
Discovery of GJC1 (Cx45) as a New Gene Underlying Congenital Heart Disease and Arrhythmias
 , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Recruitment of Research Subjects and Extraction of Genomic DNA
2.2. Genome-Wide Genotyping with Genetic Markers and Linkage Assay
2.3. Sequencing Assay of the Genes at the Located Locus
2.4. Expression Vector Construction and Site-Directed Mutagenesis
2.5. Cell Culture and Transient Transfection
2.6. Electrophysiological Analysis
2.7. Dye Uptake Analysis
2.8. Statistical Assay
3. Results
3.1. Identification of a Chinese Pedigree Affected with CHD and AVB
3.2. A Novel Genetic Locus for CHD and AVB Mapped on Chromosome 17q21.31-q21.33
3.3. Discovery of a Pathogenic Mutation in Cx45
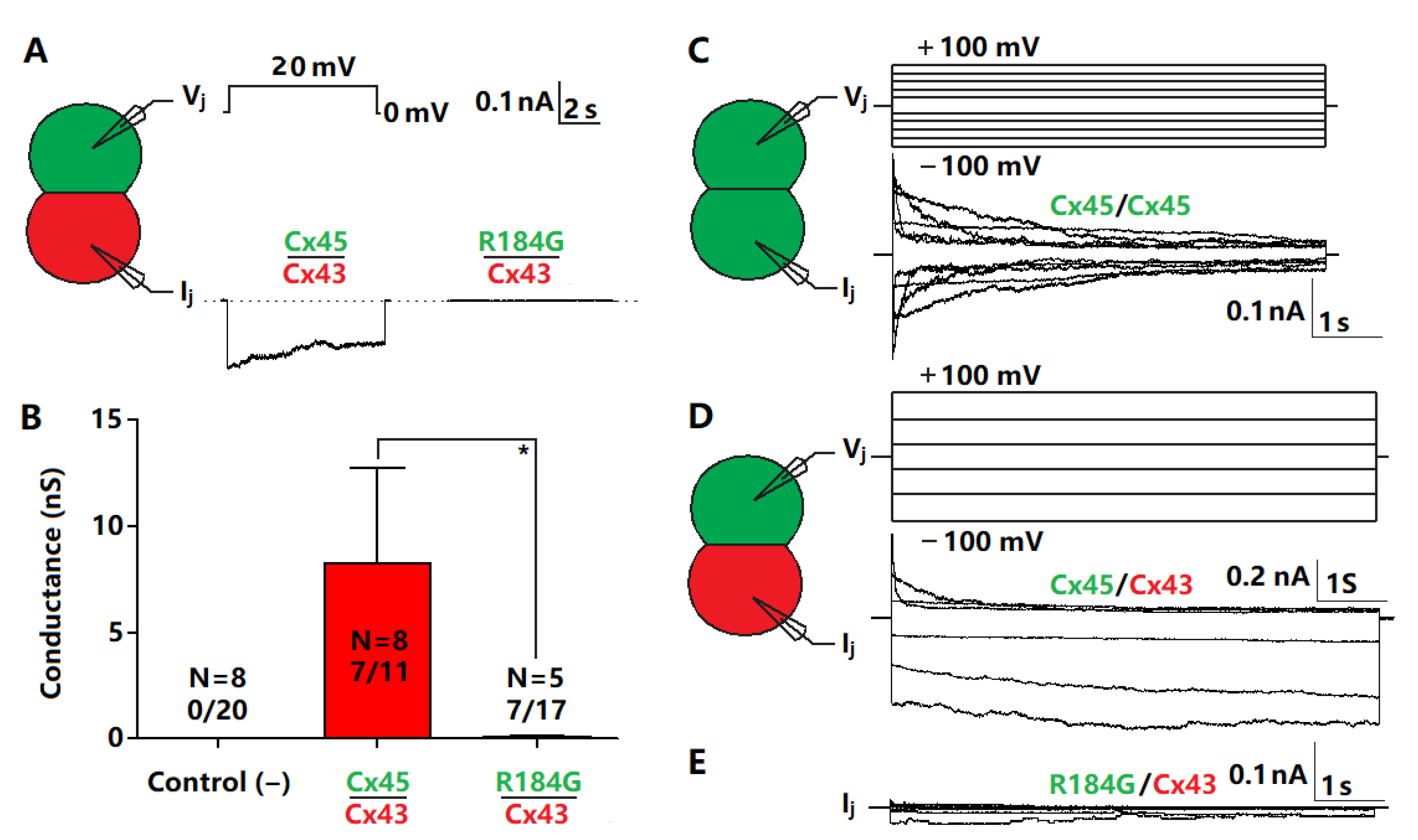
3.4. Impaired Coupling Conductance of Cells Expressing Cx45 R184G
3.5. Cx45 R184G Impaired GJ Coupling When Paired with Cx43 Expressing Cells
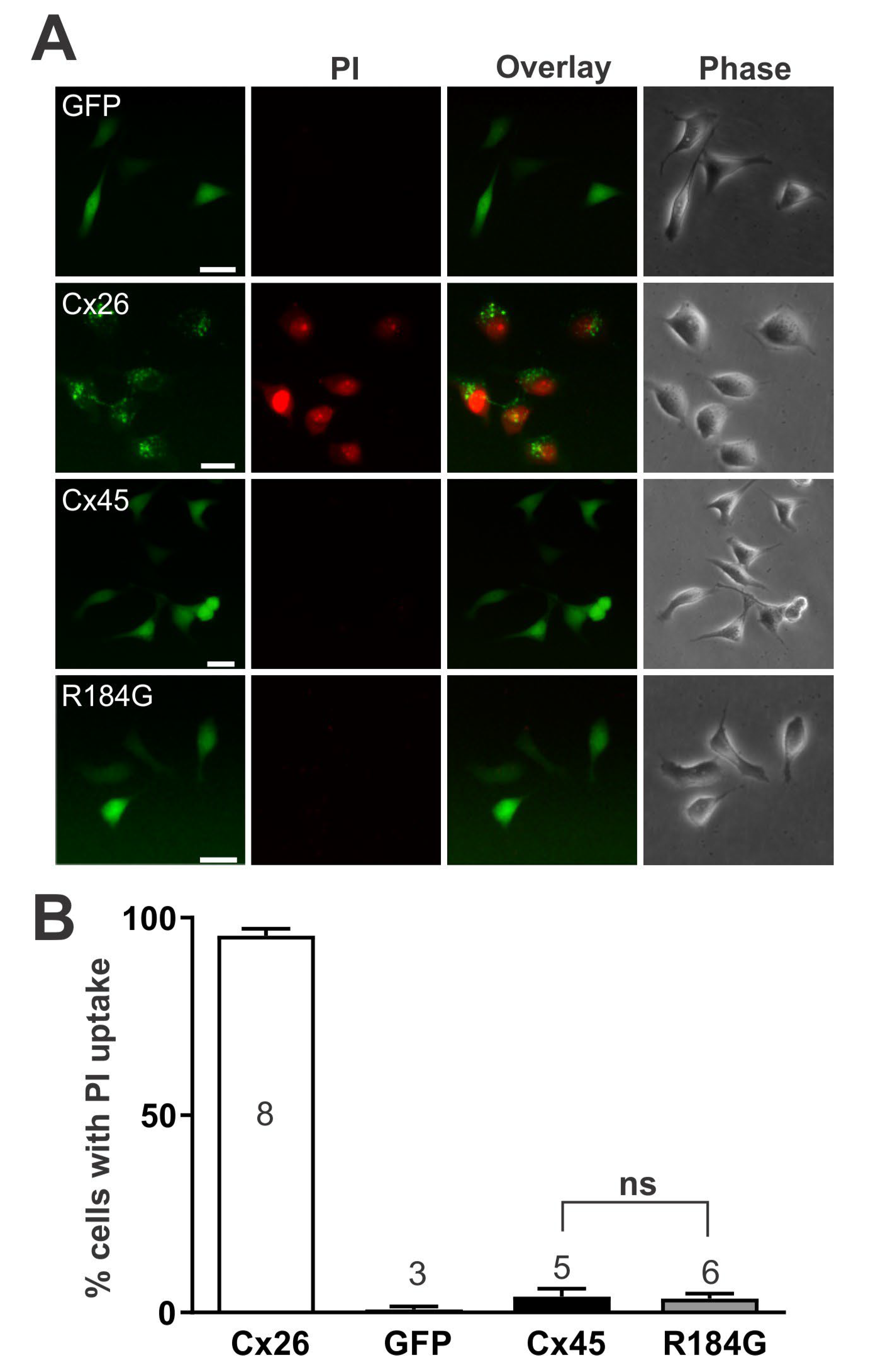
3.6. Dye Uptake of Cells Expressing Cx45 R184G Is Not Different from Wild-Type Cx45 Expressing Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DBF4B | LOC107985044 (lncRNA) | ADAM11 | GJC1 | HIGD1B |
|---|---|---|---|---|
| EFTUD2 | RN7SL405P (pseudo) | CCDC103 | GFAP | FAM187A |
| LOC105371793 (lncRNA) | KIF18B | MIR6783 (miRNA) | C1QL1 | LOC112268183 (pseudo) |
| LOC107987243 (lncRNA) | DCAKD | NMT1 | PLCD3 | MIR6784 (miRNA) |
| ACBD4 | HEXIM1 | HEXIM2 | LOC105371795 (lncRNA) | FMNL1-DT (lncRNA) |
| FMNL1 | LOC124904011 | MAP3K14-AS1 (lncRNA) | SPATA32 | MAP3K14 |
| RNA5SP443 (pseudo) | LOC105371796 (lncRNA) | ARHGAP27 | LOC124904016 (lncRNA) | PLEKHM1 |
| MIR4315-1 (miRNA) | LOC124904113 (pseudo) | LOC105369225 (lncRNA) | LRRC37A4P (pseudo) | LOC102724345 (pseudo) |
| RDM1P1 (pseudo) | DND1P1 (pseudo) | MAPK8IP1P2 (pseudo) | RPS26P8 (pseudo) | LINC02210 (lncRNA) |
| LINC02210-CRHR1 (lncRNA) | ARF2P (pseudo) | LOC105371802 (lncRNA) | LOC107985028 (lncRNA) | CRHR1 |
| MAPT-AS1 (lncRNA) | SPPL2C | MAPT | MAPT-IT1 (lncRNA) | LOC105371800 (lncRNA) |
| STH | KANSL1 | LOC107985027 (lncRNA) | KANSL1-AS1 (lncRNA) | MAPK8IP1P1 (pseudo) |
| LRRC37A | DND1P2 (pseudo) | LOC100132570 (pseudo) | LOC124904014 (lncRNA) | ARL17B |
| RN7SL656P (pseudo) | LRRC37A2 | NSFP1 (pseudo) | ARL17A | RDM1P2 (pseudo) |
| RN7SL199P (pseudo) | NSF | LOC107985026 (pseudo) | RPS7P11 (pseudo) | WNT3 |
| LOC101929777 (lncRNA) | WNT9B | LINC01974 (lncRNA) | LOC112268191 (lncRNA) | RNU6ATAC3P (pseudo) |
| GOSR2 | MIR5089 (miRNA) | RPRML | LOC101927060 (lncRNA) | LRRC37A17P (pseudo) |
| RN7SL270P (pseudo) | LOC124904015 (lncRNA) | CDC27 | LOC112268192 (pseudo) | RPS2P47 (pseudo) |
| MYL4 | ITGB3 | RNU7-186P (pseudo) | LOC107985029 (lncRNA) | EFCAB13-DT (lncRNA) |
| EFCAB13 | LOC100421523 (pseudo) | NFE2L3P2 (pseudo) | MRPL45P2 (pseudo) | NPEPPS |
| KPNB1-DT (lncRNA) | KPNB1 | TBKBP1 | TBX21 | OSBPL7 |
| MRPL10 | LRRC46 | SCRN2 | SP6 | SP2-DT (lncRNA) |
| SP2 | SP2-AS1 (lncRNA) | PNPO | PRR15L | CDK5RAP3 |
| LOC124904017 (lncRNA) | COPZ2 | MIR10226 (miRNA) | MIR152 (miRNA) | NFE2L1-DT (lncRNA) |
| NFE2L1 | CBX1 | RNU6-1201P (pseudo) | SNX11 | SKAP1 |
| LOC124904018 (lncRNA) | MIR1203 (miRNA) | RNU6-1152P (pseudo) | LSM3P1 (pseudo) | SKAP1-AS2 (lncRNA) |
| LOC124904151 (lncRNA) | LOC101927166 (lncRNA) | LOC124904019 (lncRNA) | LOC105371808 (lncRNA) | HOXB1 |
| HOXB2 | HOXB-AS1 (lncRNA) | HOXB3 | HOXB-AS2 (lncRNA) | HOXB4 |
| MIR10A (miRNA) | HOXB-AS3 (lncRNA) | HOXB5 | HOXB6 | HOXB7 |
| HOXB8 | HOXB9 | HOXB-AS4 (lncRNA) | MIR196A1 (miRNA) | LINC02086 (lncRNA) |
| RPL9P28 (pseudo) | COX6B1P2 (pseudo) | PRAC2 | PRAC1 | MIR3185 (miRNA) |
| HOXB13 | LOC105371811 (lncRNA) | LOC105371812 (lncRNA) | TTLL6 | RN7SL125P (pseudo) |
| LOC105371813 (lncRNA) | CALCOCO2 | SUMO2P17 (pseudo) | LOC105371814 (lncRNA) | LOC124900395 (lncRNA) |
| ATP5MC1 | LOC100419622 (pseudo) | UBE2Z | SNF8 | RNU1-42P (pseudo) |
| LOC124904116 (lncRNA) | GIP | IGF2BP1 | LOC124904020 (lncRNA) | RNU6-826P (pseudo) |
| B4GALNT2P1 (pseudo) | B4GALNT2 | LOC124904021 (lncRNA) | RPS10P25 (pseudo) | TRQ-TTG1-1 (tRNA) |
| GNGT2 | ABI3 | PHOSPHO1 | LOC124904022 (lncRNA) | FLJ40194 (lncRNA) |
