

Stress Factors as Possible Regulators of Pluripotent Stem Cell Survival and Differentiation

Abstract
Simple Summary
Abstract
1. Introduction
2. The Role of Stress Factors in Embryonic Growth and Proliferation
2.1. Hypoxic Stress
2.2. Osmotic Stress
2.3. Mechanical Stress
2.4. Oxidative Stress
3. Potential Stress Responses Mechanisms in PSCs
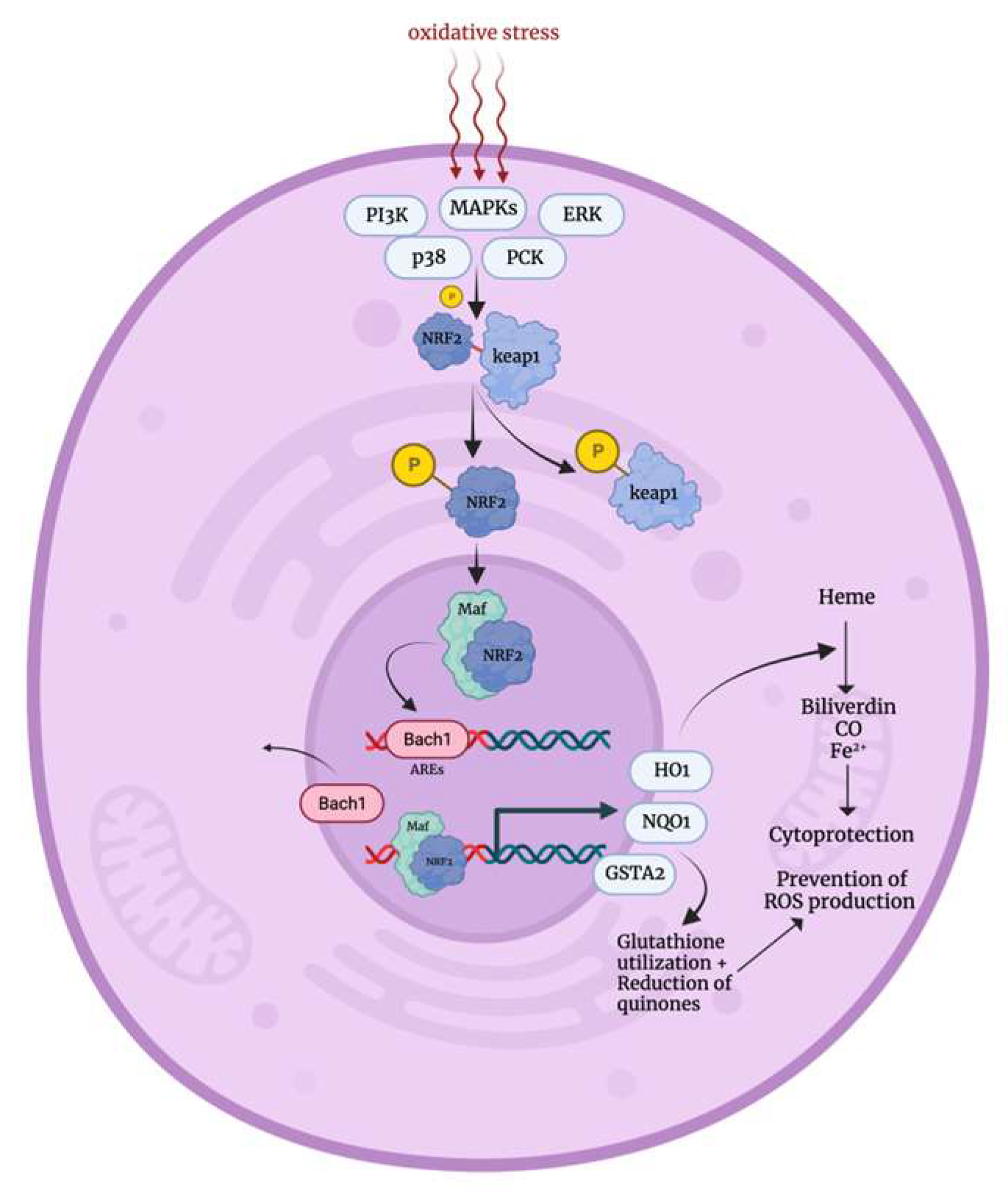
3.1. Oxidative Stress Response

3.2. Heat Shock Stress Response

3.3. Hypoxic Stress Response

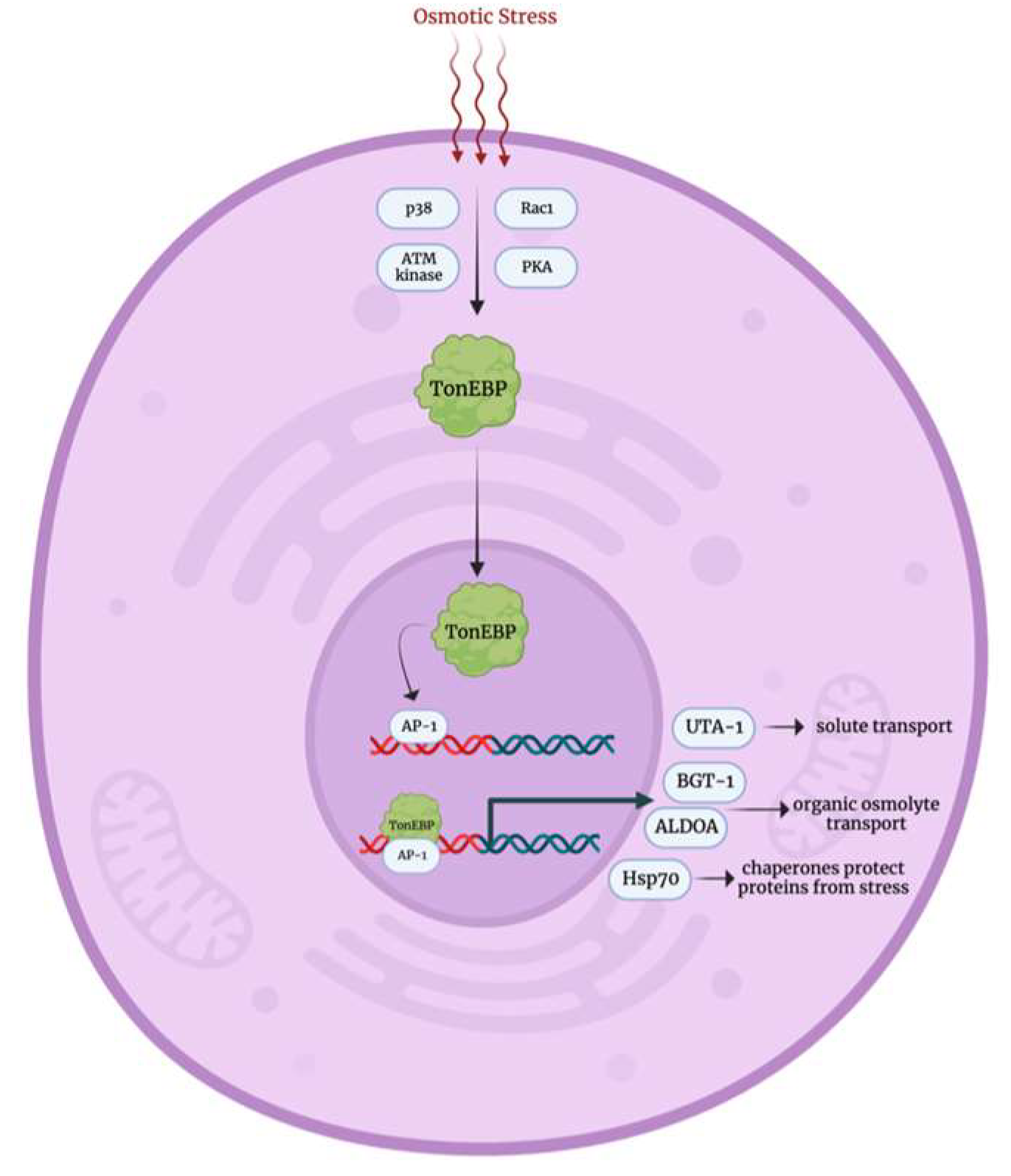
3.4. Osmotic Stress Response

3.5. DNA Damage p53 Mediated Stress Response

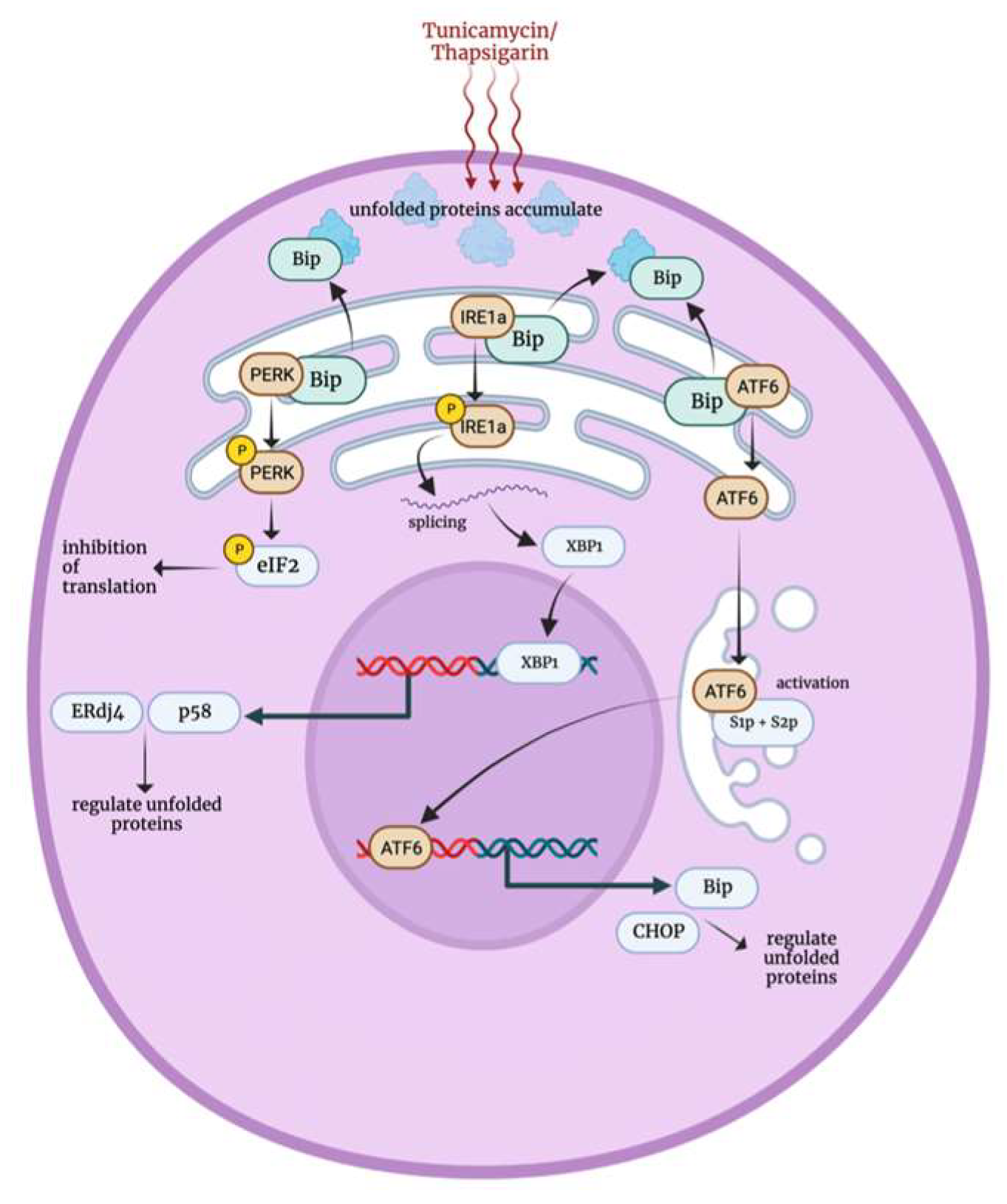
3.6. Endoplasmic Reticulum Stress Response

4. Stress Factors Role in PSCs Self Renewal and Survival
5. Stress Factors Role in iPSCs Reprogramming and Differentiation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- González, F.; Boué, S.; Izpisúa Belmonte, J.C. Methods for making induced pluripotent stem cells: Reprogramming à la carte. Nat. Rev. Genet. 2011, 12, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, P.; Takahashi, K.; Saito, M.; Yoshida, Y.; Okita, K.; Watanabe, A.; Inoue, H.; Yamashita, J.K.; Todani, M.; Nakagawa, M.; et al. Induced Pluripotent Stem Cells and Their Use in Human Models of Disease and Development. Physiol. Rev. 2019, 99, 79–114. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Simmons, S.O.; Fan, C.Y.; Ramabhadran, R. Cellular stress response pathway system as a sentinel ensemble in toxicological screening. Toxicol. Sci. 2009, 111, 202–225. [Google Scholar] [CrossRef]
- Tower, J. Stress and stem cells. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 789–802. [Google Scholar] [CrossRef]
- Ko, H.C.; Gelb, B.D. Concise review: Drug discovery in the age of the induced pluripotent stem cell. Stem Cells Transl. Med. 2014, 3, 500–509. [Google Scholar] [CrossRef]
- Kaitsuka, T.; Hakim, F. Response of Pluripotent Stem Cells to Environmental Stress and Its Application for Directed Differentiation. Biology 2021, 10, 84. [Google Scholar] [CrossRef]
- Puscheck, E.E.; Awonuga, A.O.; Yang, Y.; Jiang, Z.; Rappolee, D.A. Molecular biology of the stress response in the early embryo and its stem cells. Adv. Exp. Med. Biol. 2015, 843, 77–128. [Google Scholar] [CrossRef]
- Zhou, S.; Xie, Y.; Puscheck, E.E.; Rappolee, D.A. Oxygen levels that optimize TSC culture are identified by maximizing growth rates and minimizing stress. Placenta 2011, 32, 475–481. [Google Scholar] [CrossRef]
