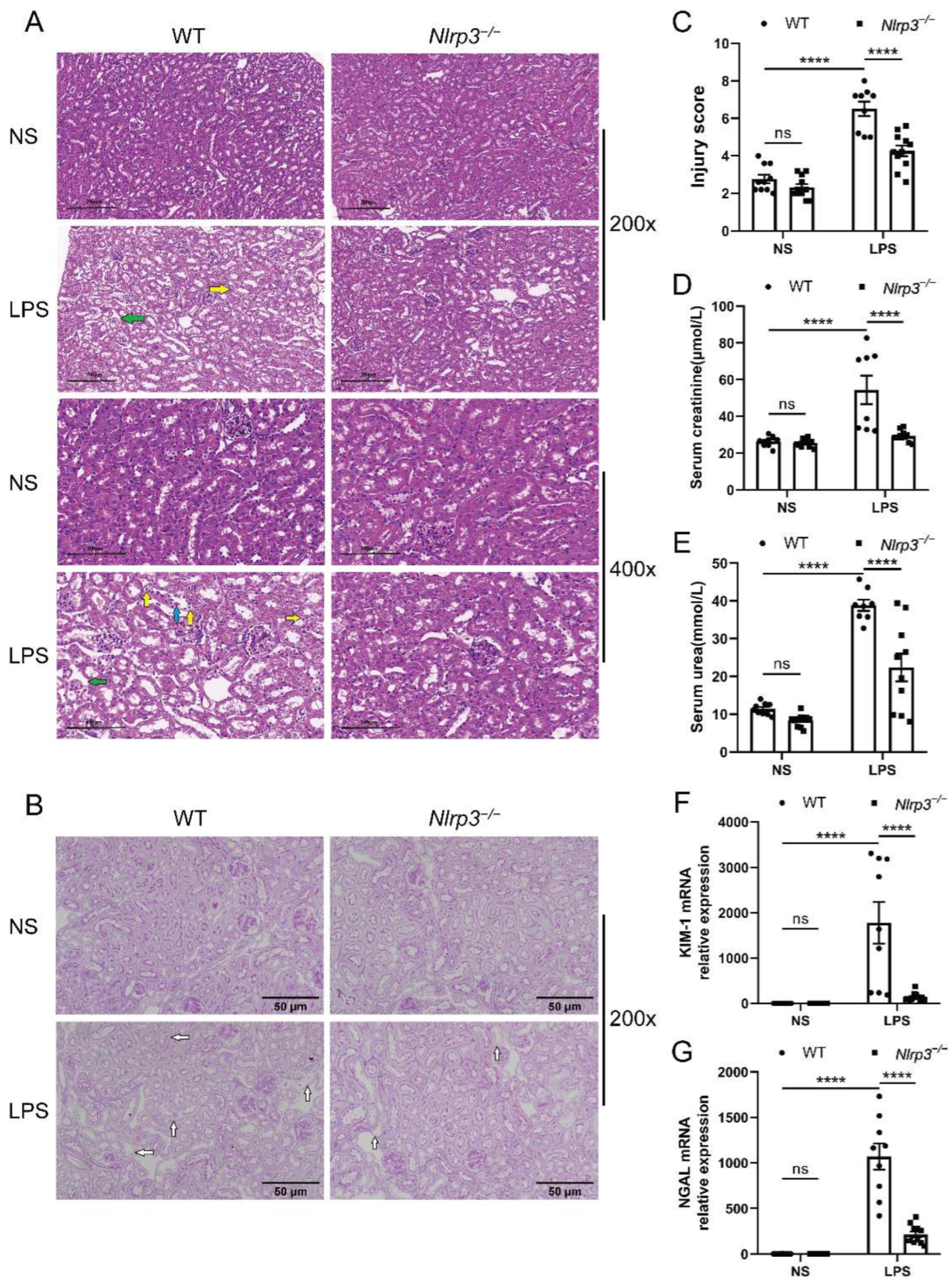
3.2. Nlrp3 Deficiency Relieves LPS-Induced Renal Injury in Mice
3.3. Nlrp3 Deficiency Alleviates Renal Inflammation in S-AKI Mice Induced by LPS
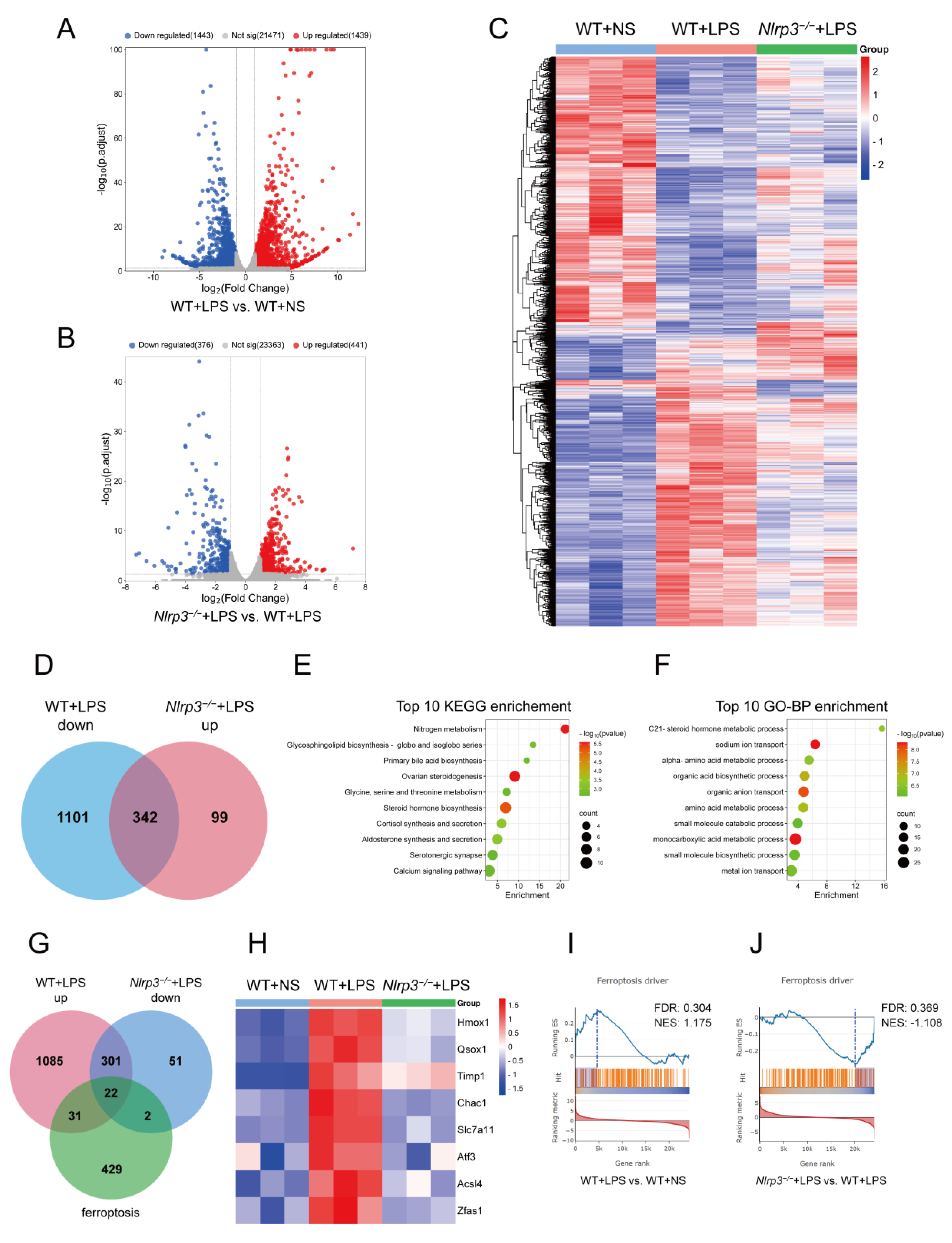
3.4. Nlrp3 Deficiency Affects Multiple Metabolic Pathways and Ferroptosis Pathways in S-AKI In Vivo
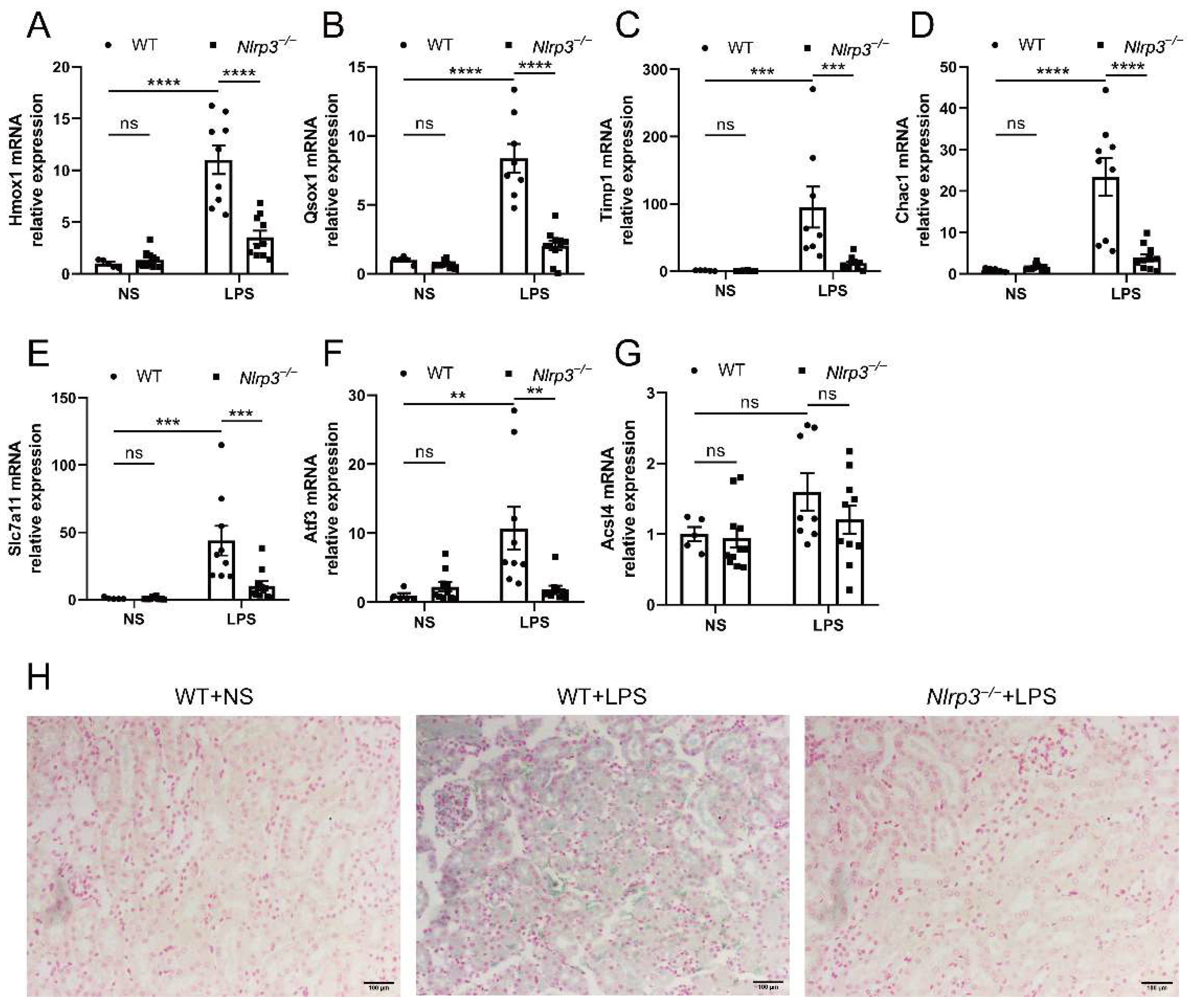
3.5. Nlrp3 Knockout Attenuates Renal Ferroptosis in S-AKI Mice Induced by LPS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoste, E.A.; Bagshaw, S.M.; Bellomo, R.; Cely, C.M.; Colman, R.; Cruz, D.N.; Edipidis, K.; Forni, L.G.; Gomersall, C.D.; Govil, D.; et al. Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med. 2015, 41, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Bagshaw, S.; Lapinsky, S.; Dial, S.; Arabi, Y.; Dodek, P.; Wood, G.; Ellis, P.; Guzman, J.; Marshall, J.; Parrillo, J.; et al. Acute kidney injury in septic shock: Clinical outcomes and impact of duration of hypotension prior to initiation of antimicrobial therapy. Intensive Care Med. 2009, 35, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Kellum, J.A.; Chawla, L.S.; Keener, C.; Singbartl, K.; Palevsky, P.M.; Pike, F.L.; Yealy, D.M.; Huang, D.T.; Angus, D.C. The Effects of Alternative Resuscitation Strategies on Acute Kidney Injury in Patients with Septic Shock. Am. J. Respir. Crit. Care Med. 2016, 193, 281–287. [Google Scholar] [CrossRef]
- Poston, J.T.; Koyner, J.L. Sepsis associated acute kidney injury. BMJ 2019, 364, k4891. [Google Scholar] [CrossRef] [PubMed]
- Ohto, U. Activation and regulation mechanisms of NOD-like receptors based on structural biology. Front. Immunol. 2022, 13, 953530. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell. Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef]