| ZNF652 | MIR6129 (miRNA) | LOC124904111 (snRNA) | ZNF652-AS1 (lncRNA) | RPL21P124 (pseudo) |
| PHB1 | EIF4EP2 (pseudo) | LINC02075 (lncRNA) | NGFR | NGFR-AS1 (lncRNA) |
| MIR6165 (miRNA) | NXPH3 | SPOP | LOC107984999 (lncRNA) | SLC35B1 |
| FAM117A | KAT7 | SRP14P3 (pseudo) | TAC4 | FLJ45513 (lncRNA) |
References
- Zaidi, S.; Brueckner, M. Genetics and genomics of congenital heart disease. Circ. Res. 2017, 120, 923–940. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics-2018 update: A report from the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef] [PubMed]
- Villaseca-Rojas, Y.; Varela-Melo, J.; Torres-Castro, R.; Vasconcello-Castillo, L.; Mazzucco, G.; Vilaró, J.; Blanco, I. Exercise Capacity in Children and Adolescents with Congenital Heart Disease: A Systematic Review and Meta-Analysis. Front. Cardiovasc. Med. 2022, 9, 874700. [Google Scholar] [CrossRef] [PubMed]
- Brudy, L.; Häcker, A.L.; Meyer, M.; Oberhoffer, R.; Hager, A.; Ewert, P.; Müller, J. Adults with Congenital Heart Disease Move Well but Lack Intensity: A Cross-Sectional Study Using Wrist-Worn Physical Activity Trackers. Cardiology 2022, 147, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Brudy, L.; Meyer, M.; Garcia-Cuenllas, L.; Oberhoffer, R.; Hager, A.; Ewert, P.; Müller, J. Objective Physical Activity Assessment in Clinical Congenital Heart Disease Research: A Systematic Review on Study Quality, Methodology, and Outcomes. Cardiology 2021, 146, 240–252. [Google Scholar] [CrossRef]
- Brudy, L.; Meyer, M.; Oberhoffer, R.; Ewert, P.; Müller, J. Move more–be happier? physical activity and health-related quality of life in children with congenital heart disease. Am. Heart J. 2021, 241, 68–73. [Google Scholar] [CrossRef]
- Liu, H.C.; Chaou, C.H.; Lo, C.W.; Chung, H.T.; Hwang, M.S. Factors Affecting Psychological and Health-Related Quality-of-Life Status in Children and Adolescents with Congenital Heart Diseases. Children 2022, 9, 578. [Google Scholar] [CrossRef]
- Sadhwani, A.; Wypij, D.; Rofeberg, V.; Gholipour, A.; Mittleman, M.; Rohde, J.; Velasco-Annis, C.; Calderon, J.; Friedman, K.G.; Tworetzky, W.; et al. Fetal Brain Volume Predicts Neurodevelopment in Congenital Heart Disease. Circulation 2022, 145, 1108–1119. [Google Scholar] [CrossRef]
- Schlatterer, S.D.; Govindan, R.B.; Murnick, J.; Barnett, S.D.; Lopez, C.; Donofrio, M.T.; Mulkey, S.B.; Limperopoulos, C.; du Plessis, A.J. In infants with congenital heart disease autonomic dysfunction is associated with pre-operative brain injury. Pediatr. Res. 2022, 91, 1723–1729. [Google Scholar] [CrossRef]
- Parekh, S.A.; Cox, S.M.; Barkovich, A.J.; Chau, V.; Steurer, M.A.; Xu, D.; Miller, S.P.; McQuillen, P.S.; Peyvandi, S. The Effect of Size and Asymmetry at Birth on Brain Injury and Neurodevelopmental Outcomes in Congenital Heart Disease. Pediatr. Cardiol. 2022, 43, 868–877. [Google Scholar] [CrossRef]
- Yeh, H.R.; Kim, E.H.; Yu, J.J.; Yun, T.J.; Ko, T.S.; Yum, M.S. Arterial ischemic stroke in children with congenital heart diseases. Pediatr. Int. 2022, 64, e15200. [Google Scholar] [CrossRef] [PubMed]
- Giang, K.W.; Fedchenko, M.; Dellborg, M.; Eriksson, P.; Mandalenakis, Z. Burden of Ischemic Stroke in Patients with Congenital Heart Disease: A Nationwide, Case-Control Study. J. Am. Heart Assoc. 2021, 10, e020939. [Google Scholar] [CrossRef]
- Rosenzweig, E.B.; Krishnan, U. Congenital Heart Disease-Associated Pulmonary Hypertension. Clin. Chest Med. 2021, 42, 9–18. [Google Scholar] [CrossRef]
- Jansen, K.; Constantine, A.; Condliffe, R.; Tulloh, R.; Clift, P.; Moledina, S.; Wort, S.J.; Dimopoulos, K. Pulmonary arterial hypertension in adults with congenital heart disease: Markers of disease severity, management of advanced heart failure and transplantation. Expert Rev. Cardiovasc. Ther. 2021, 19, 837–855. [Google Scholar] [CrossRef]