- Genbacev, O.; Zhou, Y.; Ludlow, J.W.; Fisher, S.J. Regulation of human placental development by oxygen tension. Science 1997, 277, 1669–1672. [Google Scholar] [CrossRef]
- Zhong, W.; Xie, Y.; Wang, Y.; Lewis, J.; Trostinskaia, A.; Wang, F.; Puscheck, E.E.; Rappolee, D.A. Use of hyperosmolar stress to measure stress-activated protein kinase activation and function in human HTR cells and mouse trophoblast stem cells. Reprod. Sci. 2007, 14, 534–547. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, F.; Zhong, W.; Puscheck, E.; Shen, H.; Rappolee, D.A. Shear stress induces preimplantation embryo death that is delayed by the zona pellucida and associated with stress-activated protein kinase-mediated apoptosis. Biol. Reprod. 2006, 75, 45–55. [Google Scholar] [CrossRef]
- Cao, H.; Zhi, Y.; Xu, H.; Fang, H.; Jia, X. Zearalenone causes embryotoxicity and induces oxidative stress and apoptosis in differentiated human embryonic stem cells. Toxicol. In Vitro 2019, 54, 243–250. [Google Scholar] [CrossRef]
- Hu, Q.; Khanna, P.; Ee Wong, B.S.; Lin Heng, Z.S.; Subhramanyam, C.S.; Thanga, L.Z.; Sing Tan, S.W.; Baeg, G.H. Oxidative stress promotes exit from the stem cell state and spontaneous neuronal differentiation. Oncotarget 2018, 9, 4223–4238. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef]
- Xie, Y.; Zhong, W.; Wang, Y.; Trostinskaia, A.; Wang, F.; Puscheck, E.E.; Rappolee, D.A. Using hyperosmolar stress to measure biologic and stress-activated protein kinase responses in preimplantation embryos. Mol. Hum. Reprod. 2007, 13, 473–481. [Google Scholar] [CrossRef]
- Stamenović, D.; Wang, N. Stress transmission within the cell. Compr. Physiol. 2011, 1, 499–524. [Google Scholar] [CrossRef]
- Pennimpede, T.; Cameron, D.A.; MacLean, G.A.; Li, H.; Abu-Abed, S.; Petkovich, M. The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 883–894. [Google Scholar] [CrossRef]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef]
- Kang, K.W.; Ryu, J.H.; Kim, S.G. The essential role of phosphatidylinositol 3-kinase and of p38 mitogen-activated protein kinase activation in the antioxidant response element-mediated rGSTA2 induction by decreased glutathione in H4IIE hepatoma cells. Mol. Pharmacol. 2000, 58, 1017–1025. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.W.; Lee, S.J.; Kim, S.G. Molecular mechanism of nrf2 activation by oxidative stress. Antioxid. Redox Signal. 2005, 7, 1664–1673. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U. Functional polymorphisms of the human multidrug resistance (MDR1) gene: Correlation with P glycoprotein expression and activity in vivo. In Mechanisms of Drug Resistance in Epilepsy: Novartis Foundation Symposium; John Wiley & Sons: Hoboken, NJ, USA, 2002; Volume 243, pp. 207–210; discussion 202–210. [Google Scholar] [CrossRef]