- Dai, X.; Li, Q.; Li, T.; Huang, W.; Zeng, Z.; Yang, Y.; Duan, Z.; Wang, Y.; Ai, Y. The interaction between C/EBPβ and TFAM promotes acute kidney injury via regulating NLRP3 inflammasome-mediated pyroptosis. Mol. Immunol. 2020, 127, 136–145. [Google Scholar] [CrossRef]
- Liu, R.; Wang, S.; Li, M.; Ma, X.; Jia, X.; Bu, Y.; Sun, L.; Yu, K. An Inhibitor of DRP1 (Mdivi-1) Alleviates LPS-Induced Septic AKI by Inhibiting NLRP3 Inflammasome Activation. BioMed Res. Int. 2020, 2020, 2398420. [Google Scholar] [CrossRef]
- Huang, J.; Wei, S.; Peng, Z.; Xiao, Z.; Yang, Y.; Liu, J.; Zhang, B.; Li, W. Disulfiram attenuates lipopolysaccharide-induced acute kidney injury by suppressing oxidative stress and NLRP3 inflammasome activation in mice. J. Pharm. Pharmacol. 2022, 74, 259–267. [Google Scholar] [CrossRef]
- Li, T.; Sun, H.; Li, Y.; Su, L.; Jiang, J.; Liu, Y.; Jiang, N.; Huang, R.; Zhang, J.; Peng, Z. Downregulation of macrophage migration inhibitory factor attenuates NLRP3 inflammasome mediated pyroptosis in sepsis-induced AKI. Cell Death Discov. 2022, 8, 61. [Google Scholar] [CrossRef]
- Sun, J.; Ge, X.; Wang, Y.; Niu, L.; Tang, L.; Pan, S. USF2 knockdown downregulates THBS1 to inhibit the TGF-beta signaling pathway and reduce pyroptosis in sepsis-induced acute kidney injury. Pharmacol. Res. 2022, 176, 105962. [Google Scholar] [CrossRef]
- Huang, G.; Bao, J.; Shao, X.; Zhou, W.; Wu, B.; Ni, Z.; Wang, L. Inhibiting pannexin-1 alleviates sepsis-induced acute kidney injury via decreasing NLRP3 inflammasome activation and cell apoptosis. Life Sci. 2020, 254, 117791. [Google Scholar] [CrossRef]
- Cao, Y.; Fei, D.; Chen, M.; Sun, M.; Xu, J.; Kang, K.; Jiang, L.; Zhao, M. Role of the nucleotide-binding domain-like receptor protein 3 inflammasome in acute kidney injury. FEBS J. 2015, 282, 3799–3807. [Google Scholar] [CrossRef]