- Abassi, H.; Gavotto, A.; Picot, M.C.; Bertet, H.; Matecki, S.; Guillaumont, S.; Moniotte, S.; Auquier, P.; Moreau, J.; Amedro, P. Impaired pulmonary function and its association with clinical outcomes, exercise capacity and quality of life in children with congenital heart disease. Int. J. Cardiol. 2019, 285, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Gonzalez, R.; Borgia, F.; Diller, G.P.; Inuzuka, R.; Kempny, A.; Martinez-Naharro, A.; Tutarel, O.; Marino, P.; Wustmann, K.; Charalambides, M.; et al. Abnormal lung function in adults with congenital heart disease: Prevalence, relation to cardiac anatomy, and association with survival clinical perspective. Circulation 2013, 127, 882–890. [Google Scholar] [CrossRef]
- Dimopoulos, K.; Diller, G.P.; Koltsida, E.; Pijuan-Domenech, A.; Papadopoulou, S.A.; Babu-Narayan, S.V.; Salukhe, T.V.; Piepoli, M.F.; Poole-Wilson, P.A.; Best, N.; et al. Prevalence, predictors, and prognostic value of renal dysfunction in adults with congenital heart disease. Circulation 2008, 117, 2320–2328. [Google Scholar] [CrossRef] [PubMed]
- Gillesén, M.; Fedchenko, M.; Giang, K.W.; Dimopoulos, K.; Eriksson, P.; Dellborg, M.; Mandalenakis, Z. Chronic kidney disease in patients with congenital heart disease: A nationwide, register-based cohort study. Eur. Heart J. Open 2022, 2, oeac055. [Google Scholar] [CrossRef] [PubMed]
- Kourelis, G.; Kanakis, M.; Samanidis, G.; Tzannis, K.; Bobos, D.; Kousi, T.; Apostolopoulou, S.; Kakava, F.; Kyriakoulis, K.; Bounta, S.; et al. Acute Kidney Injury Predictors and Outcomes after Cardiac Surgery in Children with Congenital Heart Disease: An Observational Cohort Study. Diagnostics 2022, 12, 2397. [Google Scholar] [CrossRef]
- Chen, H.; Ke, Q.; Weng, G.; Bao, J.; Huang, J.; Yan, L.; Zheng, F. Risk factors of postoperative acute kidney injury in patients with complex congenital heart disease and significance of early detection of serum transcription factor Nkx2.5. Am. J. Transl. Res. 2021, 13, 6468–6477. [Google Scholar]
- Cahill, T.J.; Jewell, P.D.; Denne, L.; Franklin, R.C.; Frigiola, A.; Orchard, E.; Prendergast, B.D. Contemporary epidemiology of infective endocarditis in patients with congenital heart disease: A UK prospective study. Am. Heart J. 2019, 215, 70–77. [Google Scholar] [CrossRef]
- Tutarel, O.; Alonso-Gonzalez, R.; Montanaro, C.; Schiff, R.; Uribarri, A.; Kempny, A.; Grübler, M.R.; Uebing, A.; Swan, L.; Diller, G.P.; et al. Infective endocarditis in adults with congenital heart disease remains a lethal disease. Heart 2018, 104, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Maser, M.; Freisinger, E.; Bronstein, L.; Köppe, J.; Orwat, S.; Kaleschke, G.; Baumgartner, H.; Diller, G.P.; Lammers, A. Frequency, Mortality, and Predictors of Adverse Outcomes for Endocarditis in Patients with Congenital Heart Disease: Results of a Nationwide Analysis including 2512 Endocarditis Cases. J. Clin. Med. 2021, 10, 5071. [Google Scholar] [CrossRef] [PubMed]
- Snygg-Martin, U.; Giang, K.W.; Dellborg, M.; Robertson, J.; Mandalenakis, Z. Cumulative Incidence of Infective Endocarditis in Patients with Congenital Heart Disease: A Nationwide, Case-Control Study Over Nine Decades. Clin. Infect. Dis. 2021, 73, 1469–1475. [Google Scholar] [CrossRef]
- Well, A.; Mizrahi, M.; Johnson, G.; Patt, H.; Fraser, C.D.; Mery, C.M.; Beckerman, Z. Aortic dissection and ruptures in adult congenital heart disease in Texas from 2009 to 2019. Eur. J. Cardiothorac. Surg. 2022, 61, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Arnaert, S.; De Meester, P.; Troost, E.; Droogne, W.; Van Aelst, L.; Van Cleemput, J.; Voros, G.; Gewillig, M.; Cools, B.; Moons, P.; et al. Heart failure related to adult congenital heart disease: Prevalence, outcome and risk factors. ESC Heart Fail. 2021, 8, 2940–2950. [Google Scholar] [CrossRef] [PubMed]
- Brida, M.; Lovrić, D.; Griselli, M.; Riesgo Gil, F.; Gatzoulis, M.A. Heart failure in adults with congenital heart disease. Int. J. Cardiol. 2022, 357, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Kartas, A.; Papazoglou, A.S.; Kosmidis, D.; Moysidis, D.V.; Baroutidou, A.; Doundoulakis, I.; Despotopoulos, S.; Vrana, E.; Koutsakis, A.; Rampidis, G.P.; et al. The Adult Congenital Heart Disease Anatomic and Physiological Classification: Associations with Clinical Outcomes in Patients with Atrial Arrhythmias. Diagnostics 2022, 12, 466. [Google Scholar] [CrossRef]