- Hong, F.; Sekhar, K.R.; Freeman, M.L.; Liebler, D.C. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J. Biol. Chem. 2005, 280, 31768–31775. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef]
- Westerheide, S.D.; Morimoto, R.I. Heat shock response modulators as therapeutic tools for diseases of protein conformation. J. Biol. Chem. 2005, 280, 33097–33100. [Google Scholar] [CrossRef]
- Boellmann, F.; Guettouche, T.; Guo, Y.; Fenna, M.; Mnayer, L.; Voellmy, R. DAXX interacts with heat shock factor 1 during stress activation and enhances its transcriptional activity. Proc. Natl. Acad. Sci. USA 2004, 101, 4100–4105. [Google Scholar] [CrossRef]
- Voellmy, R.; Boellmann, F. Chaperone regulation of the heat shock protein response. Adv. Exp. Med. Biol. 2007, 594, 89–99. [Google Scholar] [CrossRef]
- Holmberg, C.I.; Hietakangas, V.; Mikhailov, A.; Rantanen, J.O.; Kallio, M.; Meinander, A.; Hellman, J.; Morrice, N.; MacKintosh, C.; Morimoto, R.I.; et al. Phosphorylation of serine 230 promotes inducible transcriptional activity of heat shock factor 1. EMBO J. 2001, 20, 3800–3810. [Google Scholar] [CrossRef]
- Soncin, F.; Zhang, X.; Chu, B.; Wang, X.; Asea, A.; Ann Stevenson, M.; Sacks, D.B.; Calderwood, S.K. Transcriptional activity and DNA binding of heat shock factor-1 involve phosphorylation on threonine 142 by CK2. Biochem. Biophys Res. Commun. 2003, 303, 700–706. [Google Scholar] [CrossRef]
- Voellmy, R. Transduction of the stress signal and mechanisms of transcriptional regulation of heat shock/stress protein gene expression in higher eukaryotes. Crit. Rev. Eukaryot. Gene Expr. 1994, 4, 357–401. [Google Scholar]
- Vargas, N.; Marino, F. Heat stress, gastrointestinal permeability and interleukin-6 signaling—Implications for exercise performance and fatigue. Temperature 2016, 3, 240–251. [Google Scholar] [CrossRef]
- Satyal, S.H.; Chen, D.; Fox, S.G.; Kramer, J.M.; Morimoto, R.I. Negative regulation of the heat shock transcriptional response by HSBP1. Genes Dev. 1998, 12, 1962–1974. [Google Scholar] [CrossRef]
- Lee, K.A.; Roth, R.A.; LaPres, J.J. Hypoxia, drug therapy and toxicity. Pharmacol. Ther. 2007, 113, 229–246. [Google Scholar] [CrossRef]