- Peerapornratana, S.; Manrique-Caballero, C.L.; Gomez, H.; Kellum, J.A. Acute kidney injury from sepsis: Current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019, 96, 1083–1099. [Google Scholar] [CrossRef]
- Zhou, J.; Qian, C.; Zhao, M.; Yu, X.; Kang, Y.; Ma, X.; Ai, Y.; Xu, Y.; Liu, D.; An, Y.; et al. Epidemiology and outcome of severe sepsis and septic shock in intensive care units in mainland China. PloS ONE 2014, 9, e107181. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.; Stevens, L.; Schmid, C.; Zhang, Y.; Castro, A.; Feldman, H.; Kusek, J.; Eggers, P.; Van Lente, F.; Greene, T.; et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Radford, M.; Donadio, J.; Bergstralh, E.; Grande, J. Predicting renal outcome in IgA nephropathy. J. Am. Soc. Nephrol. JASN 1997, 8, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.; Tanaseichuk, O.; Benner, C.; Chanda, S. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Yuan, X.; Du, Q.; Zhang, Z.; Shi, X.; Bao, J.; Ning, Y.; Peng, L. FerrDb V2: Update of the manually curated database of ferroptosis regulators and ferroptosis-disease associations. Nucleic Acids Res. 2023, 51, D571–D582. [Google Scholar] [CrossRef]
- Liang, N.N.; Zhao, Y.; Guo, Y.Y.; Zhang, Z.H.; Gao, L.; Yu, D.X.; Xu, D.X.; Xu, S. Mitochondria-derived reactive oxygen species are involved in renal cell ferroptosis during lipopolysaccharide-induced acute kidney injury. Int. Immunopharmacol. 2022, 107, 108687. [Google Scholar] [CrossRef]
- Xiao, J.; Yang, Q.; Zhang, Y.; Xu, H.; Ye, Y.; Li, L.; Yang, Y.; Jin, S. Maresin conjugates in tissue regeneration-1 suppresses ferroptosis in septic acute kidney injury. Cell Biosci. 2021, 11, 221. [Google Scholar] [CrossRef] [PubMed]