- Casteigt, B.; Samuel, M.; Laplante, L.; Shohoudi, A.; Apers, S.; Kovacs, A.H.; Luyckx, K.; Thomet, C.; Budts, W.; Enomoto, J.; et al. Atrial arrhythmias and patient-reported outcomes in adults with congenital heart disease: An international study. Heart Rhythm 2021, 18, 793–800. [Google Scholar] [CrossRef]
- Wasmer, K.; Eckardt, L.; Baumgartner, H.; Köbe, J. Therapy of supraventricular and ventricular arrhythmias in adults with congenital heart disease-narrative review. Cardiovasc. Diagn. Ther. 2021, 11, 550–562. [Google Scholar] [CrossRef]
- Khairy, P.; Silka, M.J.; Moore, J.P.; DiNardo, J.A.; Vehmeijer, J.T.; Sheppard, M.N.; van de Bruaene, A.; Chaix, M.A.; Brida, M.; Moore, B.M.; et al. Sudden cardiac death in congenital heart disease. Eur. Heart J. 2022, 43, 2103–2115. [Google Scholar] [CrossRef] [PubMed]
- Van Bulck, L.; Goossens, E.; Morin, L.; Luyckx, K.; Ombelet, F.; Willems, R.; Budts, W.; De Groote, K.; De Backer, J.; Annemans, L.; et al. Last year of life of adults with congenital heart diseases: Causes of death and patterns of care. Eur. Heart J. 2022, 43, 4483–4492. [Google Scholar] [CrossRef] [PubMed]
- Constantine, A.; Costola, G.; Bianchi, P.; Chessa, M.; Giamberti, A.; Kempny, A.; Rafiq, I.; Babu-Narayan, S.V.; Gatzoulis, M.A.; Hoschtitzky, A.; et al. Enhanced Assessment of Perioperative Mortality Risk in Adults with Congenital Heart Disease. J. Am. Coll. Cardiol. 2021, 78, 234–242. [Google Scholar] [CrossRef]
- Vehmeijer, J.T.; Koyak, Z.; Leerink, J.M.; Zwinderman, A.H.; Harris, L.; Peinado, R.; Oechslin, E.N.; Robbers-Visser, D.; Groenink, M.; Boekholdt, S.M.; et al. Identification of patients at risk of sudden cardiac death in congenital heart disease: The PRospEctiVE study on implaNTable cardIOverter defibrillator therapy and suddeN cardiac death in Adults with Congenital Heart Disease (PREVENTION-ACHD). Heart Rhythm 2021, 18, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Triedman, J.K.; Newburger, J.W. Trends in congenital heart disease: The next decade. Circulation 2016, 133, 2716–2733. [Google Scholar] [CrossRef] [PubMed]
- McCracken, C.; Spector, L.G.; Menk, J.S.; Knight, J.H.; Vinocur, J.M.; Thomas, A.S.; Oster, M.E.; St Louis, J.D.; Moller, J.H.; Kochilas, L. Mortality following pediatric congenital heart surgery: An analysis of the causes of death derived from the National Death Index. J. Am. Heart Assoc. 2018, 7, e010624. [Google Scholar] [CrossRef]
- Fuchs, S.R.; Smith, A.H.; Van Driest, S.L.; Crum, K.F.; Edwards, T.L.; Kannankeril, P.J. Incidence and effect of early postoperative ventricular arrhythmias after congenital heart surgery. Heart Rhythm 2019, 16, 710–716. [Google Scholar] [CrossRef]
- Hill, M.C.; Kadow, Z.A.; Long, H.; Morikawa, Y.; Martin, T.J.; Birks, E.J.; Campbell, K.S.; Nerbonne, J.; Lavine, K.; Wadhwa, L.; et al. Integrated multi-omic characterization of congenital heart disease. Nature 2022, 608, 181–191. [Google Scholar] [CrossRef]
- Maskell, L.J.; Qamar, K.; Babakr, A.A.; Hawkins, T.A.; Heads, R.J.; Budhram-Mahadeo, V.S. Essential but partially redundant roles for POU4F1/Brn-3a and POU4F2/Brn-3b transcription factors in the developing heart. Cell Death Dis. 2017, 8, e2861. [Google Scholar] [CrossRef]
- Nie, X.; Liu, X.; Wang, C.; Wu, Z.; Sun, Z.; Su, J.; Yan, R.; Peng, Y.; Yang, Y.; Wang, C.; et al. Assessment of evidence on reported non-genetic risk factors of congenital heart defects: The updated umbrella review. BMC Pregnancy Childbirth 2022, 22, 371. [Google Scholar] [CrossRef]
- Han, X.; Wang, B.; Jin, D.; Liu, K.; Wang, H.; Chen, L.; Zu, Y. Precise Dose of Folic Acid Supplementation Is Essential for Embryonic Heart Development in Zebrafish. Biology 2021, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.B.; Foo, S.Y.R.; Chen, C.K. The Role of Epigenetics in Congenital Heart Disease. Genes 2021, 12, 390. [Google Scholar] [CrossRef]