- van den Beucken, T.; Koritzinsky, M.; Wouters, B.G. Translational control of gene expression during hypoxia. Cancer Biol. Ther. 2006, 5, 749–755. [Google Scholar] [CrossRef]
- Duyndam, M.C.; Hulscher, S.T.; van der Wall, E.; Pinedo, H.M.; Boven, E. Evidence for a role of p38 kinase in hypoxia-inducible factor 1-independent induction of vascular endothelial growth factor expression by sodium arsenite. J. Biol. Chem. 2003, 278, 6885–6895. [Google Scholar] [CrossRef]
- Gao, N.; Jiang, B.H.; Leonard, S.S.; Corum, L.; Zhang, Z.; Roberts, J.R.; Antonini, J.; Zheng, J.Z.; Flynn, D.C.; Castranova, V.; et al. p38 Signaling-mediated hypoxia-inducible factor 1alpha and vascular endothelial growth factor induction by Cr(VI) in DU145 human prostate carcinoma cells. J. Biol. Chem. 2002, 277, 45041–45048. [Google Scholar] [CrossRef]
- Yen, M.L.; Su, J.L.; Chien, C.L.; Tseng, K.W.; Yang, C.Y.; Chen, W.F.; Chang, C.C.; Kuo, M.L. Diosgenin induces hypoxia-inducible factor-1 activation and angiogenesis through estrogen receptor-related phosphatidylinositol 3-kinase/Akt and p38 mitogen-activated protein kinase pathways in osteoblasts. Mol. Pharmacol. 2005, 68, 1061–1073. [Google Scholar] [CrossRef]
- Arany, Z.; Huang, L.E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 12969–12973. [Google Scholar] [CrossRef]
- Zakrzewska, A.; Schnell, P.O.; Striet, J.B.; Hui, A.; Robbins, J.R.; Petrovic, M.; Conforti, L.; Gozal, D.; Wathelet, M.G.; Czyzyk-Krzeska, M.F. Hypoxia-activated metabolic pathway stimulates phosphorylation of p300 and CBP in oxygen-sensitive cells. J. Neurochem. 2005, 94, 1288–1296. [Google Scholar] [CrossRef]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of oxygen signaling at the consensus HRE. Sci. Signal Transduct. Knowl. Environ. 2005, 2005, re12. [Google Scholar] [CrossRef]
- Dahl, S.C.; Handler, J.S.; Kwon, H.M. Hypertonicity-induced phosphorylation and nuclear localization of the transcription factor TonEBP. Am. J. Physiol. Cell Physiol. 2001, 280, C248–C253. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.C.; Lam, A.K.; Kapus, A.; Fan, L.; Chung, S.K.; Chung, S.S. Fyn and p38 signaling are both required for maximal hypertonic activation of the osmotic response element-binding protein/tonicity-responsive enhancer-binding protein (OREBP/TonEBP). J. Biol. Chem. 2002, 277, 46085–46092. [Google Scholar] [CrossRef] [PubMed]