- Nemzek, J.; Hugunin, K.; Opp, M. Modeling sepsis in the laboratory: Merging sound science with animal well-being. Comp. Med. 2008, 58, 120–128. [Google Scholar] [PubMed]
- Kim, Y.; Kim, S.; Kim, K.; Lee, S.; Moon, J. The Role of Inflammasome-Dependent and Inflammasome-Independent NLRP3 in the Kidney. Cells 2019, 8, 1389. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Faubel, S.; Ljubanovic, D.; Mitra, A.; Falk, S.; Kim, J.; Tao, Y.; Soloviev, A.; Reznikov, L.; Dinarello, C.; et al. Endotoxemic acute renal failure is attenuated in caspase-1-deficient mice. Am. J. Physiology. Ren. Physiol. 2005, 288, F997–F1004. [Google Scholar] [CrossRef]
- Huang, W.; Wang, X.; Xie, F.; Zhang, H.; Liu, D. Serum NLRP3: A biomarker for identifying high-risk septic patients. Cytokine 2022, 149, 155725. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wu, Q.; Tang, H.; Chen, J.; Wu, Q.; Yuan, X.; Xiong, S.; Ye, Y.; Lv, H. NLRP3 Regulated CXCL12 Expression in Acute Neutrophilic Lung Injury. J. Inflamm. Res. 2020, 13, 377–386. [Google Scholar] [CrossRef]
- Haller, H.; Bertram, A.; Nadrowitz, F.; Menne, J. Monocyte chemoattractant protein-1 and the kidney. Curr. Opin. Nephrol. Hypertens. 2016, 25, 42–49. [Google Scholar] [CrossRef]
- Zimmerman, K.A.; Hopp, K.; Mrug, M. Role of chemokines, innate and adaptive immunity. Cell. Signal. 2020, 73, 109647. [Google Scholar] [CrossRef]
- Jia, P.; Xu, S.; Wang, X.; Wu, X.; Ren, T.; Zou, Z.; Zeng, Q.; Shen, B.; Ding, X. Chemokine CCL2 from proximal tubular epithelial cells contributes to sepsis-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 2022, 323, F107–F119. [Google Scholar] [CrossRef]