- García-Flores, E.; Rodríguez-Pérez, J.M.; Borgonio-Cuadra, V.M.; Vargas-Alarcón, G.; Calderón-Colmenero, J.; Sandoval, J.P.; García-Montes, J.A.; Espinoza-Gutiérrez, V.M.; Reyes-García, J.G.; Cazarín-Santos, B.G.; et al. DNA Methylation Levels of the TBX5 Gene Promoter Are Associated with Congenital Septal Defects in Mexican Paediatric Patients. Biology 2022, 11, 96. [Google Scholar] [CrossRef]
- Martin, L.J.; Benson, D.W. Focused Strategies for Defining the Genetic Architecture of Congenital Heart Defects. Genes 2021, 12, 827. [Google Scholar] [CrossRef]
- Diab, N.S.; Barish, S.; Dong, W.; Zhao, S.; Allington, G.; Yu, X.; Kahle, K.T.; Brueckner, M.; Jin, S.C. Molecular Genetics and Complex Inheritance of Congenital Heart Disease. Genes 2021, 12, 1020. [Google Scholar] [CrossRef]
- Choudhury, T.Z.; Garg, V. Molecular genetic mechanisms of congenital heart disease. Curr. Opin. Genet. Dev. 2022, 75, 101949. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, M.E.; Brueckner, M.; Chung, W.K.; Garg, V.; Lacro, R.V.; McGuire, A.L.; Mital, S.; Priest, J.R.; Pu, W.T.; Roberts, A.; et al. Genetic basis for congenital heart disease: Revisited: A scientific statement from the American Heart Association. Circulation 2018, 138, e653–e711. [Google Scholar] [CrossRef]
- Wang, C.; Lv, H.; Ling, X.; Li, H.; Diao, F.; Dai, J.; Du, J.; Chen, T.; Xi, Q.; Zhao, Y.; et al. Association of assisted reproductive technology, germline de novo mutations and congenital heart defects in a prospective birth cohort study. Cell Res. 2021, 31, 919–928. [Google Scholar] [CrossRef]
- Lahrouchi, N.; Postma, A.V.; Salazar, C.M.; De Laughter, D.M.; Tjong, F.; Piherová, L.; Bowling, F.Z.; Zimmerman, D.; Lodder, E.M.; Ta-Shma, A.; et al. Biallelic loss-of-function variants in PLD1 cause congenital right-sided cardiac valve defects and neonatal cardiomyopathy. J. Clin. Invest. 2021, 131, e142148. [Google Scholar] [CrossRef] [PubMed]
- Ekure, E.N.; Adeyemo, A.; Liu, H.; Sokunbi, O.; Kalu, N.; Martinez, A.F.; Owosela, B.; Tekendo-Ngongang, C.; Addissie, Y.A.; Olusegun-Joseph, A.; et al. Exome Sequencing and Congenital Heart Disease in Sub-Saharan Africa. Circ. Genom. Precis. Med. 2021, 14, e003108. [Google Scholar] [CrossRef]
- Audain, E.; Wilsdon, A.; Breckpot, J.; Izarzugaza, J.M.G.; Fitzgerald, T.W.; Kahlert, A.K.; Sifrim, A.; Wünnemann, F.; Perez-Riverol, Y.; Abdul-Khaliq, H.; et al. Integrative analysis of genomic variants reveals new associations of candidate haploinsufficient genes with congenital heart disease. PLoS Genet. 2021, 17, e1009679. [Google Scholar] [CrossRef]
- van Walree, E.S.; Dombrowsky, G.; Jansen, I.E.; Mirkov, M.U.; Zwart, R.; Ilgun, A.; Guo, D.; Clur, S.B.; Amin, A.S.; Savage, J.E.; et al. Germline variants in HEY2 functional domains lead to congenital heart defects and thoracic aortic aneurysms. Genet. Med. 2021, 23, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Li, R.; Lei, T.Y.; Wang, D.; Yang, X.; Han, J.; Pan, M.; Zhen, L.; Li, J.; Li, F.T.; et al. Compound heterozygous mutation of the ASXL3 gene causes autosomal recessive congenital heart disease. Hum. Genet. 2021, 140, 333–348. [Google Scholar] [CrossRef]
- Massadeh, S.; Albeladi, M.; Albesher, N.; Alhabshan, F.; Kampe, K.D.; Chaikhouni, F.; Kabbani, M.S.; Beetz, C.; Alaamery, M. Novel Autosomal Recessive Splice-Altering Variant in PRKD1 Is Associated with Congenital Heart Disease. Genes 2021, 12, 612. [Google Scholar] [CrossRef] [PubMed]
- Basel-Salmon, L.; Ruhrman-Shahar, N.; Barel, O.; Hagari, O.; Marek-Yagel, D.; Azulai, N.; Bazak, L.; Svirsky, R.; Reznik-Wolf, H.; Lidzbarsky, G.A.; et al. Biallelic variants in ETV2 in a family with congenital heart defects, vertebral abnormalities and preaxial polydactyly. Eur. J. Med. Genet. 2021, 64, 104124. [Google Scholar] [CrossRef]
- Zhao, L.; Jiang, W.F.; Yang, C.X.; Qiao, Q.; Xu, Y.J.; Shi, H.Y.; Qiu, X.B.; Wu, S.H.; Yang, Y.Q. SOX17 loss-of-function variation underlying familial congenital heart disease. Eur. J. Med. Genet. 2021, 64, 104211. [Google Scholar] [CrossRef]
- Hao, L.; Ma, J.; Wu, F.; Ma, X.; Qian, M.; Sheng, W.; Yan, T.; Tang, N.; Jiang, X.; Zhang, B.; et al. WDR62 variants contribute to congenital heart disease by inhibiting cardiomyocyte proliferation. Clin. Transl. Med. 2022, 12, e941. [Google Scholar] [CrossRef]