- Irarrazabal, C.E.; Liu, J.C.; Burg, M.B.; Ferraris, J.D. ATM, a DNA damage-inducible kinase, contributes to activation by high NaCl of the transcription factor TonEBP/OREBP. Proc. Natl. Acad. Sci. USA 2004, 101, 8809–8814. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ferraris, J.D.; Irarrazabal, C.E.; Dmitrieva, N.I.; Park, J.H.; Burg, M.B. Ataxia telangiectasia-mutated, a DNA damage-inducible kinase, contributes to high NaCl-induced nuclear localization of transcription factor TonEBP/OREBP. Am. J. Physiol. Ren. Physiol. 2005, 289, F506–F511. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, J.D.; Persaud, P.; Williams, C.K.; Chen, Y.; Burg, M.B. cAMP-independent role of PKA in tonicity-induced transactivation of tonicity-responsive enhancer/ osmotic response element-binding protein. Proc. Natl. Acad. Sci. USA 2002, 99, 16800–16805. [Google Scholar] [CrossRef] [PubMed]
- Irarrazabal, C.E.; Williams, C.K.; Ely, M.A.; Birrer, M.J.; Garcia-Perez, A.; Burg, M.B.; Ferraris, J.D. Activator protein-1 contributes to high NaCl-induced increase in tonicity-responsive enhancer/osmotic response element-binding protein transactivating activity. J. Biol. Chem. 2008, 283, 2554–2563. [Google Scholar] [CrossRef] [PubMed]
- Kültz, D. DNA damage signals facilitate osmotic stress adaptation. Am. J. Physiol. Ren. Physiol. 2005, 289, F504–F505. [Google Scholar] [CrossRef]
- Fuchs, S.Y.; Adler, V.; Pincus, M.R.; Ronai, Z. MEKK1/JNK signaling stabilizes and activates p53. Proc. Natl. Acad. Sci. USA 1998, 95, 10541–10546. [Google Scholar] [CrossRef]
- Morgan, S.E.; Kastan, M.B. p53 and ATM: Cell cycle, cell death, and cancer. Adv. Cancer Res. 1997, 71, 1–25. [Google Scholar] [CrossRef]
- Shieh, S.Y.; Ahn, J.; Tamai, K.; Taya, Y.; Prives, C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000, 14, 289–300. [Google Scholar] [CrossRef]
- Prowald, A.; Schuster, N.; Montenarh, M. Regulation of the DNA binding of p53 by its interaction with protein kinase CK2. FEBS Lett. 1997, 408, 99–104. [Google Scholar] [CrossRef]
- Wu, G.S. The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol. Ther. 2004, 3, 156–161. [Google Scholar] [CrossRef]
- Gartel, A.L.; Radhakrishnan, S.K. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005, 65, 3980–3985. [Google Scholar] [CrossRef]
- Thoma, B.S.; Vasquez, K.M. Critical DNA damage recognition functions of XPC-hHR23B and XPA-RPA in nucleotide excision repair. Mol. Carcinog. 2003, 38, 1–13. [Google Scholar] [CrossRef]
- Zhan, Q. Gadd45a, a p53- and BRCA1-regulated stress protein, in cellular response to DNA damage. Mutat. Res. 2005, 569, 133–143. [Google Scholar] [CrossRef]