- Zhang, C.; Liao, Y.; Liu, Z.; Zeng, L.; Peng, Z.; Liao, J.; Yang, Z. Mapping the Multi-Organ miRNA-mRNA Regulatory Network in LPS-Mediated Endotoxemic Mice: Exploring the Shared Underlying Key Genes and Mechanisms. Front. Mol. Biosci. 2020, 7, 573327. [Google Scholar] [CrossRef]
- van der Rijt, S.; Leemans, J.; Florquin, S.; Houtkooper, R.; Tammaro, A. Immunometabolic rewiring of tubular epithelial cells in kidney disease. Nat. Rev. Nephrol. 2022, 18, 588–603. [Google Scholar] [CrossRef] [PubMed]
- Gómez, H. Reprogramming Metabolism to Enhance Kidney Tolerance during Sepsis: The Role of Fatty Acid Oxidation, Aerobic Glycolysis, and Epithelial De-Differentiation. Nephron 2023, 147, 31–34. [Google Scholar] [CrossRef]
- Olona, A.; Leishman, S.; Anand, P.K. The NLRP3 inflammasome: Regulation by metabolic signals. Trends Immunol. 2022, 43, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Vizuete, A.F.K.; Froes, F.; Seady, M.; Zanotto, C.; Bobermin, L.D.; Roginski, A.C.; Wajner, M.; Quincozes-Santos, A.; Goncalves, C.A. Early effects of LPS-induced neuroinflammation on the rat hippocampal glycolytic pathway. J. Neuroinflammation 2022, 19, 255. [Google Scholar] [CrossRef]
- Han, S.; He, Z.; Hu, X.; Li, X.; Zheng, K.; Huang, Y.; Xiao, P.; Xie, Q.; Ni, J.; Liu, Q. Inhibiting NLRP3 Inflammasome Activation by CY-09 Helps to Restore Cerebral Glucose Metabolism in 3xTg-AD Mice. Antioxidants 2023, 12, 722. [Google Scholar] [CrossRef] [PubMed]
- Gallego, P.; Castejon-Vega, B.; Del Campo, J.A.; Cordero, M.D. The Absence of NLRP3-inflammasome Modulates Hepatic Fibrosis Progression, Lipid Metabolism, and Inflammation in KO NLRP3 Mice during Aging. Cells 2020, 9, 2148. [Google Scholar] [CrossRef]
- Kurmi, K.; Haigis, M.C. Nitrogen Metabolism in Cancer and Immunity. Trends Cell Biol. 2020, 30, 408–424. [Google Scholar] [CrossRef]
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545. [Google Scholar] [CrossRef]