- Zhou, Y.; Bai, K.; Wang, Y.; Meng, Z.; Zhou, S.; Jiang, S.; Wang, H.; Wang, J.; Yang, M.; Wang, Q.; et al. Identification of Rare Variants in Right Ventricular Outflow Tract Obstruction Congenital Heart Disease by Whole-Exome Sequencing. Front. Cardiovasc. Med. 2022, 8, 811156. [Google Scholar] [CrossRef]
- Peng, R.; Li, B.; Chen, S.; Shi, Z.; Yu, L.; Gao, Y.; Yang, X.; Lu, L.; Wang, H. Deleterious Rare Mutations of GLI1 Dysregulate Sonic Hedgehog Signaling in Human Congenital Heart Disease. Front. Cardiovasc. Med. 2022, 9, 798033. [Google Scholar] [CrossRef]
- Delea, M.; Massara, L.S.; Espeche, L.D.; Bidondo, M.P.; Barbero, P.; Oliveri, J.; Brun, P.; Fabro, M.; Galain, M.; Fernández, C.S.; et al. Genetic Analysis Algorithm for the Study of Patients with Multiple Congenital Anomalies and Isolated Congenital Heart Disease. Genes 2022, 13, 1172. [Google Scholar] [CrossRef]
- Meerschaut, I.; Steyaert, W.; Bové, T.; François, K.; Martens, T.; De Groote, K.; De Wilde, H.; Muiño Mosquera, L.; Panzer, J.; Vandekerckhove, K.; et al. Exploring the Mutational Landscape of Isolated Congenital Heart Defects: An Exome Sequencing Study Using Cardiac DNA. Genes 2022, 13, 1214. [Google Scholar] [CrossRef] [PubMed]
- Okashah, S.; Vasudeva, D.; El Jerbi, A.; Khodjet-El-Khil, H.; Al-Shafai, M.; Syed, N.; Kambouris, M.; Udassi, S.; Saraiva, L.R.; Al-Saloos, H.; et al. Investigation of Genetic Causes in Patients with Congenital Heart Disease in Qatar: Findings from the Sidra Cardiac Registry. Genes 2022, 13, 1369. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.Y.; Xie, M.S.; Yang, C.X.; Huang, R.T.; Xue, S.; Liu, X.Y.; Xu, Y.J.; Yang, Y.Q. Identification of SOX18 as a New Gene Predisposing to Congenital Heart Disease. Diagnostics 2022, 12, 1917. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.T.; Guo, Y.H.; Yang, C.X.; Gu, J.N.; Qiu, X.B.; Shi, H.Y.; Xu, Y.J.; Xue, S.; Yang, Y.Q. SOX7 loss-of-function variation as a cause of familial congenital heart disease. Am. J. Transl. Res. 2022, 14, 1672–1684. [Google Scholar]
- Xu, Z.Q.; Chen, W.C.; Li, Y.J.; Suo, M.J.; Tian, G.X.; Sheng, W.; Huang, G.Y. PTPN11 Gene Mutations and Its Association with the Risk of Congenital Heart Disease. Dis. Markers 2022, 2022, 8290779. [Google Scholar] [CrossRef]
- Gong, L.; Wang, C.; Xie, H.; Gao, J.; Li, T.; Qi, S.; Wang, B.; Wang, J. Identification of a novel heterozygous SOX9 variant in a Chinese family with congenital heart disease. Mol. Genet. Genomic. Med. 2022, 10, e1909. [Google Scholar] [CrossRef]
- Wang, Z.; Qiao, X.H.; Xu, Y.J.; Liu, X.Y.; Huang, R.T.; Xue, S.; Qiu, H.Y.; Yang, Y.Q. SMAD1 Loss-of-Function Variant Responsible for Congenital Heart Disease. Biomed Res. Int. 2022, 2022, 9916325. [Google Scholar] [CrossRef]
- Abhinav, P.; Zhang, G.F.; Zhao, C.M.; Xu, Y.J.; Wang, J.; Yang, Y.Q. A novel KLF13 mutation underlying congenital patent ductus arteriosus and ventricular septal defect, as well as bicuspid aortic valve. Exp. Ther. Med. 2022, 23, 311. [Google Scholar] [CrossRef]
- Ke, Z.P.; Zhang, G.F.; Guo, Y.H.; Sun, Y.M.; Wang, J.; Li, N.; Qiu, X.B.; Xu, Y.J.; Yang, Y.Q. A novel PRRX1 loss-of-function variation contributing to familial atrial fibrillation and congenital patent ductus arteriosus. Genet. Mol. Biol. 2022, 45, e20210378. [Google Scholar] [CrossRef]
- Li, R.G.; Xu, Y.J.; Ye, W.G.; Li, Y.J.; Chen, H.; Qiu, X.B.; Yang, Y.Q.; Bai, D. Connexin45 (GJC1) loss-of-function mutation contributes to familial atrial fibrillation and conduction disease. Heart Rhythm 2021, 18, 684–693. [Google Scholar] [CrossRef]
- Guo, X.J.; Qiu, X.B.; Wang, J.; Guo, Y.H.; Yang, C.X.; Li, L.; Gao, R.F.; Ke, Z.P.; Di, R.M.; Sun, Y.M.; et al. PRRX1 Loss-of-Function Mutations Underlying Familial Atrial Fibrillation. J. Am. Heart Assoc. 2021, 10, e023517. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.H.; Wang, J.; Guo, X.J.; Gao, R.F.; Yang, C.X.; Li, L.; Sun, Y.M.; Qiu, X.B.; Xu, Y.J.; Yang, Y.Q. KLF13 Loss-of-Function Mutations Underlying Familial Dilated Cardiomyopathy. J. Am. Heart Assoc. 2022, 11, e027578. [Google Scholar] [CrossRef] [PubMed]