- Reed, J.C. Proapoptotic multidomain Bcl-2/Bax-family proteins: Mechanisms, physiological roles, and therapeutic opportunities. Cell Death Differ. 2006, 13, 1378–1386. [Google Scholar] [CrossRef]
- Lahav, G.; Rosenfeld, N.; Sigal, A.; Geva-Zatorsky, N.; Levine, A.J.; Elowitz, M.B.; Alon, U. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet. 2004, 36, 147–150. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Schröder, M. Endoplasmic reticulum stress responses. Cell. Mol. Life Sci. 2008, 65, 862–894. [Google Scholar] [CrossRef]
- Foufelle, F.; Fromenty, B. Role of endoplasmic reticulum stress in drug-induced toxicity. Pharmacol. Res. Perspect. 2016, 4, e00211. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. Herp is dually regulated by both the endoplasmic reticulum stress-specific branch of the unfolded protein response and a branch that is shared with other cellular stress pathways. J. Biol. Chem. 2004, 279, 13792–13799. [Google Scholar] [CrossRef]
- Zhou, J.; Dong, X.H.; Zhang, F.Z.; Zhu, H.M.; Hao, T.; Jiang, X.X.; Zheng, W.B.; Zhang, T.; Wang, P.Z.; Li, H.; et al. Real microgravity condition promoted regeneration capacity of induced pluripotent stem cells during the TZ-1 space mission. Cell Prolif. 2019, 52, e12574. [Google Scholar] [CrossRef]
- Chen, Y.; Tristan, C.A.; Chen, L.; Jovanovic, V.M.; Malley, C.; Chu, P.H.; Ryu, S.; Deng, T.; Ormanoglu, P.; Tao, D.; et al. A versatile polypharmacology platform promotes cytoprotection and viability of human pluripotent and differentiated cells. Nat. Methods 2021, 18, 528–541. [Google Scholar] [CrossRef]
- Julian, L.; Olson, M.F. Rho-associated coiled-coil containing kinases (ROCK): Structure, regulation, and functions. Small GTPases 2014, 5, e29846. [Google Scholar] [CrossRef]
- Chen, G.; Hou, Z.; Gulbranson, D.R.; Thomson, J.A. Actin-myosin contractility is responsible for the reduced viability of dissociated human embryonic stem cells. Cell Stem Cell 2010, 7, 240–248. [Google Scholar] [CrossRef]
- Vallabhaneni, H.; Lynch, P.J.; Chen, G.; Park, K.; Liu, Y.; Goehe, R.; Mallon, B.S.; Boehm, M.; Hursh, D.A. High Basal Levels of γH2AX in Human Induced Pluripotent Stem Cells Are Linked to Replication-Associated DNA Damage and Repair. Stem Cells 2018, 36, 1501–1513. [Google Scholar] [CrossRef]
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef]
- Salloum-Asfar, S.; Engelke, R.; Mousa, H.; Goswami, N.; Thompson, I.R.; Palangi, F.; Kamal, K.; Al-Noubi, M.N.; Schmidt, F.; Abdulla, S.A.; et al. Hyperosmotic Stress Induces a Specific Pattern for Stress Granule Formation in Human-Induced Pluripotent Stem Cells. Stem Cells Int. 2021, 2021, 8274936. [Google Scholar] [CrossRef]