- Zhang, N.; Zhou, Z.; Huang, Y.; Wang, G.; Tang, Z.; Lu, J.; Wang, C.; Ni, X. Reduced hydrogen sulfide production contributes to adrenal insufficiency induced by hypoxia via modulation of NLRP3 inflammasome activation. Redox Rep. 2023, 28, 2163354. [Google Scholar] [CrossRef]
- Wang, C.; Liu, Y.; Duan, G.; Zhao, W.; Li, X.; Zhu, X.; Ni, X. CBS and CSE are critical for maintenance of mitochondrial function and glucocorticoid production in adrenal cortex. Antioxid. Redox Signal. 2014, 21, 2192–2207. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Duan, G.; Liu, Y.; Yu, Q.; Tang, X.; Zhao, W.; Li, X.; Zhu, X.; Ni, X. Overproduction of nitric oxide by endothelial cells and macrophages contributes to mitochondrial oxidative stress in adrenocortical cells and adrenal insufficiency during endotoxemia. Free. Radic. Biol. Med. 2015, 83, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.S.; Singh, S.P.; Schuster, M.; Grinenko, T.; Bornstein, S.R.; Kanczkowski, W. RNA-seq analysis of LPS-induced transcriptional changes and its possible implications for the adrenal gland dysregulation during sepsis. J. Steroid Biochem. Mol. Biol. 2019, 191, 105360. [Google Scholar] [CrossRef]
- Wu, Z.; Deng, J.; Zhou, H.; Tan, W.; Lin, L.; Yang, J. Programmed Cell Death in Sepsis Associated Acute Kidney Injury. Front. Med. 2022, 9, 883028. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wu, C.; Ba, D.; Wang, N.; Wang, Y.; Li, X.; Li, Q.; Zhao, G. Ferroptosis contribute to neonicotinoid imidacloprid-evoked pyroptosis by activating the HMGB1-RAGE/TLR4-NF-kappaB signaling pathway. Ecotoxicol. Environ. Saf. 2023, 253, 114655. [Google Scholar] [CrossRef]