- Aboagye, E.T.; Adadey, S.M.; Esoh, K.; Jonas, M.; de Kock, C.; Amenga-Etego, L.; Awandare, G.A.; Wonkam, A. Age Estimate of GJB2-p.(Arg143Trp) Founder Variant in Hearing Impairment in Ghana, Suggests Multiple Independent Origins across Populations. Biology 2022, 11, 476. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.G.; Yue, B.; Aoyama, H.; Kim, N.K.; Cameron, J.A.; Chen, H.; Bai, D. Junctional delay, frequency, and direction-dependent uncoupling of human heterotypic Cx45/Cx43 gap junction channels. J. Mol. Cell. Cardiol. 2017, 111, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Santos-Miranda, A.; Chen, H.; Chen, R.C.; Odoko-Ishimoto, M.; Aoyama, H.; Bai, D. The amino terminal domain plays an important role in transjunctional voltage-dependent gating kinetics of Cx45 gap junctions. J. Mol. Cell. Cardiol. 2020, 143, 71–84. [Google Scholar] [CrossRef]
- Garcia-Vega, L.; O’Shaughnessy, E.M.; Albuloushi, A.; Martin, P.E. Connexins and the Epithelial Tissue Barrier: A Focus on Connexin 26. Biology 2021, 10, 59. [Google Scholar] [CrossRef]
- Zhu, Y. Gap Junction-Dependent and -Independent Functions of Connexin43 in Biology. Biology 2022, 11, 283. [Google Scholar] [CrossRef]
- Zhang, Y.; Khan, S.; Liu, Y.; Siddique, R.; Zhang, R.; Yong, V.W.; Xue, M. Gap Junctions and Hemichannels Composed of Connexins and Pannexins Mediate the Secondary Brain Injury Following Intracerebral Hemorrhage. Biology 2021, 11, 27. [Google Scholar] [CrossRef]
- Torrisi, F.; Alberghina, C.; Lo Furno, D.; Zappalà, A.; Valable, S.; Li Volti, G.; Tibullo, D.; Vicario, N.; Parenti, R. Connexin 43 and Sonic Hedgehog Pathway Interplay in Glioblastoma Cell Proliferation and Migration. Biology 2021, 10, 767. [Google Scholar] [CrossRef]
- Li, X. Seeing Is Believing: Gap Junctions in Motion. Biology 2021, 10, 494. [Google Scholar] [CrossRef]
- Peng, B.; Xu, C.; Wang, S.; Zhang, Y.; Li, W. The Role of Connexin Hemichannels in Inflammatory Diseases. Biology 2022, 11, 237. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.H.; Yang, Y.Q. Atrial Fibrillation: Focus on Myocardial Connexins and Gap Junctions. Biology 2022, 11, 489. [Google Scholar] [CrossRef] [PubMed]
- Severs, N.J.; Bruce, A.F.; Dupont, E.; Rothery, S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc. Res. 2008, 80, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Kumai, M.; Nishii, K.; Nakamura, K.; Takeda, N.; Suzuki, M.; Shibata, Y. Loss of connexin45 causes a cushion defect in early cardiogenesis. Development 2000, 127, 3501–3512. [Google Scholar] [CrossRef]
- Krüger, O.; Plum, A.; Kim, J.S.; Winterhager, E.; Maxeiner, S.; Hallas, G.; Kirchhoff, S.; Traub, O.; Lamers, W.H.; Willecke, K. Defective vascular development in connexin 45-deficient mice. Development 2000, 127, 4179–4193. [Google Scholar] [CrossRef] [PubMed]
- Nishii, K.; Seki, A.; Kumai, M.; Morimoto, S.; Miwa, T.; Hagiwara, N.; Shibata, Y.; Kobayashi, Y. Connexin45 contributes to global cardiovascular development by establishing myocardial impulse propagation. Mech. Dev. 2016, 140, 41–52. [Google Scholar] [CrossRef]
- Frank, M.; Wirth, A.; Andrié, R.P.; Kreuzberg, M.M.; Dobrowolski, R.; Seifert, G.; Offermanns, S.; Nickenig, G.; Willecke, K.; Schrickel, J.W. Connexin45 provides optimal atrioventricular nodal conduction in the adult mouse heart. Circ. Res. 2012, 111, 1528–1538. [Google Scholar] [CrossRef]
- Krüger, O.; Maxeiner, S.; Kim, J.S.; van Rijen, H.V.; de Bakker, J.M.; Eckardt, D.; Tiemann, K.; Lewalter, T.; Ghanem, A.; Lüderitz, B.; et al. Cardiac morphogenetic defects and conduction abnormalities in mice homozygously deficient for connexin40 and heterozygously deficient for connexin45. J. Mol. Cell. Cardiol. 2006, 41, 787–797. [Google Scholar] [CrossRef]
- Seki, A.; Ishikawa, T.; Daumy, X.; Mishima, H.; Barc, J.; Sasaki, R.; Nishii, K.; Saito, K.; Urano, M.; Ohno, S.; et al. Progressive atrial conduction defects associated with bone malformation caused by a connexin-45 mutation. J. Am. Coll. Cardiol. 2017, 70, 358–370. [Google Scholar] [CrossRef]