- Palangi, F.; Samuel, S.M.; Thompson, I.R.; Triggle, C.R.; Emara, M.M. Effects of oxidative and thermal stresses on stress granule formation in human induced pluripotent stem cells. PLoS ONE 2017, 12, e0182059. [Google Scholar] [CrossRef]
- Hakim, F.; Kaitsuka, T.; Raeed, J.M.; Wei, F.Y.; Shiraki, N.; Akagi, T.; Yokota, T.; Kume, S.; Tomizawa, K. High oxygen condition facilitates the differentiation of mouse and human pluripotent stem cells into pancreatic progenitors and insulin-producing cells. J. Biol. Chem. 2014, 289, 9623–9638. [Google Scholar] [CrossRef]
- Ayabe, H.; Anada, T.; Kamoya, T.; Sato, T.; Kimura, M.; Yoshizawa, E.; Kikuchi, S.; Ueno, Y.; Sekine, K.; Camp, J.G.; et al. Optimal Hypoxia Regulates Human iPSC-Derived Liver Bud Differentiation through Intercellular TGFB Signaling. Stem Cell Rep. 2018, 11, 306–316. [Google Scholar] [CrossRef]
- Correia, C.; Serra, M.; Espinha, N.; Sousa, M.; Brito, C.; Burkert, K.; Zheng, Y.; Hescheler, J.; Carrondo, M.J.; Sarić, T.; et al. Combining hypoxia and bioreactor hydrodynamics boosts induced pluripotent stem cell differentiation towards cardiomyocytes. Stem Cell Rev. Rep. 2014, 10, 786–801. [Google Scholar] [CrossRef]
- Okada, R.; Onodera, K.; Ito, T.; Doyu, M.; Okano, H.J.; Okada, Y. Modulation of oxygen tension, acidosis, and cell density is crucial for neural differentiation of human induced pluripotent stem cells. Neurosci. Res. 2021, 163, 34–42. [Google Scholar] [CrossRef]
- Jha, R.; Wu, Q.; Singh, M.; Preininger, M.K.; Han, P.; Ding, G.; Cho, H.C.; Jo, H.; Maher, K.O.; Wagner, M.B.; et al. Simulated Microgravity and 3D Culture Enhance Induction, Viability, Proliferation and Differentiation of Cardiac Progenitors from Human Pluripotent Stem Cells. Sci. Rep. 2016, 6, 30956. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Zhang, F.; Dong, X.; Hao, T.; Jiang, X.; Zheng, W.; Zhang, T.; Chen, X.; Wang, P.; et al. Spaceflight Promoted Myocardial Differentiation of Induced Pluripotent Stem Cells: Results from Tianzhou-1 Space Mission. Stem Cells Dev. 2019, 28, 357–360. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stress | Cell Type | Mechanism | Title 4 | Ref. | |
|---|---|---|---|---|---|
| Hypoxia (O2) | 2% 20% | Mouse TSC 1 | pSAPK, CDX2, Id2, Errβ increased | Multipotency | [9] |
| 0.5% | pSAPK, Gem1, Tpbpa, Hand1 increased | Differentiation | |||