- Cao, Z.; Qin, H.; Huang, Y.; Zhao, Y.; Chen, Z.; Hu, J.; Gao, Q. Crosstalk of pyroptosis, ferroptosis, and mitochondrial aldehyde dehydrogenase 2-related mechanisms in sepsis-induced lung injury in a mouse model. Bioengineered 2022, 13, 4810–4820. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Indicators | Control (n = 5) | S-AKI (n = 4) | p Value |
|---|---|---|---|
| Gender (number of female) | 2 | 3 | 0.5238 |
| Age (years) | 29.6 ± 20.2 | 49.8 ± 13.6 | 0.1753 |
| Serum creatinine (µmol/L) | 59.1 ± 15.6 | 357.2 ± 311.7 | 0.1017 |
| Serum urea (mmol/L) * | 5.0 ± 1.2 | 12.2 ± 4.8 | 0.0252 |
| eGFR (mL/min/1.73 m2) *** | 114.8 ± 12.0 | 25.8 ± 16.4 | 0.0003 |
| White blood cells (×109/L) | 8.6 ± 2.2 | 12.6 ± 4.5 | 0.1628 |
| NEUT% (%) | 57.4 ± 20.6 | 82.1 ± 5.3 | 0.0773 |
| C-reactive protein (mg/L) | 2.0 ± 1.6 | 101.0 ± 100.0 | 0.1039 |
| ESR (mm/h) | 57.0 ± 16.1 | 77.0 ± 43.0 | a |
| Procalcitonin (ng/mL) | <0.05 # | 6.8 ± 10.3 | |
| Urinary protein (g/24 h) | 5.3 ± 4.7 | 0.8 ± 0 | a |
| Urine protein-to-creatinine ratio (g/g) | 5.3 ± 3.7 | 2.1 ± 1.6 | a |
| Gene | Primer Sequences | |
|---|---|---|
| β-actin | Forward | 5′-CACTGTCGAGTCGCGTCC-3′ |
| Reverse | 5′-TCATCCATGGCGAACTGGTG-3′ | |
| KIM-1 | Forward | 5′-TTAGGTGCTAGGAGGAGACAA-3′ |
| Reverse | 5′-TATCACACCTGCAAATAGGACT-3′ | |
| NGAL | Forward | 5′-AATTACCCTGTATGGAAGAACC-3′ |
| Reverse | 5′-CAGAGAAGATGATGTTGTCGTC-3′ | |
| IL-6 | Forward | 5′-TAGTCCTTCCTACCCCAATTTCC-3′ |
| Reverse | 5′-TTGGTCCTTAGCCACTCCTTC-3′ | |
| TNF-α | Forward | 5′-CCTGTAGCCCACGTCGTAG-3′ |
| Reverse | 5′-GGGAGTAGACAAGGTACAACCC-3′ | |
| CCL2 | Forward | 5′-TTAAAAACCTGGATCGGAACCAA-3′ |
| Reverse | 5′-GCATTAGCTTCAGATTTACGGGT-3′ | |
| CCL5 | Forward | 5′-ATATGGCTCGGACACCACTC-3′ |
| Reverse | 5′-ACTGCAAGATTGGAGCACTT-3′ | |
| CXCL9 | Forward | 5′-GGAGTTCGAGGAACCCTAGTG-3′ |
| Reverse | 5′-GGGATTTGTAGTGGATCGTGC-3′ | |