- Gu, H.; Smith, F.C.; Taffet, S.M.; Delmar, M. High incidence of cardiac malformations in connexin40-deficient mice. Circ. Res. 2003, 93, 201–206. [Google Scholar] [CrossRef]
- Reaume, A.G.; de Sousa, P.A.; Kulkarni, S.; Langille, B.L.; Zhu, D.; Davies, T.C.; Juneja, S.C.; Kidder, G.M.; Rossant, J. Cardiac malformation in neonatal mice lacking connexin43. Science 1995, 267, 1831–1834. [Google Scholar] [CrossRef] [PubMed]
- Britz-Cunningham, S.H.; Shah, M.M.; Zuppan, C.W.; Fletcher, W.H. Mutations of the Connexin43 gap-junction gene in patients with heart malformations and defects of laterality. N. Engl. J. Med. 1995, 332, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- Molica, F.; Meens, M.J.; Morel, S.; Kwak, B.R. Mutations in cardiovascular connexin genes. Biol. Cell 2014, 106, 269–293. [Google Scholar] [CrossRef] [PubMed]
- Lübkemeier, I.; Bosen, F.; Kim, J.S.; Sasse, P.; Malan, D.; Fleischmann, B.K.; Willecke, K. Human Connexin43E42K mutation from a sudden infant death victim leads to impaired ventricular activation and neonatal death in mice. Circ. Cardiovasc. Genet. 2015, 8, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.A.; van Veen, T.A.; de Bakker, J.M.; van Rijen, H.V. Cardiac connexins and impulse propagation. J. Mol. Cell. Cardiol. 2010, 48, 76–82. [Google Scholar] [CrossRef]
- Donahue, J.K. Connexin gene transfer preserves conduction velocity and prevents atrial fibrillation. Circulation 2012, 125, 216–225. [Google Scholar] [CrossRef]





| Family Member Information | Cardiac Phenotypes | |||
|---|---|---|---|---|
| Identity (Family 1) | Sex | Years of Age | Cardiac Developmental Deformations | Arrhythmias |
| I-1 II-1 II-6 III-1 III-4 III-7 III-9 III-17 IV-1 IV-2 IV-4 IV-9 IV-14 | M M F M F M M M F M M F M | 62 * 57 * 67 47 45 38 35 41 1 * 23 22 12 15 | ASD, VSD ASD, VSD ASD ASD ASD ASD ASD ASD ASD, TOF ASD, VSD ASD ASD ASD | AVB (third-degree), SB AVB (third-degree), SB AVB (third-degree), SB AVB (third-degree) AVB (third-degree) AVB (second-degree) AVB (second-degree) AVB (second-degree) AVB (first-degree) AVB (first-degree) AVB (second-degree) AVB (first-degree) AVB (first-degree) |
| Marker | Scores of the Two-Point Logarithm of Odds for the Markers at θ = | ||||||
|---|---|---|---|---|---|---|---|
| 0.00 | 0.01 | 0.05 | 0.10 | 0.20 | 0.30 | 0.40 | |
| D17S1802 | (−∞) | 1.8523 | 2.3017 | 2.2781 | 1.8844 | 1.3013 | 0.6230 |
| D17S951 | (−∞) | 0.0723 | 0.6292 | 0.7465 | 0.6597 | 0.4246 | 0.1479 |
| D17S1861 | (−∞) | 1.5557 | 2.0228 | 2.0228 | 1.6803 | 1.1552 | 0.5438 |
| D17S791 | 4.2141 | 4.1399 | 3.8356 | 3.4376 | 2.5823 | 1.6632 | 0.7496 |
| D17S1868 | 3.6124 | 3.5427 | 3.2597 | 2.8978 | 2.1540 | 1.3958 | 0.6421 |
| D17S1795 | (−∞) | -0.4400 | 0.7442 | 1.0686 | 1.0783 | 0.7872 | 0.3677 |
| D17S1869 | (−∞) | 1.5513 | 2.0007 | 1.9777 | 1.5901 | 1.0332 | 0.4347 |
| D17S956 | (−∞) | 0.6657 | 1.1867 | 1.2570 | 1.0680 | 0.7168 | 0.3063 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.-J.; Wang, J.; Ye, W.G.; Liu, X.-Y.; Li, L.; Qiu, X.-B.; Chen, H.; Xu, Y.-J.; Yang, Y.-Q.; Bai, D.; et al. Discovery of GJC1 (Cx45) as a New Gene Underlying Congenital Heart Disease and Arrhythmias. Biology 2023, 12, 346. https://doi.org/10.3390/biology12030346
Li Y-J, Wang J, Ye WG, Liu X-Y, Li L, Qiu X-B, Chen H, Xu Y-J, Yang Y-Q, Bai D, et al. Discovery of GJC1 (Cx45) as a New Gene Underlying Congenital Heart Disease and Arrhythmias. Biology. 2023; 12(3):346. https://doi.org/10.3390/biology12030346
Chicago/Turabian StyleLi, Yan-Jie, Juan Wang, Willy G. Ye, Xing-Yuan Liu, Li Li, Xing-Biao Qiu, Honghong Chen, Ying-Jia Xu, Yi-Qing Yang, Donglin Bai, and et al. 2023. "Discovery of GJC1 (Cx45) as a New Gene Underlying Congenital Heart Disease and Arrhythmias" Biology 12, no. 3: 346. https://doi.org/10.3390/biology12030346
APA StyleLi, Y.-J., Wang, J., Ye, W. G., Liu, X.-Y., Li, L., Qiu, X.-B., Chen, H., Xu, Y.-J., Yang, Y.-Q., Bai, D., & Huang, R.-T. (2023). Discovery of GJC1 (Cx45) as a New Gene Underlying Congenital Heart Disease and Arrhythmias. Biology, 12(3), 346. https://doi.org/10.3390/biology12030346