| 2% | Human TSC | Cyclin B + cell count increased | Proliferation | [10] | |
| 20% | p21Cip1/Waf1 increased | Cell cycle arrest | |||
| Osmotic | 50 mM sorbitol | Human TSC + mouse TSC | pSAPK increased + decreased cell count | Decreased proliferation | [11] |
| 150 + 200 mM sorbitol | pSAPK increased + No cell growth | Stress marker | |||
| 400 + 600 mM sorbitol | pSAPK increased | Apoptosis | |||
| Mechanical | 1.2 dynes/cm2 of shear stress | Mouse embryos | MAPK8/9 | Apoptosis | [12] |
| Oxidative | Zearalenone 1, 2 and 4 μg/mL | hESC-derived EBs 2 | p53, Bax, caspase-9 and caspase-3 + Bax/Bcl-2 ratio increased | Apoptosis | [13] |
| Paraquat 75–100 μM | NT2 cells 3 | Pax6, Gfra1, Hoxa1 and Ncam increased | Differentiation | [14] | |
| Stress | Cell Type | Markers Expressed | Directed Cell Type | Ref. | |
|---|---|---|---|---|---|
| Hyperoxia | 60% O2 | hiPSCs 1 | INS increased | Islet endocrine cells: C-peptide and glucagon positive cells | [77] |
| NGN3 and VEGFA increased HES1 decreased | Ductal epithelium | ||||
| Hypoxia | 10% O2 | hiPSC-LBs 2 | Albumin, vitronectin and urea increased CYP3A4 activity increased | Hepatocytes and Cholangiocytes | [78] |
| 4% O2 | Murine iPSCs | eGFP and cell number increased beating activity detected | Cardiomyocytes | [79] | |
| 20% O2 + 3% CO2 | hiPSCs | Pax6, Sox1, OLIG2 and NGN2 increased | Motor neuron progenitor cells | [80] | |
| 20% + 5% CO2 | |||||
| 5% O2 + 5% CO2 | |||||
| Gravity | Microgravity (μg) | hiPSCs | MESP1 increased KDR, PDGFRa, CD13 increased | Cardiomyocytes | [81] |
| Mouse iPSC-EB 3 | GFP increased | Myocardial progenitor cells | [82] | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darwish, T.; Swaidan, N.T.; Emara, M.M. Stress Factors as Possible Regulators of Pluripotent Stem Cell Survival and Differentiation. Biology 2023, 12, 1119. https://doi.org/10.3390/biology12081119
Darwish T, Swaidan NT, Emara MM. Stress Factors as Possible Regulators of Pluripotent Stem Cell Survival and Differentiation. Biology. 2023; 12(8):1119. https://doi.org/10.3390/biology12081119
Chicago/Turabian StyleDarwish, Toqa, Nuha Taysir Swaidan, and Mohamed M. Emara. 2023. "Stress Factors as Possible Regulators of Pluripotent Stem Cell Survival and Differentiation" Biology 12, no. 8: 1119. https://doi.org/10.3390/biology12081119
APA StyleDarwish, T., Swaidan, N. T., & Emara, M. M. (2023). Stress Factors as Possible Regulators of Pluripotent Stem Cell Survival and Differentiation. Biology, 12(8), 1119. https://doi.org/10.3390/biology12081119