| Hmox1 | Forward | 5′-AAGCCGAGAATGCTGAGTTCA-3′ |
| Reverse | 5′-GCCGTGTAGATATGGTACAAGGA-3′ | |
| Qsox1 | Forward | 5′-TGGCGCTAATGTGCAGACTC-3′ |
| Reverse | 5′-CACTGCCACAGCATGGTACT-3′ | |
| Timp1 | Forward | 5′-GCAACTCGGACCTGGTCATAA-3′ |
| Reverse | 5′-CGGCCCGTGATGAGAAACT-3′ | |
| Chac1 | Forward | 5′-CTTGGTGGCTATGACACTAAGG-3′ |
| Reverse | 5′-CCTCGGCAAGCAAGGATCTG-3′ | |
| Slc7a11 | Forward | 5′-GGCACCGTCATCGGATCAG-3′ |
| Reverse | 5′-CTCCACAGGCAGACCAGAAAA-3′ | |
| Atf3 | Forward | 5′-CCTCAGAAGTCAGTGCGACC-3′ |
| Reverse | 5′-CATCCGATGGCAGAGGTGTT-3′ | |
| Acsl4 | Forward | 5′-CTCACCATTATATTGCTGCCTGT-3′ |
| Reverse | 5′-TCTCTTTGCCATAGCGTTTTTCT-3′ | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Wang, X.; Peng, Y.; Yin, H.; Yu, S.; Zhang, W.; Ni, X. Nlrp3 Deficiency Alleviates Lipopolysaccharide-Induced Acute Kidney Injury via Suppressing Renal Inflammation and Ferroptosis in Mice. Biology 2023, 12, 1188. https://doi.org/10.3390/biology12091188
Li Z, Wang X, Peng Y, Yin H, Yu S, Zhang W, Ni X. Nlrp3 Deficiency Alleviates Lipopolysaccharide-Induced Acute Kidney Injury via Suppressing Renal Inflammation and Ferroptosis in Mice. Biology. 2023; 12(9):1188. https://doi.org/10.3390/biology12091188
Chicago/Turabian StyleLi, Zhilan, Xuan Wang, Yi Peng, Hongling Yin, Shenyi Yu, Weiru Zhang, and Xin Ni. 2023. "Nlrp3 Deficiency Alleviates Lipopolysaccharide-Induced Acute Kidney Injury via Suppressing Renal Inflammation and Ferroptosis in Mice" Biology 12, no. 9: 1188. https://doi.org/10.3390/biology12091188
APA StyleLi, Z., Wang, X., Peng, Y., Yin, H., Yu, S., Zhang, W., & Ni, X. (2023). Nlrp3 Deficiency Alleviates Lipopolysaccharide-Induced Acute Kidney Injury via Suppressing Renal Inflammation and Ferroptosis in Mice. Biology, 12(9), 1188. https://doi.org/10.3390/biology12091188